

Abstract
Time series data can provide valuable insight into the complexity of biological reactions. Such information can be obtained by mass-spectrometry-based approaches that measure pre-steady-state kinetics. These methods are based on a mixing device that rapidly mixes the reactants prior to the on-line mass measurement of the transient intermediate steps. Here, we describe an improved continuous-flow mixing apparatus for real-time electrospray mass spectrometry measurements. Our setup was designed to minimize metal–solution interfaces and provide a sheath flow of nitrogen gas for generating stable and continuous spray that consequently enhances the signal-to-noise ratio. Moreover, the device was planned to enable easy mounting onto a mass spectrometer replacing the commercial electrospray ionization source. We demonstrate the performance of our apparatus by monitoring the unfolding reaction of cytochrome C, yielding improved signal-to-noise ratio and reduced experimental repeat errors.
Keywords: kinetics, mass spectrometry, on-line measurements, proteins, rapid mixing device, time-resolved experiments, structure characterization of biomolecules
Introduction
Biological reactions are dynamic. Therefore, complete understanding of reaction mechanisms involves elucidation of the various intermediate steps along the reaction path and their rate. These kinetics aspects have been studied by a variety of time-dependent techniques including nuclear magnetic resonance (NMR),[1] circular dichroism (CD),[2] fluorescence spectroscopy[3] and X-ray absorption spectroscopy.[4] Mass spectrometry (MS), the focus of this work, is an additional method that has been widely employed in kinetic studies.[5]
The application of mass spectrometry for the study of time-dependent reactions has several distinct advantages:[5a] its conceptual simplicity, its applicability to proteins assemblies of unlimited size, and its ability to detect in a single spectrum multiple reactive species. MS also enables the use of relatively low concentrations of sample (picomolar), and the speed of analysis allows capturing non-equilibrium states. Typically, reactions are monitored by MS without artificial labeling, as this method takes advantage of the fact that reactants, products and intermediate species often have different masses. As a result, mass measurements can be employed to detect small mass shifts reflecting ligand binding, catalytic processes, or changes in composition and assembly states.[6] Binding affinities can be estimated from ion intensity ratios of the protein bound and unbound states.[7] In addition, considering that compact protein structure generates low charge states, whereas unfolded proteins produce wide distributions of highly protonated ions,[8] the conformational state of the protein components can be probed by their charge state distribution. Taken as a whole, MS measurements present an attractive approach for monitoring the temporal sequence of events during biomolecular interactions.
To provide time-dependent MS measurements, experimental setups usually employ a rapid-mixing apparatus to efficiently combine the components before on-line infusion into the mass spectrometer.[5b] A range of MS-coupled rapid-mixing devices have been devised using either a stopped-flow apparatus,[9] capillary setup with a static mixer,[10] an adjustable position mixer for continuous reaction monitoring,[11] or a microfluidic chip.[12] Various biological processes were characterized using these devices including protein folding and unfolding reactions,[13] enzymatic catalysis[14] and subunit association.[15]
The continued improvement of on-line mixing techniques remains an important avenue towards progress in MS-based kinetics studies. Here, we describe the construction and validation of a simple mixing apparatus for real-time analysis of biological reactions. Our experimental setup is based on the use of a capillary apparatus designed by Wilson and coworkers[16] in which several new features were introduced to allow continuous ion flow, enhanced desolvation and improved signal-to-noise ratios. As a proof of principle, the performance of the device is demonstrated by measuring the unfolding kinetics of cytochrome C (Cyt c).
Results and Discussion
A capillary mixer device for characterizing biological reactions at the subsecond time scale
The basic design of the mixer was first described by Wilson and Konermann[11, 14a and was optimized by Liuni et al.[16] Here we describe modifications that we have introduced to the system in order to improve the stability of the ion current and enable simple mounting of the device in front of the mass spectrometer orifice (Figure 1).
Figure 1.
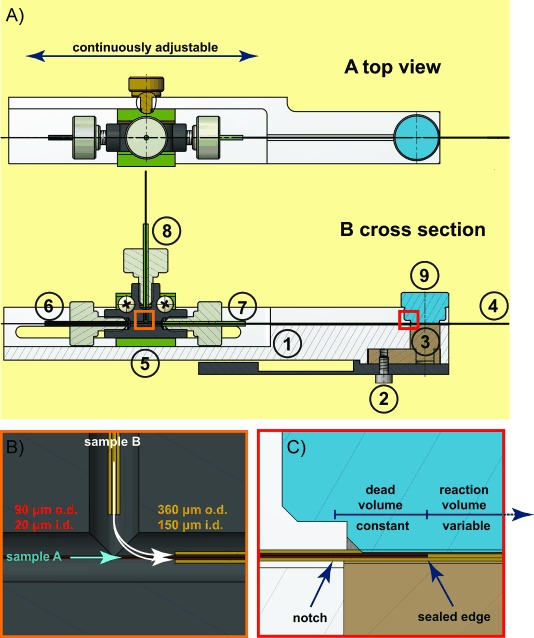
Schematic cross-sectional diagram of the experimental device used in this study for time-resolved ESI-MS experiments. A) To enable easy replacement of the manufactures sample sprayer with the devised mixer, the capillary system was embraced within a Delrin fabricated adapter (1), which is directly connected to the instrument through the three-axis manipulator (2). The stainless steel component of the adaptor (3) provides the electrical contact that is required for the electrospray process (4). Flexibility in the positioning of the device in respect to the instrument orifice is achieved via an adjustable connection unit (5). The inner (6) and outer (7) capillaries introduce the reactants, samples A and B, respectively. Mixing of the two solutions initiates the reaction of interest. Sample B is introduced to the system through a third capillary, which is connected to the remaining inlet of the tee (8). The positioning of the mixing capillary is fixed by a non-conductive screw (9). The orange and red boxes that designate the position of the connection tee and mixing unit are enlarged in panels B and C, respectively. B) The mixing unit is built from a mixing tee that connects the inner, outer and supply capillaries. C) Within the mixing unit, reactants flowing through the inner capillary exit through the notch and are forced into the narrow space between the inner and outer capillaries, where they are mixed with the reactants from the outer capillary. The observed reaction time is adjusted by changing the position of the inner capillary with respect to the fixed outer capillary. Early reaction times are observed when the inner capillary is positioned close to the ESI outlet. Increasing the gap between the inner and outer capillaries correspond to larger reaction volumes and in turn later reaction times.
In general, the setup consists of two concentric capillaries, an inner capillary which is inserted through an outer capillary of larger diameter. Two reactants are supplied, one in each capillary (shown as sample A and B in Figure 1). A small notch, approximately 3 mm away from the plugged tip of the inner capillary, enables the solution from the inner capillary to escape into the intercapillary space and mix rapidly with the solution in the outer capillary. The reaction time depends on the applied flow rate as well as the distance between the mixing point and the tip of the outer capillary. While the flow rate is set to a constant value, the distance between the capillaries is modulated by repositioning the inner capillary with respect to the steady position of the outer capillary (Figure 1). When the tip of the inner capillary is close to the ending of the outer capillary, then the internal volume after mixing is small and the reaction time is short. Longer reaction times are obtained when the end of the inner capillary is far from the outer capillary, this increases the reaction volume and the reaction time is extended. Unlike the variable reaction volume, the dead volume—the intercapillary volume between the facilitated notch and sealed tip of the inner capillary—remains constant throughout the experiments.
To facilitate simple and straightforward configuration of the capillary mixing device, we have fabricated an adaptor that embraces the above-described capillary set-up (Figure 1, item 1 and Figure 2). The adaptor is designed to enable easy mounting onto the quadrupole/time-of-flight (Q-TOF) Synapt mass spectrometer by using the existing platform of the standard nanoflow electrospray ionization (ESI) source. Specifically, the adaptor replaces the manufactures sample sprayer (either glass capillary or nano-LC) and is screwed onto the three-axis manipulator (XYZ stage) (Figure 1, item 2 and Figure 2). Moreover, by using an adaptable fitting, the location of the device in respect to the mass spectrometer inlet is adjustable enabling flexible positioning (Figure 1, item 5).
Figure 2.

Images of the capillary mixing device mounted on to the Synapt mass spectrometer. A) The capillary apparatus is embedded within a Delrin fabricated adaptor, which is attached to the standard ionization source three-axis manipulator (XYZ-stage). The image shows how the reactant solutions, sample A and B, are introduced, the voltage and gas supply sources, as well as the positioning of the mixing tee. For clarity, the electrospray and gas supply regions highlighted in panel A) by red and yellow boxes are enlarged in panel B) and C), respectively. B) This picture displays the metal syringe needle, which holds the outer capillary and provides the voltage supply for the electrospray process as well as the teflon tubing that surrounds it supplying the nitrogen gas. C) The image exemplifies the split flow of the nitrogen nebulizing gas (see red arrows). The metal needle is pierced a few millimeters before the tip to direct the nitrogen flow between both the needle and the teflon tubing as well as through the interface between the needle and the outer capillary.
Our design took into account the fact that during the electrospray ionization process, when a current flows through the electrospray emitter, electrochemical reactions take place at metal–solution interfaces.[17] These reactions, besides altering the composition of the solution and affecting the analyte chemical stability,[18] may also generate gas bubbles that could interrupt the spray.[19] To overcome these undesirable side effects and achieve a steady spray, our device is composed predominantly of non-conductive material. The adapter is manufactured from polyoxymethylene (Delrin) (Figure 1, item 1) and fused silica is used for both the inner and outer capillaries (Figure 1, item 6 and 7). High voltage is supplied to the tip of the outer capillary (Figure 1, item 4) through a metal syringe needle (Figure 2 right panel) that encloses the capillary assembly and is connected to the conductive stainless-steel component embedded within the adaptor (Figure 1, item 3). To ensure the steadiness of the electrical connectivity, the tip of the outer capillary and the metal syringe needle are joined by silver paint (see Experimental Section). Overall, this strategy significantly improves the spray stability and improves upon previous applications[16] in which a metal outer capillary was employed.
To facilitate the electrospray process, we introduced a nitrogen supply to act as a nebulizer and desolvation gas. This was achieved by covering the metal syringe needle with teflon tubing supplied with nitrogen gas (Figure 2). We pierced the metal needle a few millimeters away from the tip to direct the gas flow into two paths (Figure 2). The gas flows in the gap between the metal needle and the teflon tubing as well as is in the interspace between the metal needle and the outer capillary. Both gas flows progress into the same direction facilitating solvent evaporation and preventing leakage of the sample into the gap between the needle and the outer capillary due to capillary forces. The nitrogen gas flow helps to direct the spray emerging from the capillary tip towards the mass spectrometer orifice.
Assessing the capillary mixing device performance
We initially examined the electrospray stability of the device by measuring the total ion current (TIC). Figure 3 shows a typical TIC trace of Cyt c. The measurements were performed using a flow rate of 2.75 μL min−1 and the minimal reaction time of 0.4 s, corresponding to the dead volume. Over 20 min of data collection, the normalized TIC signal intensity was stable ranging between 0.8 to 1.0, emphasizing the added benefit of minimizing metal contacts and reducing gas bubble formation.
Figure 3.
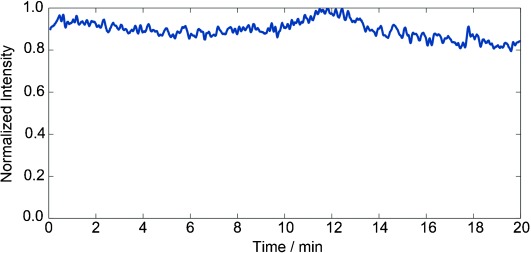
High signal stability of the capillary mixer was recorded over a period of 20 min. The total ion count for a 5 μm solution of Cyt c in 50 mm NH4OAc. The flow rate was 2.75 μL min−1 and inner capillary positioning was at 0.4 s to allow minimal reaction time. The maximum signal corresponds to 6.2×104 ion counts.
To test the effectiveness of the system, we chose to monitor the unfolding reaction of Cyt c, which has been extensively analyzed by MS,[12, 20] making it a convenient model system for assessing new MS-based methods. In this type of reaction, the transition between the native folded state and the denatured conformation is accompanied by changes in the charge state distribution.[21] Unfolded states will show higher multiply charged ion envelopes (lower m/z value) than the corresponding folded or compact forms. During the experiment, acid-induced unfolding of Cyt c was monitored by operating the capillary mixing device in ‘spectral mode’ in which the mixer is fixed at various positions within the main channel.[5b] Spectral mode allows the reaction to be monitored indefinitely at fixed time points, facilitating the acquisition of high signal-to-noise mass spectra.
Figure 4 shows the kinetic profile of Cyt c unfolding. At the initial time, a rather narrow distribution of charge states between 7+ to 10+ exists, representing the compact folded state of the protein (Figure 4). Between 1 s and 5 s, there is a decrease in the intensity of the 8–10+ charge states and an increase in the intensities of peaks centered at 15+, corresponding to the unfolded conformation—reflecting the decay of the folded form and formation of the unfolded state. After 5 s, the unfolding reaction reached steady state and no significant change in the charge state distribution was observed. Interestingly, we do not detect significant population of an unfolding intermediate, which has been reported previously.[12] This may be due to the slightly lower time resolution of the current device, but may also reflect a lack of side reactions occurring in the source due to production of additional H+ ions. Our results therefore suggest two-state unfolding for Cyt c, which is in agreement with fluorescence-based T-jump data.[22]
Figure 4.
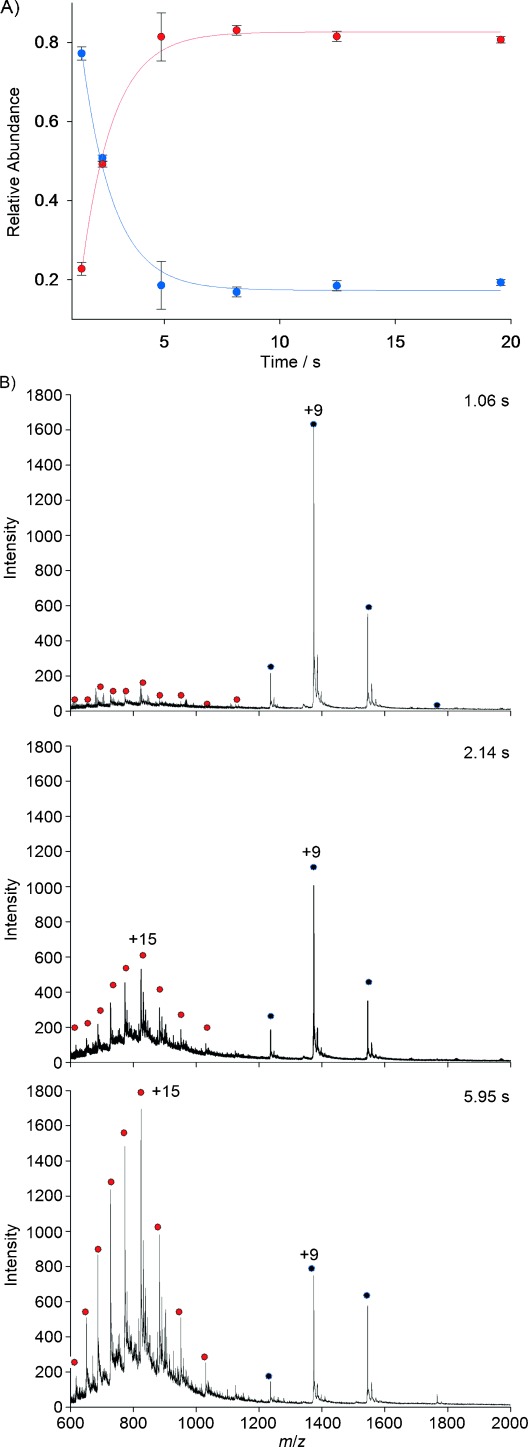
Unfolding kinetics of Cyt c monitored by time-resolved mass spectrometry. A) The kinetic profile of acid induced denaturation of Cyt c. The depletion of folded Cyt c ( ) and the formation of unfolded species (
) and the formation of unfolded species ( ) is demonstrated. The plot was generated by measuring the signal intensity of charge states 7–20+ at different time points after mixing Cyt c with 5.0 % AcOH. Error bars represent the average error of three independent experiments. Solid lines are fits to the experimental data. B) Representative ESI-MS spectra recorded at different time points, 1.06, 2.14 and 5.95 s, during unfolding.
) is demonstrated. The plot was generated by measuring the signal intensity of charge states 7–20+ at different time points after mixing Cyt c with 5.0 % AcOH. Error bars represent the average error of three independent experiments. Solid lines are fits to the experimental data. B) Representative ESI-MS spectra recorded at different time points, 1.06, 2.14 and 5.95 s, during unfolding.
Notably, our results show a significant reduction in the experimental repeat errors, with an average of 4.2 % (Figure 4), compared to an average of approximately 15 % for conductive capillary mixing devices.[11] We attribute this almost threefold improvement to the high signal-to-noise ratio of the spectra recorded with our setup.
Conclusions
Time-resolved electrospray ionization mass spectrometry employs rapid mixing devices to access short-timescale processes in biological reactions. In the last decade, this approach was extensively applied in combination with pulse labeling and hydrogen exchange methods to follow protein folding and unfolding processes[13a, 23] as well as enzymatic reactions.[14] Here, we describe the application of an improved rapid mixing device for time-resolved measurements in the sub-second region.
The mixing device we devised has the key advantage that metal–solution interfaces were minimized (Figure 1). As a consequence, spray disruption and capillary clogging due to gas bubble formation, were drastically reduced. Moreover, our capillary mixing system is embedded within a plastic fabricated adaptor, enabling easy mounting onto the Synapt electrospray stage and rapid replacement of the manufacturers sprayer (Figures 1 and 2). Additionally, steady spray is attained by adding nitrogen supply to the surrounding of the outer capillary improving the desolvation and nebulizing process (Figure 2). The experimental setup was tested by monitoring the unfolding reaction of Cyt c. We observe significant improvement in both signal-to-noise ratio and in the error between experimental repeats.
While the device introduced in this study was mounted on a Synapt instrument, we did not take advantage of the ion-mobility capabilities of the instrument. In future studies, we plan to harness this feature and record mass spectrometry ion-mobility data sets to follow not only shifts in mass associated with biological reactions but also structural transitions reflected by drift time measurements.[24] In addition, in the current study, we have recorded the data exercising spectral mode analysis, in which the inner capillary was withdrawn in discrete steps, to allow longer acquisition time in each time point. In upcoming experiments, by connecting the inner capillary to a separate syringe pump and continually increasing the distance between the inner and outer capillaries, while monitoring the abundance of selected ions, kinetic mode analysis may be introduced providing detailed kinetics profiles.[25] Overall, we anticipate that this improved rapid mixing device will open many new possibilities for mechanistic characterization of dynamic enzymatic reactions, assembly pathways and subunit exchange reactions.
Experimental Section
Reagents
AcOH (≥99.7 %), NH4OAc and horse heart cytochrome c (Cyt c) were obtained from Sigma–Aldrich. Methanol (HPLC supra reagent) was purchase from Bio-Lab (Jerusalem, Israel). Deionized H2O was obtained from a Direct-Q3 system (Millipore, France).
Time-resolved capillary mixer
The device was constructed in-house using fused silica capillaries (Polymicro Technologies, Phoenix, AZ, USA), a PEEK tee with a 0.02′′ hole size (Upchurch Scientific, Oak Harbor, WA, USA), a fingertight fitting F-300 1/16′′ OD (Upchurch Scientific), a sealing sleeve 1/16x.0155×1.55′’ (Upchurch Scientific), a syringe needle 21G 1 1/2“ (Microlance 3, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) and silver paint (Agar Scientific, Stansted, UK). The tee is fitted within a fabricated Delrin adaptor manufactured at the Weizmann Institute warehouse.
Initially, the inner capillary (i.d. 20 μm, o.d. 90 μm, about 40 cm length) was sealed at its end and a notch was etched 3 mm upstream the sealed edge using a Versa-Laser engraving device (Universal Laser, Scottsdale, AZ, USA). The following parameters were used: speed 25 %, pulses per inch 500 and thickness 0.1 mm. The power altered between 7 % and 100 % depending whether a notch or seal were introduced, respectively. Only 1–2 exposures were required to seal the edge, while 4–5 repetitions were applied for notch formation. The capillary was pre-conditioned with deionized H2O prior to the laser treatment.
The treated inner capillary was passed through the mixing tee and inserted into the 15 cm long outer capillary (o.d. 360 μm, i.d. 150 μm). The outer capillary was fixed to the adaptor by threading it into a sealing sleeve that was pre-attached to the Delrin adaptor. Flexibility in the positioning of the inner capillary was achieved using a fingertight fitting. The tip of the outer capillary was passed into a syringe needle and connections were attached with silver paint (Agar Scientific). A third capillary (o.d. 360 μm, i.d. 150 μm) was inserted into the remaining vertical port of the tee in order to deliver the solution to the outer capillary. Both inner and supply capillaries were connected to 100 μL Hamilton syringes via Luer-lock systems (Upchurch Scientific). The syringes were fitted into the same syringe pump (infusion 400, Chemyx, Stafford, USA) using a flow rate of 2.75 μL min−1 giving a dead time of 0.4 s. Nitrogen supply was introduced by teflon tubing encompassing the syringe needle.
Sample preparation
For verifying system performance, the unfolding reaction of Cyt c was examined. Solution flowing in the inner capillary contained 5 % AcOH (v/v) in deionized H2O. The outer capillary solvent was comprised of 10 μm Cyt c dissolved in deionized H2O, pH 7.0.
MS experimental conditions
ESI MS measurements were carried out on a Synapt HDMS system (Waters Corp., UK) adapted for high-mass measurements.[26] Typical settings were: capillary voltage, 3.5 kV; cone voltage, 70 V; extraction cone voltage 2 V; trap collision energy, 5 V; transfer collision energy 6 V; bias 40 V and backing pressure, 2.0–2.8 mbar. All ESI-MS experiments were carried out at RT.
Reactions were initiated by mixing the two solutions at the outlet of the inner capillary. The data was obtained for 20 different time points, ranging from 1.06 s to 28.29 s. Spectra were recorded using the spectral mode by monitoring the entire mass spectrum of the reaction at fixed distances between the inner and outer capillaries.[11] For every time point, three independent experiments were acquired, in which 180 s acquisition time was summed and analyzed without smoothing.
The calculated reaction time is the sum of the dead time and increasing mixing volume as a function of the solutions linear velocity at a fixed volume for each time point. Relative abundances were assessed from the peak heights of the species being monitored. Line-fitting was achieved using Sigmaplot v11.0.0.77 by Systat Software Inc. (San Jose, CA, USA) and error bars represent standard deviations from the mean.
Acknowledgments
M.S. is thankful for the financial support of a Starting Grant from the European Research Council (ERC) (FP7/2007–2013/ERC Grant Agreement no. 239679) and a grant from the Israel Science Foundation (Grant no. 220/10). M.S. is the incumbent of the Elaine Blond Career Development Chair. A.K.S was supported by a Weizmann Dean Fellowship.
References
- 1a.Udgaonkar JB, Baldwin RL. Nature. 1988;335:694–9. doi: 10.1038/335694a0. [DOI] [PubMed] [Google Scholar]
- 1b.Nabuurs SM, van Mierlo CP. J. Biol. Chem. 2010;285:4165–72. doi: 10.1074/jbc.M109.087932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2a.Greenfield NJ. Methods Mol. Biol. 2004;261:55–78. doi: 10.1385/1-59259-762-9:055. [DOI] [PubMed] [Google Scholar]
- 2b.Schlepckow K, Wirmer J, Bachmann A, Kiefhaber T, Schwalbe H. J. Mol. Biol. 2008;378:686–98. doi: 10.1016/j.jmb.2008.02.033. [DOI] [PubMed] [Google Scholar]
- 2c.Chen E, Goldbeck RA, Kliger DS. Curr. Protein Pept. Sci. 2009;10:464–75. doi: 10.2174/138920309789352001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a.Kolygo K, Ranjan N, Kress W, Striebel F, Hollenstein K, Neelsen K, Steiner M, Summer H, Weber-Ban E. J. Struct. Biol. 2009;168:267–277. doi: 10.1016/j.jsb.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 3b.Suhling K, French PM, Phillips D. Photochem. Photobiol. Sci. 2005;4:13–22. doi: 10.1039/b412924p. [DOI] [PubMed] [Google Scholar]
- 4.Solomon A, Akabayov B, Frenkel A, Milla ME, Sagi I. Proc. Natl. Acad. Sci. USA. 2007;104:4931–6. doi: 10.1073/pnas.0700066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5a.Ben-Nissan G, Sharon M. Chem. Soc. Rev. 2011;40:3627–37. doi: 10.1039/c1cs15052a. [DOI] [PubMed] [Google Scholar]
- 5b.Rob T, Wilson DJ. Eur. J. Mass Spectrom. 2012;18:205–14. doi: 10.1255/ejms.1176. [DOI] [PubMed] [Google Scholar]
- 5c.Liu J, Konermann L. Biochemistry. 2013;52:1717. doi: 10.1021/bi301693g. [DOI] [PubMed] [Google Scholar]
- 6a.Painter AJ, Jaya N, Basha E, Vierling E, Robinson CV, Benesch JL. Chem. Biol. 2008;15:246–53. doi: 10.1016/j.chembiol.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 6b.Bothner B, Chavez R, Wei J, Strupp C, Phung Q, Schneemann A, Siuzdak G. J. Biol. Chem. 2000;275:13455–9. doi: 10.1074/jbc.275.18.13455. [DOI] [PubMed] [Google Scholar]
- 6c.Fabris D. Mass Spectrom. Rev. 2005;24:30–54. doi: 10.1002/mas.20007. [DOI] [PubMed] [Google Scholar]
- 6d.Lee ED, Mück W, Henion JD, Covey TR. J Am Chem. Soc. 1989;111:4600–4604. [Google Scholar]
- 6e.van den Heuvel RH, Gato S, Versluis C, Gerbaux P, Kleanthous C, Heck AJ. Nucleic Acids Res. 2005;33:e96. doi: 10.1093/nar/gni099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Liu J, Konermann L. J. Am. Soc. Mass Spectrom. 2011;22:408–17. doi: 10.1007/s13361-010-0052-1. [DOI] [PubMed] [Google Scholar]
- 7b.Wang W, Kitova EN, Klassen JS. Anal. Chem. 2003;75:4945–55. doi: 10.1021/ac034300l. [DOI] [PubMed] [Google Scholar]
- 7c.Cubrilovic D, Biela A, Sielaff F, Steinmetzer T, Klebe G, Zenobi R. J. Am. Soc. Mass Spectrom. 2012;23:1768–77. doi: 10.1007/s13361-012-0451-6. [DOI] [PubMed] [Google Scholar]
- 8a.Chowdhury SK, Katta V, Chait BT. J. Am. Chem. Soc. 1990;112:9012–9013. [Google Scholar]
- 8b.Testa L, Brocca S, Grandori R. Anal. Chem. 2011;83:6459–63. doi: 10.1021/ac201740z. [DOI] [PubMed] [Google Scholar]
- 8c.Kaltashov IA, Abzalimov RR. J. Am. Soc. Mass Spectrom. 2008;19:1239–46. doi: 10.1016/j.jasms.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 8d.Borysik AJ, Radford SE, Ashcroft AE. J. Biol. Chem. 2004;279:27069–77. doi: 10.1074/jbc.M401472200. [DOI] [PubMed] [Google Scholar]
- 8e.Kim MY, Maier CS, Reed DJ, Deinzer ML. Protein Sci. 2002;11:1320–9. doi: 10.1110/ps.3140102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Kolakowski BM, Simmons DA, Konermann L. Rapid Commun. Mass Spectrom. 2000;14:772–6. doi: 10.1002/(SICI)1097-0231(20000515)14:9<772::AID-RCM942>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 9b.Kolakowski BM, Konermann L. Anal. Biochem. 2001;292:107–14. doi: 10.1006/abio.2001.5062. [DOI] [PubMed] [Google Scholar]
- 10a.Konermann L, Rosell FI, Mauk AG, Douglas DJ. Biochemistry. 1997;36:6448–54. doi: 10.1021/bi970353j. [DOI] [PubMed] [Google Scholar]
- 10b.Paiva AA, Tilton RF, Jr, Crooks GP, Huang LQ, Anderson KS. Biochemistry. 1997;36:15472–6. doi: 10.1021/bi971883i. [DOI] [PubMed] [Google Scholar]
- 11.Wilson DJ, Konermann L. Anal. Chem. 2003;75:6408–14. doi: 10.1021/ac0346757. [DOI] [PubMed] [Google Scholar]
- 12.Rob T, Wilson DJ. J. Am. Soc. Mass Spectrom. 2009;20:124–30. doi: 10.1016/j.jasms.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 13a.Vahidi S, Stocks BB, Liaghati-Mobarhan Y, Konermann L. Anal. Chem. 2013;85:8618. doi: 10.1021/ac401148z. [DOI] [PubMed] [Google Scholar]
- 13b.Wu L, Lapidus LJ. Anal. Chem. 2013;85:4920–4. doi: 10.1021/ac3033646. [DOI] [PubMed] [Google Scholar]
- 13c.Pan Y, Brown L, Konermann L. J. Mol. Biol. 2011;410:146–58. doi: 10.1016/j.jmb.2011.04.074. [DOI] [PubMed] [Google Scholar]
- 13d.Keppel TR, Weis DD. Anal. Chem. 2013;85:5161–8. doi: 10.1021/ac4004979. [DOI] [PubMed] [Google Scholar]
- 14a.Wilson DJ, Konermann L. Anal. Chem. 2004;76:2537–43. doi: 10.1021/ac0355348. [DOI] [PubMed] [Google Scholar]
- 14b.Liuni P, Olkhov-Mitsel E, Orellana A, Wilson DJ. Anal. Chem. 2013;85:3758–64. doi: 10.1021/ac400191t. [DOI] [PubMed] [Google Scholar]
- 15.Stocks BB, Rezvanpour A, Shaw GS, Konermann L. J. Mol. Biol. 2011;409:669–79. doi: 10.1016/j.jmb.2011.04.028. [DOI] [PubMed] [Google Scholar]
- 16.Liuni P, Jeganathan A, Wilson DJ. Angew. Chem. 2012;124:9804–9807. doi: 10.1002/anie.201204903. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:9666–9. doi: 10.1002/anie.201204903. [DOI] [PubMed] [Google Scholar]
- 17.Abonnenc M, Qiao L, Liu B, Girault HH. Annu. Rev. Anal. Chem. 2010;3:231–54. doi: 10.1146/annurev.anchem.111808.073740. [DOI] [PubMed] [Google Scholar]
- 18a.Konermann L, Silva EA, Sogbein OF. Anal. Chem. 2001;73:4836–44. doi: 10.1021/ac010545r. [DOI] [PubMed] [Google Scholar]
- 18b.Van Berkel GJ, Zhoub F, Aronson JT. Int. J. Mass Spectrom. Ion Processes. 1997;162:55–62. [Google Scholar]
- 19a.Smith AD, Moini M. Anal. Chem. 2001;73:240–6. doi: 10.1021/ac0007940. [DOI] [PubMed] [Google Scholar]
- 19b.Moini M, Cao P, Bard AJ. Anal. Chem. 1999;71:1658–61. doi: 10.1021/ac9811266. [DOI] [PubMed] [Google Scholar]
- 20a.Yan X, Watson J, Ho PS, Deinzer ML. Mol. Cell. Proteomics. 2004;3:10–23. doi: 10.1074/mcp.R300010-MCP200. [DOI] [PubMed] [Google Scholar]
- 20b.Konermann L, Douglas DJ. Biochemistry. 1997;36:12296–12302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]
- 20c.Konermann L, Douglas DJ. J. Am. Soc. Mass Spectrom. 1998;9:1248–54. doi: 10.1016/S1044-0305(98)00103-2. [DOI] [PubMed] [Google Scholar]
- 21.Kaltashov IA, Eyles SJ. Mass Spectrom. Rev. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 22.Panda M, Benavides-Garcia MG, Pierce MM, Nall BT. Protein Sci. 2000;9:536–543. doi: 10.1110/ps.9.3.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23a.Stefanowicz P, Petry-Podgorska I, Kowalewska K, Jaremko L, Jaremko M, Szewczuk Z. Biosci. Rep. 2010;30:91–9. doi: 10.1042/BSR20090015. [DOI] [PubMed] [Google Scholar]
- 23b.Pan J, Han J, Borchers CH, Konermann L. Anal. Chem. 2010;82:8591–7. doi: 10.1021/ac101679j. [DOI] [PubMed] [Google Scholar]
- 23c.Pan J, Rintala-Dempsey AC, Li Y, Shaw GS, Konermann L. Biochemistry. 2006;45:3005–13. doi: 10.1021/bi052349a. [DOI] [PubMed] [Google Scholar]
- 23d.Konermann L, Pan J, Liu YH. Chem. Soc. Rev. 2011;40:1224–34. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 24a.Hyung SJ, Ruotolo BT. Proteomics. 2012;12:1547–64. doi: 10.1002/pmic.201100520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b.Konijnenberg A, Butterer A, Sobott F. Biochim. Biophys. Acta. 2013;1834:1239–56. doi: 10.1016/j.bbapap.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 25.Wilson DJ, Rafferty SP, Konermann L. Biochemistry. 2005;44:2276–83. doi: 10.1021/bi047684y. [DOI] [PubMed] [Google Scholar]
- 26.Michaelevski M, Kirshenbaum N, Sharon S. J. Vis. Exp. 2010;41:1985. doi: 10.3791/1985. [DOI] [PMC free article] [PubMed] [Google Scholar]