

Abstract
Hydrophobic bile acids like deoxycholic acid (DCA), which cause oxidative DNA damage and activate NF-κB in Barrett's metaplasia, might contribute to carcinogenesis in Barrett's esophagus. We have explored mechanisms whereby ursodeoxycholic acid (UDCA, a hydrophilic bile acid) protects against DCA-induced injury in vivo in patients and in vitro using nonneoplastic, telomerase-immortalized Barrett's cell lines. We took biopsies of Barrett's esophagus from 21 patients before and after esophageal perfusion with DCA (250 μM) at baseline and after 8 wk of oral UDCA treatment. DNA damage was assessed by phospho-H2AX expression, neutral CometAssay, and phospho-H2AX nuclear foci formation. Quantitative PCR was performed for antioxidants including catalase and GPX1. Nrf2, catalase, and GPX1 were knocked down with siRNAs. Reporter assays were performed using a plasmid construct containing antioxidant responsive element. In patients, baseline esophageal perfusion with DCA significantly increased phospho-H2AX and phospho-p65 in Barrett's metaplasia. Oral UDCA increased GPX1 and catalase levels in Barrett's metaplasia and prevented DCA perfusion from inducing DNA damage and NF-κB activation. In cells, DCA-induced DNA damage and NF-κB activation was prevented by 24-h pretreatment with UDCA, but not by mixing UDCA with DCA. UDCA activated Nrf2 signaling to increase GPX1 and catalase expression, and protective effects of UDCA pretreatment were blocked by siRNA knockdown of these antioxidants. UDCA increases expression of antioxidants that prevent toxic bile acids from causing DNA damage and NF-κB activation in Barrett's metaplasia. Elucidation of this molecular pathway for UDCA protection provides rationale for clinical trials on UDCA for chemoprevention in Barrett's esophagus.
Keywords: bile acids, chemoprevention, esophageal adenocarcinoma, catalase, GPX1
the frequency of adenocarcinoma of the esophagus has increased more than sevenfold over the past several decades in the United States (25). Major risk factors for this deadly cancer are gastroesophageal reflux disease (GERD) and its complication, Barrett's esophagus, the condition in which metaplastic columnar epithelium predisposed to malignancy replaces squamous epithelium of the distal esophagus (29). It has been estimated that 2–7% of adults in Western countries have Barrett's esophagus. Clearly, a safe and effective chemopreventive agent for these patients would be highly desirable.
The cancer-prevention strategy currently recommended for Barrett's esophagus involves endoscopic surveillance for dysplasia and treatment of the underlying GERD with proton pump inhibitors (PPIs) (30). The benefit of surveillance for these patients has never been established, and one recent, high-quality study found that endoscopic surveillance was not associated with a significant reduction in the risk of death from esophageal adenocarcinoma (5). PPIs are highly effective at decreasing gastric acid production and healing GERD. Despite the widespread use of PPIs, however, the incidence of esophageal adenocarcinoma continues to climb (25), suggesting that factors other than refluxed hydrochloric acid contribute to the development of this cancer. There are reasons to suspect that bile acids might have an important role in carcinogenesis in Barrett's esophagus (22).
The gastroesophageal reflux of bile acids occurs frequently in patients with Barrett's esophagus (8, 13, 24, 32). Esophageal monitoring studies using a spectrophotometric system to detect bile reflux have demonstrated bile in the esophagus of normal subjects for <2% of the day, whereas patients with complicated Barrett's have bile in the esophagus for a median 46% of the day (range 20.8–77.5%) (32). Studies in which material aspirated from the esophagus is analyzed by high-performance liquid chromatography confirm that bile acid reflux is common in Barrett's patients (8, 13, 24). For example, Nehra et al. (24) found refluxed bile acids in concentrations >200 μM in 50% of Barrett's patients, some of whom had refluxed deoxycholic acid (DCA, a toxic, hydrophobic bile acid) in concentrations as high as 282 μM.
In an earlier report, we showed that DCA exposure caused Barrett's cells to produce reactive oxygen species (ROS) that induced DNA damage detectable by phospho-H2AX expression (11). In those same cells, DCA also activated nuclear factor (NF)-κB, which prevented the apoptosis often triggered by severe DNA damage. Furthermore, we documented DNA damage and NF-κB activation in biopsies of Barrett's metaplasia from five patients whose esophagus was perfused with DCA (250 μM) for only 5 min. These studies showed that DCA induces ROS production with genotoxic injury while simultaneously activating NF-κB to prevent apoptosis in Barrett's cells. Thus refluxed hydrophobic bile acids might contribute to carcinogenesis in Barrett's esophagus.
Hydrophilic bile acids such as ursodeoxycholic acid (UDCA) can protect against injuries caused by hydrophobic bile acids and might protect against cancers whose development can involve exposure to bile acids (1, 9). In HeLa cells exposed to bile, DNA damage declines as the concentration of UDCA increases (21). UDCA prevents colon cancer in animal models (33) and, in a rat model of Barrett's esophagus, rats treated with a combination of UDCA and aspirin develop fewer esophageal adenocarcinomas (26). UDCA is used to treat chronic liver diseases (18), and UDCA appears to prevent colon cancer in patients with primary sclerosing cholangitis and inflammatory bowel disease (27). In earlier studies, furthermore, we found that UDCA did not damage DNA or activate NF-κB in Barrett's metaplasia (11). These data suggest a potential chemopreventive role for UDCA in Barrett's esophagus. In nonneoplastic Barrett's cell lines and in patients with Barrett's esophagus, we have now performed translational studies to explore mechanisms whereby UDCA protects against genotoxic effects of hydrophobic bile acids.
METHODS
Patients.
This study was approved by the institutional review board of the Dallas VA Medical Center and is registered on www.clinicaltrials.gov under the ClinicalTrials.gov number NCT00858858. Patients with Barrett's esophagus (specialized intestinal metaplasia involving >2 cm of the distal esophagus) without dysplasia who were scheduled for elective endoscopy were invited to participate (Fig. 1). All patients were treated with PPIs for at least 4 wk before the first endoscopy and all were maintained on omeprazole 20 mg bid for the duration of the study. Aspirin and nonsteroidal anti-inflammatory medications were withheld for at least 8 days before each endoscopy. During endoscopy, six biopsies of Barrett's metaplasia were taken by use of jumbo biopsy forceps (Olympus FB-50K-1) before and after perfusion of the distal esophagus with either 10 ml of 250 μM DCA or 10 ml of 250 μM UDCA over 5 min as described previously (11). A sealed-envelope strategy was used to randomly assign patients to receive either DCA or UDCA perfusion during the first endoscopy. One year later, patients underwent a second endoscopy, during which the esophagus was perfused with the bile acid not used in the first endoscopy; biopsies were obtained as described above. After the second endoscopy, patients were treated with oral UDCA (10 mg/kg) for 8 wk, after which they returned for a final endoscopy, during which the esophagus was perfused with 250 μM DCA and biopsy specimens were taken as described above.
Fig. 1.
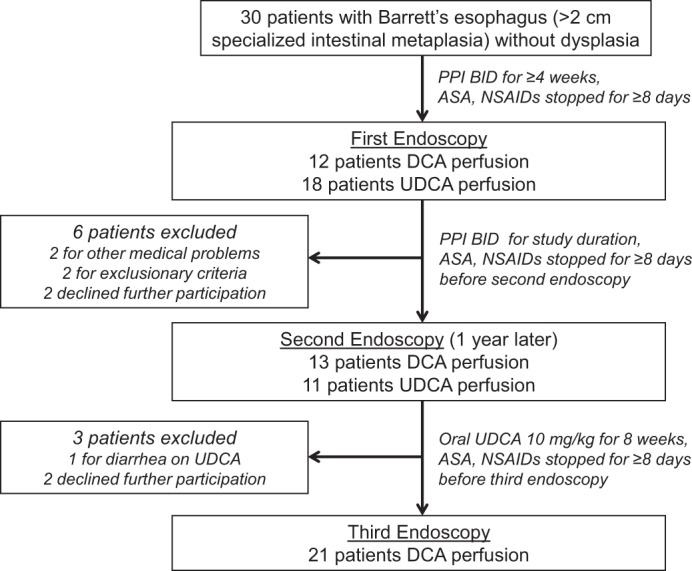
Flow diagram of the clinical trial. DCA, deoxycholic acid; UDCA, ursodeoxycholic acid.
Cell lines.
We used two nonneoplastic, telomerase-immortalized Barrett's epithelial cell lines (BAR-T, BAR-10T) created from endoscopic biopsy specimens of nondysplastic Barrett's esophagus (14, 35). The Barrett's cells were cocultured with a fibroblast feeder layer and maintained in growth medium as previously described (14, 35). For individual experiments, the Barrett's cells were seeded into collagen IV-coated wells (BD Biosciences, San Jose, CA) and maintained in growth medium. All cells were maintained at 37°C in a 5% CO2 incubator. We selected to use the BAR-T line for all experiments (unless otherwise indicated) because this cell line has been extensively characterized by our laboratory (11, 14, 34, 35).
Bile salt exposure.
For individual experiments, Barrett's cells were treated with experimental medium (pH 7.2) containing DCA alone (125 or 250 μM, Sigma, St. Louis, MO), experimental medium (pH 7.2) containing DCA mixed with UDCA (125 or 250 μM, Calbiochem, San Diego, CA), or control medium (pH 7.2) without bile acids. We selected to use DCA, which has been measured in the refluxed gastric juice of Barrett's patients in concentrations as high as 282 μM, to explore mechanisms whereby UDCA protects against the genotoxic effects of a physiologically relevant, toxic, hydrophobic bile acid (24). Media were added for 5 min to equally seeded wells of subconfluent cells, then removed and replaced with growth medium. Cells were pretreated with UDCA (300 μM) for 24 h prior to exposure to 250 μM DCA for 5 min. Cells were also treated with UDCA (300 μM) for times ranging from 0.5–24 h.
Detection of intracellular ROS.
ROS were detected by using the OxiSelect Intracellular ROS assay kit (Cell Biolabs, San Diego, CA) per manufacturer's instructions. Equally seeded wells of cells were pretreated with or without 300 μM UDCA for 24 h and then washed twice with wash buffer and incubated with 100 μl of 1× DCFH-DA for 45 min at 37°C while protected from light. Cells were then exposed to medium containing 250 μM DCA for 5 min, after which the assay was terminated by adding 100 μl of 2× cell lysis buffer. The lysates were analyzed by a fluorometric plate reader, and fluorescence intensity was immediately measured at 480/530 nm by using the POLARstar Omega software (BMG Labtech, Cary, NC). A higher florescence reading indicates greater production of intracellular ROS.
Protein extraction and immunoblotting.
For Barrett's epithelial cell lines, total protein was extracted using 200 μl of 1× cell lysis buffer supplemented with 0.5 mM phenylmethylsulfonyl fluoride (PMSF) according to manufacturer's instructions (Cell Signaling Technology, Danvers, MA). Nuclear extracts were isolated from cells using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Fisher, Rockford, IL) per manufacturer's instructions. For the biopsy specimens, total protein was extracted in lysis buffer containing 150 mmol/l NaCl, 1% NP-40, 1% deoxycholate, 20 mmol/l Tris·HCl (pH 7.5), 1 mmol/l EDTA, 1 mmol/l EGTA, 2.5 mmol/l sodium pyrophosphate, 1 mmol/l β-glycerophosphate, 1 mmol/l sodium orthovanadate, 1 μg/ml leupeptin, 1 or 0.1 mmol/l phenylmethylsulfonyl fluoride, and one Protease Inhibitor Cocktail Tablet per 50 ml lysis buffer (Roche Applied Science, Indianapolis, IN). Protein concentrations were determined by use of the BCA-200 Protein Assay kit (Pierce, Rockford, IL). Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and incubated with primary antibodies (Table 1) overnight at 4°C. Secondary antibody was either goat anti-rabbit or horse anti-mouse IgG conjugated with horseradish peroxidase (Cell Signaling Technology), and chemiluminescence was determined by using the ECL detection system (Pierce, Rockford, IL). The membranes were stripped and reprobed with mouse anti-β-tubulin (Sigma, St. Louis, MO) as a loading control; TFIID (Santa Cruz, Santa Cruz, CA) was used as a loading control for nuclear fractions. Proteins were quantified by densitometry (ImageQuant Version 5, Amersham Biosciences), and the relative quantity of protein with respect to the loading control was calculated.
Table 1.
Antibodies used
| Antibody | Source Information (catalog no., vendor) | Dosage | Usage |
|---|---|---|---|
| Catalase | Ab52477, Abcam | 1:800 dilution | WB |
| GPX1 | Ab22604, Abcam | 1:800 dilution | WB |
| p-Nrf2 | Ab76026, Abcam | 1:2,000 dilution | WB |
| Total-Nrf2 | Ab62352, Abcam | 1:800 dilution | WB |
| p-H2AX | no. 2577, Cell Signaling | 1:100 dilution, | IHC |
| 1:1,000 dilution | WB | ||
| 1:100 dilution | IF | ||
| p65 | no. 4764, Cell Signaling | 1:1,000 dilution | WB |
| p-p65 | no. 3033, Cell Signaling | 1:1,000 dilution | WB |
| TFIID | sc-273, Santa Cruz | 1:3,000 | WB |
| β-Tubulin | T-5293, Sigma | 1:3,000 | WB |
WB, Western blot; IHC, immunohistochemistry; IF, immunofluorescence.
qPCR.
Quantitative reverse transcription polymerase chain reaction (qPCR) was performed for catalase, glutathione peroxidase 1 (GPX1), and cyclophilin mRNAs. Total RNAs were isolated by using the RNeasy Mini kit (Qiagen, Valencia, CA) per manufacturer's instructions and quantitated by using the Nanophotometer (IMPLEN, Westlake Village, CA). Reverse transcription was performed using QuantiTect Reverse Transcription kit (Qiagen) per manufacturer's instructions. The primer sequences (Table 2) were designed with Primer Express (Applied BioSystems, Foster City, CA) and manufactured by Integrated DNA Technologies (Coralville, IA). Real-time PCRs for mRNA expression of the catalase or GPX1 were carried out with the StepOnePlus Real-Time PCR System and SYBR Green mix (Applied Biosystems) as previously described (4). The reference gene cyclophilin served as an internal control. The relative quantity of mRNA was normalized to cyclophilin.
Table 2.
Oligonucleotide primers
| Primer | Sequence (5′ to 3′) | Location | Use |
|---|---|---|---|
| Catalase-5′ | CAT TCG ATC TCA CCA AGG TTT GGC C | sense | qPCR |
| Catalase-3′ | AGC AGG TAG GGA CAG TTC ACA GG | antisense | qPCR |
| GPX1-5′ | CCC GTC CAA CCA GTT TGG | sense | qPCR |
| GPX1-3′ | TGA GGG AAT TCA GAA TCT CTT CGT | antisense | qPCR |
| Cyclophillin-5′ | CCC ACC GTG TTC TTC GAC AT | sense | qPCR |
| Cyclophillin-3′ | CCA GTG CTC AGA GCA CGA AA | antisense | qPCR |
siRNA inhibition of Nrf2, catalase, and GPX1.
BAR-T cells were equally plated in six-well tissue culture plates. After 24 h, the cells were transfected using Lipofectamine LTX (Invitrogen, Carlsbad, CA) with 75 pmol of the SMARTpool ON-TARGETplus NFE2L2, catalase, or GPX1 siRNA (Thermo Scientific, Waltham, MA), for 5 h per the manufacturer's instructions. As a control, cells were transfected with ON-TARGETplus Nontargeting siRNA no. 1 (Thermo Scientific). After transfection, the medium was removed and replaced with growth medium overnight. For the NF-E2-related factor 2 (Nrf2) siRNA experiments, cells were treated the next day with 300 μM UDCA or control medium for 24 h. For the GPX1 and catalase siRNA experiments, cells were treated the next day with and without 24 h of pretreatment with 300 μM UDCA and then exposed to 250 μM DCA for 5 min.
Reporter assays.
For reporter assays, we used a plasmid construct containing the antioxidant-responsive element (ARE; SA Biosciences, Valencia, CA) or four repeats of the NF-κB consensus binding site (generous gift of Zhiping Liu, University of Texas Southwestern) upstream of a luciferase reporter; the Renilla reporter pRL-TK (Promega, Madison, WI) plasmid was used to equalize for transfection efficiency. Cells were transfected for 5 h with 500 ng/well of either the ARE or the NF-κB reporter constructs along with 25 ng pRL-TK by using Lipofectamine LTX (Invitrogen) per manufacturer's instructions. After transfection, the medium was removed and replaced with growth medium overnight. For the NF-κB reporter assays, cells were pretreated with 300 μM UDCA for 24 h, and then exposed to 250 μM DCA for 5 min. At 24 h after the DCA exposure, cell extracts were assayed for NF-κB luciferase activities using a Dual Luciferase kit (Promega, Fitchburg, WI). After transfection of the ARE reporter, cells were treated with 300 μM UDCA for 6 h, and luciferase activities were determined using the Dual Luciferase kit (Promega). Data are expressed as relative light units for firefly luciferase normalized to Renilla. A higher luciferase reading indicates greater activity of the NF-κB or the ARE reporter construct.
Neutral single-cell gel electrophoresis (CometAssay).
DNA damage was detected using the neutral CometAssay (Cell Biolabs), which mainly detects DNA double-strand breaks, as previously described (34). Slides were stained with Green DNA Dye and comet “tails” were visualized with fluorescence microscope (Nikon, Melville, NY), respectively. The comet extent tail moment values were quantitated from slides by using the CometScore software (TriTek, Sumerduck, VA) from a minimum of 50 individual cells. BAR-T cells treated with 200 μM hydrogen peroxide (H2O2) for 10 min served as positive controls.
Phospho-H2AX immunofluorescence.
Cells were equally seeded onto glass coverslips and exposed to 300 μM UDCA or control medium for 24 h, after which the medium was removed and the cells were washed in PBS followed by exposure to 250 μM DCA or control medium for 5 min. Cells were then washed with cold PBS and immediately fixed with cold methanol (−20°C) for 10 min followed by incubation in 3% bovine serum albumin in PBS for 45 min to block the nonspecific binding sites. Primary antibody to phospho-H2AX was added for 1 h at room temperature. Secondary antibodies were conjugated with Alexa Fluor 488 and used at 1:1,000 dilutions of goat anti-rabbit for phospho-H2AX. The slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for analysis. BAR-T cells treated with 200 μM H2O2 for 10 min served as positive controls for phospho-H2AX.
Power calculations.
For the patient studies, the primary outcome was to determine increases in the levels of p-H2AX and p-p65 after perfusion with DCA. In our pilot study on this issue (11), we used densitometry to quantitate data on DCA perfusion for five study subjects (Table 3).
Table 3.
Increases in the levels of p-H2AX and p-p65 after perfusion with deoxycholic acid
| Effect |
|||
|---|---|---|---|
| Preperfusion | Postperfusion | Size, d | |
| p-H2AX | 0.38 ± 0.13 | 1.07 ± 0.61 | 1.56 |
| p-p65 | 0.62 ± 0.33 | 1.41 ± 0.58 | 1.67 |
Values are means ± SD in relative intensity units.
For these demonstrated effect sizes, we estimated that only seven total subjects would be required for paired-samples t-tests with a study alpha of 0.05 and a beta of 0.20. However, to account for patient dropout and the unknown effect sizes for a number of the other biochemical determinations, we elected to recruit 30 total patients into the trial.
Data analyses.
All data were collected from at least two independent experiments. Quantitative data are expressed as means ± SE. Statistical analyses were performed using a paired or unpaired Student's t-test with the Instat for Windows or Prism statistical software package (GraphPad Software, San Diego, CA). For multiple comparisons, an ANOVA and the Student-Newman-Keuls multiple-comparisons test was performed with the Instat for Windows statistical software package (GraphPad). P values ≤0.05 were considered significant for all analyses.
RESULTS
Clinical characteristics of study subjects.
Thirty patients were enrolled and had the first endoscopy. During the course of the study, two experienced medical problems precluding further participation, one developed diarrhea on UDCA, four declined further participation, and two developed exclusionary criteria (Fig. 1). Thus 21 patients (19 male; mean age 58 ± 2.3 yr, range 38–82 yr; 20 Caucasian, 1 African-American) completed all phases of the study, and all 21 patients were included in the molecular analyses described below.
In patients with Barrett's esophagus, oral UDCA prevents DCA-induced DNA damage and activation of NF-κB subunit p65.
Esophageal perfusion with DCA for 5 min caused a significant increase in phospho-H2AX and phospho-p65 (relative to total p65) in Barrett's metaplasia (Fig. 2, A and D). In contrast, esophageal perfusion with UDCA had no significant effect on phospho-H2AX and phospho-p65 levels in those same patients (Fig. 2, B and D). The DCA-induced increase in phospho-p65/total p65 was due to a significant increase in phospho-p65 levels; total p65 levels did not change significantly (data not shown). UDCA perfusion did not significantly alter either phospho-p65 or total p65 levels (data not shown).
Fig. 2.

Oral UDCA treatment prevents DCA-induced phosphorylation of H2AX and p65 in Barrett's esophagus. A: DCA perfusion of the esophagus increases phospho-H2AX and phospho/total p65 expression. B: UDCA perfusion does not alter phospho-H2AX and phospho/total p65 expression. C: 8 wk of oral UDCA treatment prevents the increase in phospho-H2AX and phospho-p65/total p65 levels following DCA perfusion. Horizontal dotted line beside each column represents the mean for 21 patients. D: representative Western blots from Barrett's mucosa of 2 patients demonstrating phospho-H2AX, phospho-p65, and total p65 before (pre) and after (post) esophageal perfusion with DCA, UDCA, or DCA after 8 wk of oral UDCA.
In contrast to the experiments performed before UDCA treatment, esophageal perfusion with DCA did not increase phospho-H2AX or phospho-p65/total p65 expression in biopsy specimens of Barrett's metaplasia taken after patients were treated with UDCA for 8 wk (Fig. 2, C and D). This demonstrates that oral treatment with UDCA protects against bile acid-induced DNA damage and NF-κB activation in the metaplastic mucosa of Barrett's patients.
Mixing DCA and UDCA together does not prevent DNA damage or activation of p65 in Barrett's cells.
We treated BAR-T and BAR-10T cells with DCA alone (at concentrations of 250 or 125 μM), or with a mixture of DCA and UDCA (both at concentrations of 250 or 125 μM, resulting in total bile acid concentrations of 250 and 500 μM), and determined effects on phospho-H2AX and phospho-p65 expression. In both cell lines, there were no apparent differences in phospho-H2AX and phospho-p65 expression between DCA treatment alone and DCA mixed with UDCA at either dose (Fig. 3A). These findings suggest that UDCA does not interfere directly with the ability of DCA to cause DNA damage and activate NF-κB in Barrett's cells.
Fig. 3.

Pretreatment with UDCA prevents DCA-induced phosphorylation of H2AX and p65. Representative Western blots for phospho-H2AX, phospho-p65, and total p65 in (A) BAR-T and BAR-10T cells exposed to DCA alone at concentrations of 250 and 125 μM, or to the same concentrations of DCA mixed with UDCA at concentrations of 250 and 125 μM, and (B) BAR-T and BAR-10T cells exposed to 250 μM DCA with and without 24-h pretreatment (Pre-Tx) with UDCA. Numbers represent the relative quantity of protein with respect to the loading control. C: representative CometAssay of BAR-T cells exposed to DCA with and without 24-h pretreatment with UDCA at low (×4) and high (×10) magnification; cells treated with hydrogen peroxide served as a positive control. Mean comet tail moment +SE from a minimum of 50 individual BAR-T cells. (***P ≤ 0.001 compared with control; +++P ≤ 0.001 compared with DCA treatment alone). D: representative CometAssays of BAR-10T cells exposed to DCA with and without 24-h pretreatment with UDCA at low (×4) and high (×10) magnification. Mean comet tail moment +SE from a minimum of 50 individual BAR-10T cells. (**P ≤ 0.01 compared with control; ++P ≤ 0.01 compared with DCA treatment alone). E: representative experiments of phospho-H2AX nuclear foci formation in BAR-T and BAR-10T cells exposed to DCA with and without 24-h pretreatment with UDCA; DAPI shows the total number of cell nuclei in the same field. Cells treated with hydrogen peroxide served as a positive control.
UDCA pretreatment prevents DCA-induced DNA damage and activation of p65 in Barrett's cells.
We pretreated Barrett's cells with UDCA for 24 h, then exposed them to 250 μM DCA. Exposure of untreated cells to DCA increased phospho-H2AX and phospho-p65 expression (Fig. 3B). In contrast, 24-h pretreatment with UDCA decreased phospho-H2AX and phospho-p65 expression after DCA exposure in both cell lines (Fig. 3B). We also assessed DNA damage after DCA exposure using the CometAssay at neutral conditions, which detects double strand breaks (DSBs). DCA induced a significant increase in DNA damage (detected in comet “tails”), which was significantly reduced by pretreatment with UDCA (Fig. 3, C and D). To confirm DSBs, we used immunofluorescence to detect nuclear foci of phospho-H2AX (which are more specific for DSBs). DCA induced nuclear foci of phospho-H2AX, which were eliminated by pretreating the cells with UDCA (Fig. 3E). These findings demonstrate that DCA causes DNA damage, including DSBs, and causes activation of the NF-κB pathway, both of which can be prevented by pretreating the cells with UDCA.
UDCA pretreatment blocks ROS production and reduces NF-κB transcriptional activity induced by DCA in Barrett's cells.
In earlier experiments, we showed that DCA induced production of ROS, and that those ROS were responsible for DNA damage and NF-κB activation in Barrett's epithelial cells (11). Now, we determined effects of UDCA pretreatment on DCA-induced ROS production and on NF-κB transcriptional activity. DCA significantly increased production of ROS in both Barrett's cell lines, and this increase was blocked by pretreatment with UDCA (Fig. 4A). As expected, DCA significantly increased the activity of the NF-κB reporter, which also decreased when cells were pretreated with UDCA (Fig. 4B).
Fig. 4.

Pretreatment with UDCA reduces fluorescence from the intracellular reactive oxygen species (ROS) production assay (A) and luciferase production from the NF-κB reporter (B) in BAR-T and BAR-10T cells exposed to DCA. Data are means + SE of 2 separate experiments. *P ≤ 0.05 compared with control; **P ≤ 0.01 compared with control; ***P ≤ 0.001 compared with corresponding control; ++P ≤ 0.01 compared with DCA treatment alone; +++P ≤ 0.001 compared with DCA treatment alone.
UDCA treatment increases mRNA and protein expression of antioxidants GPX1 and catalase in Barrett's cells.
Since UDCA pretreatment suppressed ROS production induced by DCA, we next determined whether UDCA pretreatment increased expression of antioxidants including GPX1, catalase, superoxide dismutase (SOD)1 and SOD2. Treatment of both Barrett's cell lines with UDCA for 24 h increased expression of GPX1 and catalase, but not SOD1 or SOD2 (Fig. 5A). We then determined whether the increase in antioxidant protein levels was associated with increased mRNA expression. By 6 h of UDCA treatment, we found that both cell lines exhibited significant elevations in expression of GPX1 and catalase mRNAs by qPCR (Fig. 5B).
Fig. 5.

UDCA treatment increases protein and mRNA expression of the antioxidants GPX1 and catalase in Barrett's cells. A: representative experiments of Western blotting for GPX1, catalase, SOD1, and SOD2 protein. Numbers represent the relative quantity of protein with respect to the loading control. B: representative experiments of qPCR for GPX1 and catalase mRNA. *P ≤ 0.05 compared with control; **P ≤ 0.01 compared with control.
UDCA treatment increases activity of ARE reporter, increases expression of phospho-Nrf2, and causes nuclear translocation of phospho-Nrf2 in Barrett's cells.
Antioxidant proteins are expressed through a coordinated response regulated by ARE in the promoters of target genes including GPX1 and catalase (16, 28). We determined whether UDCA activates the ARE reporter in Barrett's cells. After transient transfection with ARE reporter, we treated cells with UDCA for 6 h, which significantly increased ARE reporter activity (Fig. 6A).
Fig. 6.

A: UDCA treatment increases luciferase production from the antioxidant-responsive element (ARE) reporter in BAR-T and BAR-10T cells. Data are means + SE of 2 separate experiments. **P ≤ 0.01 compared with control; ***P ≤ 0.001 compared with corresponding control. B: UDCA treatment increases phospho-Nrf2 expression and causes its nuclear translocation in BAR-T cells. Representative Western blots demonstrating increased cytoplasmic and nuclear levels of phospho-Nrf2 accompanied by decreased cytoplasmic and increased nuclear levels of total Nrf2 by 30 min of UDCA treatment. C: Nrf2 siRNA inhibits the UDCA-induced increase in protein expression of GPX1 and catalase in BAR-T cells. Representative Western blots of phospho-Nrf2, total Nrf2, catalase, and GPX1 in BAR-T cells containing either control or Nrf2 siRNA.
Most known ARE-responsive, target-cytoprotective genes are transcriptionally regulated by Nrf2 (16). Activation and phosphorylation of Nrf2 leads to its nuclear translocation. As shown in Fig. 6B, 30 min of UDCA treatment increased cytoplasmic and nuclear expression of phospho-Nrf2, accompanied by decreased cytoplasmic and increased nuclear expression of total Nrf2 in BAR-T cells. These data suggest that the Nrf2 pathway is activated by UDCA in Barrett's cells.
Inhibition of the Nrf2 pathway suppresses the effect of UDCA on GPX1 and catalase protein expression in Barrett's cells.
To explore whether Nrf2 activation contributes to UDCA-induced increases in GPX1 and catalase protein expression, we inhibited Nrf2 using a specific siRNA and then treated the BAR-T cells with UDCA for 24 h; Western blotting was also performed for Nrf2 to determine the efficiency of siRNA for inhibiting Nrf2 expression. In agreement with our earlier findings, UDCA treatment caused an increase in GPX1 and catalase protein in BAR-T cells containing control siRNA (Fig. 6C). In Nrf2 knockdown cells, in contrast, treatment with UDCA did not increase GPX1 or catalase protein expression (Fig. 6C). These findings demonstrate that Nrf2 activation contributes to the UDCA-induced increase in protein expression of GPX1 and catalase in Barrett's cells.
Inhibition of GPX1 or catalase reduces the protective effect of UDCA against DCA-induced DNA damage.
To determine whether upregulation of GPX1 or catalase by UDCA contributes to protection from DCA-induced DNA damage, we inhibited GPX1 or catalase using specific siRNAs and then exposed the BAR-T cells to 5 min of DCA with and without 24-h pretreatment with UDCA. Western blotting was performed for GPX1 and catalase to determine the efficiency of siRNA for inhibiting expression of these antioxidants. We noted a substantial reduction of GPX1 and complete elimination of catalase protein expression after transfection with specific siRNAs in BAR-T cells both at baseline and after treatment with UDCA for 24 h (Fig. 7A). Treatment with DCA caused an increase in phospho-H2AX expression in cells containing control siRNA, GPX1 siRNA, and catalase siRNA (Fig. 7, B and C). In GPX1 knockdown cells, there were no apparent differences in the amount of phospho-H2AX induced by DCA between BAR-T cells with and without UDCA pretreatment (Fig. 7B). Likewise, in catalase knockdown cells, there were no apparent differences in the amount of phospho-H2AX induced by DCA between cells with and without UDCA pretreatment (Fig. 7C). These findings suggest that increased expression of GPX1 and catalase protein contributes to the protective effect of UDCA in reducing DNA damage caused by DCA in Barrett's cells.
Fig. 7.

GPX1 and catalase knockdown prevents the UDCA-induced decrease in phospho-H2AX after DCA exposure in BAR-T cells. A: representative Western blot demonstrating GPX1 and catalase knockdown by siRNA. BAR-T cells were exposed to DCA with and without 24-h pretreatment with UDCA. Representative Western blots demonstrating that GPX1 siRNA (B) and catalase siRNA (C) prevented the UDCA-induced decease in phospho-H2AX expression after DCA exposure.
In patients with Barrett's esophagus, 8 wk of oral UDCA treatment significantly increases expression of GPX1 and catalase protein in Barrett's metaplasia.
After demonstrating in our Barrett's cells that UDCA increases expression of GPX1 and catalase, and that these antioxidants contribute to the protective effect of UDCA in reducing DNA damage after DCA exposure, we sought to confirm that these same effects occur in patients with Barrett's esophagus who are treated with oral UDCA for 8 wk. In agreement with our in vitro data, we found significantly increased protein levels of GPX1 and catalase in biopsy specimens of Barrett's metaplasia from our 21 patients (Fig. 8, A and B).
Fig. 8.

Oral UDCA treatment for 8 wk increases expression of GPX1 and catalase protein in Barrett's metaplasia. A: left, representative Western blots of Barrett's mucosa from 2 patients demonstrating GPX1 expression before (pre) and after (post) 8 wk of oral UDCA. Right, oral UDCA increases GPX1 in the esophagus of patients with Barrett's mucosa. B: left, representative Western blots from Barrett's mucosa from 2 patients demonstrating catalase expression before (pre) and after (post) 8 wk of oral UDCA. Right, oral UDCA increases catalase in Barrett's esophagus. Horizontal line in each column represents the mean for all 21 patients.
DISCUSSION
In patients with Barrett's esophagus, we found that esophageal perfusion with DCA for only 5 min significantly increased phospho-H2AX and phospho-p65 expression in Barrett's metaplasia. In those same patients, 8 wk of oral treatment with UDCA prevented DNA damage and NF-κB activation induced by DCA perfusion. Using Barrett's cell lines to explore molecular mechanisms underlying these effects, we found that UDCA activated Nrf2 signaling to increase intracellular levels of GPX1 and catalase antioxidants, and that pretreating cells with UDCA prevented DCA from inducing DNA damage and NF-κB activation. This protective effect of UDCA pretreatment could be blocked in vitro by siRNA knockdown of GPX1 and catalase and, in patients, we demonstrated that oral UDCA treatment increased protein levels of these antioxidants in Barrett's metaplasia. Thus we have shown that UDCA increases expression of antioxidants that prevent DNA damage and NF-κB activation in Barrett's metaplasia exposed to a toxic bile acid found in refluxed gastric juice. These findings suggest a potential chemopreventive role for UDCA in Barrett's esophagus.
In Barrett's epithelial cells, we found that DCA caused DNA damage with DSBs. DSBs are dangerous mutations because they can cause genomic instability contributing to carcinogenesis, and agents that cause DSBs are considered carcinogens (2, 23). Since DCA can be considered a carcinogen in Barrett's esophagus, an agent that can counter DCA's DNA-damaging effects might have a role in chemoprevention. Our findings suggest that UDCA might be such an agent. As in our earlier studies (11), we found that a 5-min esophageal perfusion with UDCA caused no DNA damage and no NF-κB activation in Barrett's metaplasia for any study patient. We treated our patients with oral UDCA in a dose of 10 mg/kg because this dosage has been used safely to treat patients with liver diseases and, on this dosage, UDCA replaces hydrophobic bile acids to comprise 40–50% of the bile salt pool (18). Thus we anticipated that oral UDCA treatment would decrease concentrations of toxic bile acids like DCA in refluxed gastric juice and increase its concentration of the cytoprotective bile acid UDCA.
We found that oral UDCA treatment prevented DCA from inducing DNA damage and NF-κB activation in Barrett's metaplasia, and we performed experiments to delineate mechanisms underlying these effects. In cell-free systems, UDCA has antioxidant properties, scavenging ROS such as superoxide and hydroxyl radicals (19, 20). Other studies have suggested that UDCA protects cells by binding to and stabilizing their plasma membranes (9). Cell protection due to antioxidant and cell membrane stabilizing effects would be expected to be manifest immediately upon exposure to UDCA, but we observed no differences in phospho-H2AX and phospho-p65 expression between cells treated with DCA alone and cells treated with an equimolar mixture of DCA and UDCA. In contrast, Goldman et al. (7) found that CPA Barrett's cells exposed to acidic bile salts developed less DNA damage when UDCA was added to the medium. It is not clear why our findings differ from Goldman's, but a number of differences in design of these studies might account for discrepant results. For example, we exposed cells to a single hydrophobic bile acid (DCA) at neutral pH, whereas Goldman used a bile acid “cocktail” of conjugated bile acids and DCA in a medium acidified to pH 4. In addition, we used a telomerase-immortalized Barrett's cell line that has no neoplastic features, whereas the CPA cell line used by Goldman exhibits invasive features in 3D organotypic culture (7, 17).
After finding that mixing UDCA with DCA did not prevent DNA damage, our next experiments involved pretreating Barrett's cells with UDCA for 24 h prior to DCA exposure. UDCA pretreatment prevented DCA-induced ROS production, DNA damage, and NF-κB activation. Protection from oxidative stress might be explained by increases in intracellular antioxidant levels, and we found that UDCA pretreatment indeed upregulated mRNA and protein expression of GPX1 and catalase. The expression of antioxidant proteins is regulated by the ARE binding site in promoter regions of antioxidant target genes (16, 28). Using an ARE reporter assay, we found that UDCA increased activity of the ARE reporter, confirming that UDCA upregulates transcription of antioxidant genes in Barrett's cells.
The induction of antioxidants by chemopreventive agents often involves Nrf2, an ARE-binding transcription factor and member of the basic leucine zipper (bZIP) protein family (10). Nrf2 normally is sequestered in cytoplasm by binding to Keap1. With phosphorylation, Nrf2 dissociates from Keap1, heterodimerizes with other bZIP proteins, translocates to the nucleus, and binds to ARE that regulate transcription of antioxidants (10). In Barrett's cells, we have shown that UDCA increases phospho-Nrf2 and causes its nuclear translocation. In addition, we have shown that knockdown of Nrf2 by siRNA prevents upregulation of GPX1 and catalase by UDCA. These findings show that UDCA treatment activates Nrf2 signaling to increase intracellular levels of antioxidants.
To confirm the physiological importance of antioxidant upregulation in UDCA protection from oxidative stress, we studied the effects of UDCA pretreatment on DCA-induced DNA damage in cells transfected with siRNA against GPX1 or catalase. We found that transfection with either siRNA abolished the protective effect of UDCA. In patients with Barrett's esophagus, furthermore, we found that 8 wk of oral treatment with UDCA increased protein levels of GPX1 and catalase in Barrett's metaplasia. These data indicate that UDCA protects against oxidative stress induced by toxic bile acids in Barrett's esophagus by upregulating expression of GPX1 and catalase antioxidants.
All patients in our study were given omeprazole before and during UDCA treatment, and there is plausible rationale for using PPIs in chemoprevention for Barrett's esophagus. PPIs heal reflux esophagitis and thus might protect against cancer-promoting effects of chronic esophageal inflammation. PPIs reduce esophageal exposure to acid that, like bile acids, can produce DNA DSBs (34). In earlier studies, we showed that PPIs block esophageal epithelial cells from secreting IL-8, a proinflammatory and proproliferative cytokine, through effects on NF-κB and activating protein-1 that are independent of effects on gastric acid secretion (12). Finally, observational studies suggest that PPIs protect against neoplasia in Barrett's esophagus (15).
Despite their numerous beneficial effects, PPIs do nothing to correct the underlying reflux diathesis in Barrett's esophagus, and gastroesophageal reflux of acidic and nonacidic gastric material occurs frequently in Barrett's patients taking PPIs (6, 32). In Barrett's patients, furthermore, PPI treatment increases gastric luminal concentrations of toxic, unconjugated bile acids like DCA, which can be especially damaging at the neutral pH levels common in gastric juice of patients on PPIs (31). Ongoing reflux of toxic bile acids during PPI therapy might contribute to carcinogenesis despite the otherwise beneficial actions of these agents. Thus a combination of PPIs and UDCA might be more effective for chemoprevention than either agent alone.
To our knowledge, only one clinical study has explored the role of UDCA combined with PPIs for chemoprevention in Barrett's esophagus (3). That study found no effects of this combination therapy on inflammation, dysplasia, proliferation, differentiation, and p53 and p16 abnormalities. However, the study included only nine patients who took UDCA for only 6 mo and was not adequately powered to detect changes in some of the markers studied. Furthermore, these markers are of questionable validity as indexes for protection from oxidative DNA damage, the protective mechanism that we have demonstrated for UDCA. Our study provides strong rationale for future studies on UDCA combined with PPIs for chemoprevention in Barrett's esophagus.
In conclusion, we have shown that oral treatment with UDCA prevents a toxic bile acid from causing DNA damage and NF-κB activation in the metaplastic mucosa of patients with Barrett's esophagus. Using Barrett's epithelial cells in vitro, we have shown that UDCA activates Nrf2 to upregulate expression of GPX1 and catalase antioxidants, which prevent DCA-induced ROS generation, DNA damage, and NF-κB activation. Finally, we have verified this in vitro finding by demonstrating upregulation of GPX1 and catalase in biopsy specimens of Barrett's metaplasia taken from patients after oral UDCA treatment. These data elucidate a molecular pathway whereby treatment with UDCA protects against bile-acid induced oxidative injury in Barrett's esophagus and provide the rationale for clinical trials of UDCA for chemoprevention in patients with this common disorder.
GRANTS
This work was supported by the Office of Medical Research, Departments of Veterans Affairs (R. F. Souza, S. J. Spechler, D. H. Wang), the National Institutes of Health (R01-DK63621 and R01-CA134571 to R. F. Souza and S. J. Spechler, R01-DK097340 to D. H. Wang, K12 HD-068369-01 to E. Cheng), the American Gastroenterological Association Institute (Fellow to Faculty Transition Award to E. Cheng), and NASPGHAN Foundation/AstraZeneca Award (E. Cheng).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.P., X.H., Q.Z., X.Z., C.Y., K.A., E.C., T.H.P., D.H.W., M.C., R.F.S., and S.J.S. conception and design of research; S.P., X.H., D.R., R.F.S., and S.J.S. performed experiments; S.P., X.H., R.F.S., and S.J.S. analyzed data; S.P., X.H., R.F.S., and S.J.S. interpreted results of experiments; S.P., X.H., R.F.S., and S.J.S. prepared figures; S.P., X.H., R.F.S., and S.J.S. drafted manuscript; S.P., X.H., D.R., Q.Z., X.Z., C.Y., K.A., E.C., T.H.P., D.H.W., R.F.S., and S.J.S. edited and revised manuscript; S.P., X.H., D.R., Q.Z., X.Z., C.Y., K.A., E.C., T.H.P., D.H.W., M.C., R.F.S., and S.J.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Daisha J. Cipher, Associate Professor, University of Texas at Arlington School of Nursing, for her contribution to the power calculations for our patient studies.
REFERENCES
- 1. Araki Y, Andoh A, Bamba H, Yoshikawa K, Doi H, Komai Y, Higuchi A, Fujiyama Y. The cytotoxicity of hydrophobic bile acids is ameliorated by more hydrophilic bile acids in intestinal cell lines IEC-6 and Caco-2. Oncol Rep 10: 1931–1936, 2003 [PubMed] [Google Scholar]
- 2. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer 8: 957–967, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bozikas A, Marsman WA, Rosmolen WD, van Baal JW, Kulik W, Ten Kate FJ, Krishnadath KK, Bergman JJ. The effect of oral administration of ursodeoxycholic acid and high-dose proton pump inhibitors on the histology of Barrett's esophagus. Dis Esophagus 21: 346–354, 2008 [DOI] [PubMed] [Google Scholar]
- 4. Cheng E, Zhang X, Huo X, Yu C, Zhang Q, Wang DH, Spechler SJ, Souza RF. Omeprazole blocks eotaxin-3 expression by oesophageal squamous cells from patients with eosinophilic oesophagitis and GORD. Gut 62: 824–832, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corley DA, Mehtani K, Quesenberry C, Zhao W, de Boer J, Weiss NS. Impact of endoscopic surveillance on mortality from Barrett's esophagus-associated esophageal adenocarcinomas. Gastroenterology 145: 312–319, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frazzoni M, Conigliaro R, Melotti G. Weakly acidic refluxes have a major role in the pathogenesis of proton pump inhibitor-resistant reflux oesophagitis. Aliment Pharmacol Ther 33: 601–606, 2011 [DOI] [PubMed] [Google Scholar]
- 7. Goldman A, Condon A, Adler E, Minnella M, Bernstein C, Bernstein H, Dvorak K. Protective effects of glycoursodeoxycholic acid in Barrett's esophagus cells. Dis Esophagus 23: 83–93, 2010 [DOI] [PubMed] [Google Scholar]
- 8. Gotley DC, Morgan AP, Ball D, Owen RW, Cooper MJ. Composition of gastro-oesophageal refluxate. Gut 32: 1093–1099, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guldutuna S, Zimmer G, Imhof M, Bhatti S, You T, Leuschner U. Molecular aspects of membrane stabilization by ursodeoxycholate [see comment]. Gastroenterology 104: 1736–1744, 1993 [DOI] [PubMed] [Google Scholar]
- 10. Hu R, Saw CL, Yu R, Kong AN. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid Redox Signal 13: 1679–1698, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED, Wang JY, Cheng E, Meyer F, Wang DH, Zhang Q, Spechler SJ, Souza RF. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-κB activation in benign Barrett's epithelial cells. Am J Physiol Gastrointest Liver Physiol 301: G278–G286, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huo X, Zhang X, Yu C, Zhang Q, Cheng E, Wang DH, Pham TH, Spechler SJ, Souza RF. In oesophageal squamous cells exposed to acidic bile salt medium, omeprazole inhibits IL-8 expression through effects on nuclear factor-kappaB and activator protein-1. Gut. 2013. September 18. 10.1136/gutjnl-2013-305533 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iftikhar SY, Ledingham S, Steele RJ, Evans DF, Lendrum K, Atkinson M, Hardcastle JD. Bile reflux in columnar-lined Barrett's oesophagus. Ann R Coll Surg Engl 75: 411–416, 1993 [PMC free article] [PubMed] [Google Scholar]
- 14. Jaiswal KR, Morales CP, Feagins LA, Gandia KG, Zhang X, Zhang HY, Hormi-Carver K, Shen Y, Elder F, Ramirez RD, Sarosi GA, Jr, Spechler SJ, Souza RF. Characterization of telomerase-immortalized, nonneoplastic, human Barrett's cell line (BAR-T). Dis Esophagus 20: 256–264, 2007 [DOI] [PubMed] [Google Scholar]
- 15. Kastelein F, Spaander MC, Steyerberg EW, Biermann K, Valkhoff VE, Kuipers EJ, Bruno MJ. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett's esophagus. Clin Gastroenterol Hepatol 11: 382–388, 2013 [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal 7: 385–394, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Kosoff RE, Gardiner KL, Merlo LM, Pavlov K, Rustgi AK, Maley CC. Development and characterization of an organotypic model of Barrett's esophagus. J Cell Physiol 227: 2654–2659, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kowdley KV. Ursodeoxycholic acid therapy in hepatobiliary disease. Am J Med 108: 481–486, 2000 [DOI] [PubMed] [Google Scholar]
- 19. Lapenna D, Ciofani G, Festi D, Neri M, Pierdomenico SD, Giamberardino MA, Cuccurullo F. Antioxidant properties of ursodeoxycholic acid. Biochem Pharmacol 64: 1661–1667, 2002 [DOI] [PubMed] [Google Scholar]
- 20. Ljubuncic P, Abu-Salach O, Bomzon A. Ursodeoxycholic acid and superoxide anion. World J Gastroenterol 11: 4875–4878, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masamune K, Kunitomo K, Sasaki K, Yagi K, Komi N, Tashiro S. Bile-induced DNA strand breaks and biochemical analysis of bile acids in an experimental model of anomalous arrangement of the pancreaticobiliary ducts. J Med Invest 44: 47–51, 1997 [PubMed] [Google Scholar]
- 22. McQuaid KR, Laine L, Fennerty MB, Souza R, Spechler SJ. Systematic review: the role of bile acids in the pathogenesis of gastro-oesophageal reflux disease and related neoplasia. Aliment Pharmacol Ther 34: 146–165, 2011 [DOI] [PubMed] [Google Scholar]
- 23. Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev 194: 77–95, 2003 [DOI] [PubMed] [Google Scholar]
- 24. Nehra D, Howell P, Williams CP, Pye JK, Beynon J. Toxic bile acids in gastro-oesophageal reflux disease: influence of gastric acidity. Gut 44: 598–602, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pohl H, Sirovich B, Welch HG. Esophageal adenocarcinoma incidence: are we reaching the peak? Cancer Epidemiol Biomarkers Prev 19: 1468–1470, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Rizvi S, DeMars CJ, Comba A, Gainullin VG, Rizvi Z, Almada LL, Wang K, Lomberk G, Fernandez-Zapico ME, Buttar NS. Combinatorial chemoprevention reveals a novel smoothened-independent role of GLI1 in esophageal carcinogenesis. Cancer Res 70: 6787–6796, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singh S, Khanna S, Pardi DS, Loftus EV, Jr, Talwalkar JA. Effect of ursodeoxycholic acid use on the risk of colorectal neoplasia in patients with primary sclerosing cholangitis and inflammatory bowel disease: a systematic review and meta-analysis. Inflamm Bowel Dis 19: 1631–1638, 2013 [DOI] [PubMed] [Google Scholar]
- 28. Soyalan B, Minn J, Schmitz HJ, Schrenk D, Will F, Dietrich H, Baum M, Eisenbrand G, Janzowski C. Apple juice intervention modulates expression of ARE-dependent genes in rat colon and liver. Eur J Nutr 50: 135–143, 2011 [DOI] [PubMed] [Google Scholar]
- 29. Spechler SJ. Barrett esophagus and risk of esophageal cancer: a clinical review. JAMA 310: 627–636, 2013 [DOI] [PubMed] [Google Scholar]
- 30. Spechler SJ, Sharma P, Souza RF, Inadomi JM, Shaheen NJ. American Gastroenterological Association technical review on the management of Barrett's esophagus. Gastroenterology 140: e18–e52, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Theisen J, Nehra D, Citron D, Johansson J, Hagen JA, Crookes PF, DeMeester SR, Bremner CG, DeMeester TR, Peters JH. Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial overgrowth and deconjugation of bile acids. J Gastrointest Surg 4: 50–54, 2000 [DOI] [PubMed] [Google Scholar]
- 32. Vaezi MF, Richter JE. Role of acid and duodenogastroesophageal reflux in gastroesophageal reflux disease. Gastroenterology 111: 1192–1199, 1996 [DOI] [PubMed] [Google Scholar]
- 33. Wali RK, Frawley BP, Jr, Hartmann S, Roy HK, Khare S, Scaglione-Sewell BA, Earnest DL, Sitrin MD, Brasitus TA, Bissonnette M. Mechanism of action of chemoprotective ursodeoxycholate in the azoxymethane model of rat colonic carcinogenesis: potential roles of protein kinase C-alpha, -beta II, and -zeta. Cancer Res 55: 5257–5264, 1995 [PubMed] [Google Scholar]
- 34. Zhang HY, Hormi-Carver K, Zhang X, Spechler SJ, Souza RF. In benign Barrett's epithelial cells, acid exposure generates reactive oxygen species that cause DNA double-strand breaks. Cancer Res 69: 9083–9089, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang X, Yu C, Wilson K, Zhang H, Melton SD, Huo X, Wang DH, Genta RM, Spechler SJ, Souza RF. Malignant transformation of nonneoplastic Barrett's epithelial cells through well-defined genetic manipulations. PLoS One 5: e13093, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]