

Abstract
This study reviewed the clinical characteristics of 112 pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL) patients with TCF3-PBX1 fusion treated according to the Japan Association of Childhood Leukemia Study (JACLS) ALL02 protocol (n = 82) and Children's Cancer and Leukemia Study Group (CCLSG) ALL 2004 protocol (n = 30). The 3-year event-free survival (EFS) and overall survival (OS) rates were 85.4 ± 3.9% and 89.0 ± 3.5% in JACLS cohort, and the 5-year EFS and OS were 82.8 ± 7.0% and 86.3 ± 6.4% in CCLSG cohort, respectively, which are comparable to those reported in western countries. Conventional prognostic factors such as age at onset, initial white blood cell count, and National Cancer Institute risk have also no impact on OS in both cohorts. Surprisingly, the pattern of relapse in JACLS cohort, 9 of 82 patients, was unique: eight of nine patients relapsed during the maintenance phase and one patient had primary induction failure. However, bone marrow status and assessment of minimal residual disease on days 15 and 33 did not identify those patients. Interestingly, the two patients with IKZF1 deletion eventually relapsed in JACLS cohort, as did one patient in CCLSG cohort. International collaborative study of larger cohort is warranted to clarify the impact of the IKZF1 deletion on the poor outcome of TCF3-PBX1 positive BCP-ALL.
Keywords: IKZF1 deletion, pediatric acute lymphoblastic leukemia, TCF3-PBX1
Introduction
The translocation t(1;19)(q23;p13) and its unbalanced variant der(19)t(1;19)(q23;p13) are well-known chromosomal abnormalities in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL) 1,2. This translocation results in the fusion of TCF3 on 19p13 with PBX1 on 1q23, generating the fusion gene TCF3-PBX1 on derivative chromosome 19 3.
Although t(1;19)(q23;p13) was initially associated with poor prognosis in pediatric BCP-ALL, the recent development of intensified chemotherapy regimens has improved the outcome of this subgroup, resulting in a 5-year event-free survival (EFS) rate of ∼85−90% in western countries, which is similar to that of TEL-AML1 positive or high hyperdiploid BCP-ALL 2,4–6. However, ∼10% of patients experience relapse with dismal prognosis 2,4, underscoring the importance of identifying reliable prognostic markers to improve the treatment of these patients. In the last decades, several studies have attempted to identify prognostic markers for this subgroup of pediatric BCP-ALL with unsatisfactory results 4,5,7. Classic prognostic factors, such as age at onset, initial white blood cell (WBC) count, National Cancer Institute (NCI) risk group, and type of chromosomal abnormality [balanced t(1;19) and unbalanced t(1;19)], did not have prognostic value in recent studies 4,5. Genetic analysis to identify alterations related to poor prognosis in pediatric BCP-ALL patients with TCF3-PBX1 fusion has not been performed to date, with the exception of one study that analyzed the relationship between TP53 mutation and poor prognosis in a small number of patients 8. Herein, we reviewed the clinical data of 112 pediatric BCP-ALL patients with TCF3-PBX1 fusion, which is the largest such cohort reported to date. Additionally, we performed genetic analyses, including IKZF1 and TP53, to determine the prognostic value of these genetic alterations in pediatric BCP-ALL with TCF3-PBX1.
Design and Methods
Patient cohorts and samples
From April 2002 to May 2008, 1138 patients aged 1–18 years with newly diagnosed BCP-ALL were enrolled in the Japan Association of Childhood Leukemia Study (JACLS) ALL02 study 9–11. The diagnosis of BCP-ALL was based on morphological findings of bone marrow (BM) aspirates and immuno-phenotypic analysis of leukemic cells by flow cytometry. Conventional cytogenetic analysis using G-banding was performed as part of the routine workup. Molecular studies using quantitative RT-PCR (RQ-PCR) for the detection of TCF3-PBX1 were also performed as part of the routine workup (Table S1). Ph + ALL and infantile ALL patients were excluded from the study. Patients with Down syndrome were also excluded.
Bone marrow smears were examined under the microscope on days 15 and 33 (at the end of the induction phase) to evaluate the treatment response. M1, M2, and M3 marrow were defined as fewer than 5%, 5−25%, and more than 25% blast cells in the BM aspirate, respectively. Complete remission (CR) was defined as the absence of blast cells in the peripheral blood, fewer than 5% blast cells in the BM aspirate, normal cellularity and trilineage hematopoiesis, and absence of blast cells in the cerebrospinal fluid and elsewhere. RQ-PCR for TCF3-PBX1 was also performed on days 15, 33, and 71 (at the end of consolidation) to determine minimal residual disease (MRD). The GAPDH gene was amplified as an internal control of RNA quality.
An independent validation cohort of 30 pediatric BCP-ALL patients with TCF3-PBX1 fusion was enrolled from the Children's Cancer and Leukemia Study Group (CCLSG) ALL 2004 protocol between June 2004 and May 2009 12. The diagnosis of BCP-ALL was based on morphological and immuno-phenotypic analyses as described for the JACLS cohort. Patients with t(1;19)/der(19)t(1;19) determined by G-banding analysis or TCF3-PBX1 fusion determined by RQ-PCR in the JACLS or CCLSG cohorts were enrolled in this analysis. Informed consent was obtained from the patients' guardians according to the Declaration of Helsinki; treatment and genetic study protocols were approved by the Institutional Review Boards of the participating institutions.
Determination of IKZF1 deletion by multiplex ligation-dependent probe amplification analysis
Genomic DNA was isolated from diagnostic BM or peripheral blood samples using the Qiagen DNeasy tissue and blood kit according to the manufacturer's instructions (Qiagen, Venio, the Netherlands). DNA specimens of 53 patients in the JACLS cohort and 22 patients in the CCLSG cohort were analyzed using the SALSA multiplex ligation-dependent probe amplification (MLPA) kit P335-A4 according to the manufacturer's instructions (MRC Holland, Amsterdam, the Netherlands) as described elsewhere 11,13.
JAK2 and TP53 mutations analysis
To screen for the JAK2 mutation in exons 16, 20, and 21 (accession number NM 004972), genomic DNA was extracted from diagnostic BM samples of two patients in the JACLS cohort harboring IKZF1 deletions. Because the frequency of JAK2 mutation has been reported to be quite rare and clustered in the patients with IKZF1 deletion 11, we analyzed JAK2 mutation in the patients with IKZF1 deletion. To screen for TP53 mutations in exons 5, 6, 7, 8, and 9 (accession number NM 000546), genomic DNA was also extracted from diagnostic BM samples of eight patients in the JACLS cohort who experienced relapse. Because TP53 mutation was associated with relapsed TCF3-PBX1 positive ALL 8, genomic DNA extracted from relapsed BM samples of four of the eight cases in the JACLS cohort was also used for TP53 mutation screening. The primers used for JAK2 mutation screening were as previously described 11. The primers used for TP53 mutation screening are listed in Table S1. The PCR product was analyzed by direct sequencing using a BigDye Terminator sequencing kit (Applied Biosystems, Foster City, CA).
Statistical analysis
Estimation of survival distributions was performed using the Kaplan–Meier method and differences were compared using the log-rank test. A P-value of <0.05 (two-sided) was considered significant. EFS and overall survival (OS) were defined as the times from diagnosis to event (any death, relapse, secondary malignancy, and failure to therapy) and from diagnosis to death from any cause or the last follow up. Patients without an event of interest were censored at the date of last contact. The median follow-up time for EFS and OS was 5.2 years. Hazard ratios for probability of relapse between subgroups were calculated using univariate Cox models. Other comparisons were performed using the χ2 test, Fisher's exact test, and Mann–Whitney U-test, as appropriate.
Results
Patient characteristics and basic cytogenetic data in JACLS cohort
The fusion transcript of TCF3-PBX1 was detected in 82 (7.2%) of the 1138 patients in the JACLS cohort. The characteristics of these 82 patients are summarized in Table 1 and 2. The median age at diagnosis was 6 years (range, 1−15 years), and 38 were males and 44 were females. The median leukocyte count at diagnosis was 21,750 × 109/L (range 2,700−183,300). Forty-nine patients were classified as NCI-SR and 33 patients as NCI-HR. Seventy-three patients were included in the prednisolone good responder (PGR) group and nine patients in the prednisolone poor responder (PPR) group. The translocation was balanced in 17 (20.1%) cases and unbalanced in 25 (30%) cases. Either balanced or unbalanced translocation was not determined in 17 cases, including seven cases with karyotypic failure. Twenty-three (28.1%) cases with normal karyotype and seven (8.5%) cases with karyotypic failure were identified as TCF3-PBX1 positive by RT-PCR. Fifty-three (64.6%) of the 82 patients were selected for further genetic analysis on the basis of the availability of material for testing. A comparison of the clinical characteristics of patients with and without available DNA/RNA specimens is shown in Table 1. No major differences were observed between the analyzed and nonanalyzed cohorts except for initial WBC count and NCI risk group.
Table 1.
Comparison of the characteristics of 112 BCP-ALL patients with TCF3-PBX1 fusion between those included and not included in the genetic analyses
| Study protocol | JACLS ALL02 | CCLSG ALL2004 | ||||
|---|---|---|---|---|---|---|
| Number of patients | 53 | 29 | P-value | 22 | 8 | |
| Analyzed | Nonanalyzed | Analyzed | Nonanalyzed | P-value | ||
| Sex (male/female) | 28/25 | 10/19 | 0.16 | 9/13 | 3/5 | 1.0 |
| Age (years) at diagnosis, median (range) | 5 (1−14) | 6 (1−15) | 0.27 | 7 (1−14) | 9 (2−14) | 0.57 |
| WBC count, cells/μL median (range) | 24,500 (4700−183,300) | 16,900 (4700−55,220) | <0.01 | 17,100 (3200−118,000) | 27,800 (9000−137,360) | 0.26 |
| NCI risk group, SR/HR | 27/26 | 22/7 | 0.04 | 10/12 | 3/5 | 1.0 |
| Chromosome | 0.23 | 0.90 | ||||
| Normal karyotype | 13 | 10 | 3 | 1 | ||
| t(1;19)(q23;p13) | 11 | 6 | 6 | 3 | ||
| der t(1;19)(q23;p13) | 20 | 5 | 12 | 4 | ||
| Unknown | 9 | 8 | 1 | 0 | ||
| SCT in 1st CR (n) | 2 | 0 | ND | ND | ||
| Observation period, median (range) | 5.7 (1.6−8.7) | 4.7 (0.1−8.8) | 0.47 | 6.4 (2.3−7.7) | 4.5 (3.5−7.4) | 0.02 |
| Relapse, n (%) | 8 (15.1) | 1 (3.4) | 0.15 | 4 (18.2) | 2 (25.0) | 0.65 |
| Survival | 1.0 | 1.0 | ||||
| Alive, n (%) | 45 (84.9) | 25 (86.2) | 20 (90.1) | 7 (87.5) | ||
| Dead, n (%) | 8 (15.1) | 4 (13.8) | 2 (9.9) | 1 (12.5) | ||
JACLS, Japan Association of Childhood Leukemia Study; CCLSG, Children's Cancer and Leukemia Study Group; WBC, white blood cell; NCI, National Cancer Institute; SR, standard risk; HR, high risk. SCT, stem cell transplantation; CR, complete remission; ND, not determined.
Table 2.
Summary of the overall results of clinical trials of pediatric BCP-ALL with TCF3-PBX1 from major research groups
| n | Sex (male/female) | Age (years) at diagnosis, median (range) | WBC count, cells/μL, median (range) | CNS relapse (%) | EFS (y) | OS (y) | Reference | |
|---|---|---|---|---|---|---|---|---|
| I-BFM (I-ALL90, 96, 12-ALLIC02) | 48 | 22/26 | ND (0.8−16) | ND (1400−434,000) | 0 | 85 | ND | 6 |
| SJCRH XIIIa-XV | 41 | 18/23 | ND | ND | 9 | 84 (5) | 96.4 (5) | 5 |
| MRC-ALL97/99 | 50 | ND | ND | ND | 6 | 80 (5) | 84 (5) | 2 |
| NOPHO-ALL1992 and 2000 | 47 | 21/26 | 7 (1−18) | 16,000 (1300−159,000) | 0 | 79 (5) | 85 (5) | 4 |
| CCLSG ALL2004 | 30 | 12/18 | 7 (1−14) | 17,550 (3200−137,360) | 6.7 | 82.8 (5) | 86.3 (5) | Present study |
| JACLS ALL02 | 82 | 38/44 | 6 (1−15) | 21,750 (2700−183,300) | 1.2 | 85.4 (3) | 89.0 (3) | Present study |
CNS, central nervous system; EFS, event-free survival; OS, overall survival; I-BFM, International Berlin-Frankfurt-Munster; SJCRH, St. Jude Children's Research Hospital; MRC, Medical Research Council; NOPHO, Nordic Society of Pediatric Hematology Oncology; JACLS, Japan Association of Childhood Leukemia Study; CCLSG, Children's Cancer and Leukemia Study Group ND, not described.
Patient characteristics and basic cytogenetic data in CCLSG cohort
The fusion transcript of TCF3-PBX1 was detected in 30 (11.4%) of the 264 patients in the CCLSG cohort. The characteristics of these 30 patients are also summarized in Table 1 and 2. The median age at diagnosis was 7 years (range, 1−14 years), and 12 were males and 18 were females. The median leukocyte count at diagnosis was 17,550 × 109/L (range 3200−137,360). Thirteen patients were classified as NCI-SR and 17 patients as NCI-HR. All evaluable 27 patients were included in the PGR group. The translocation was balanced in 9 (30%) cases and unbalanced in 16 (53.3%) cases. Four (13.3%) cases with normal karyotype and 1 (0.03%) case with karyotypic failure were identified as TCF3-PBX1 positive by RT-PCR. Twenty-two (73.3%) of the 30 patients were selected for further genetic analysis on the basis of the availability of material for testing. A comparison of the clinical characteristics of patients with and without available DNA/RNA specimens is shown in Table 1. No major differences were observed between the analyzed and nonanalyzed cohorts.
Conventional adverse prognostic factors were not associated with poor OS in the patients with TCF3-PBX1 fusion
The predicted 3-year EFS and OS rates in the 82 TCF3-PBX1 positive patients were 85.4 ± 3.9% and 89.0 ± 3.5% in JACLS cohort, and the 5-yerar EFS and OS were 82.8 ± 7.0% and 86.3 ± 6.4% in CCLSG cohort, respectively, which were comparable to the rates reported in western countries (Fig. 1 and Table 2). In JACLS cohort, nine (11%) of the 82 patients experienced relapse. The site of relapse was the BM in eight patients and the central nervous system (CNS) in one patient. Eight of nine patients experienced relapse during maintenance therapy, and one after allogeneic stem cell transplantation (allo-SCT) during the first CR. Four of the nine patients died of disease progression. The remaining five patients received allo-SCT. However, two of them died of transplant-related complications, and the remaining three eventually experienced relapse and died of disease progression. A comparison of the characteristics of patients according to the occurrence of relapse is shown in Table 3. The results of univariate analysis showed that none of the conventional prognostic factors, including age at onset, initial WBC count, NCI risk group, and BM status on days 15 and 33, were associated with poor EFS/OS in the 82 patients (Table S2). Although data on MRD on days 15 and 33 were not available for all patients, no statistically significant differences in MRD on days 15 and 33 were detected between nonrelapsed and relapsed patients (Figure S1).
Figure 1.
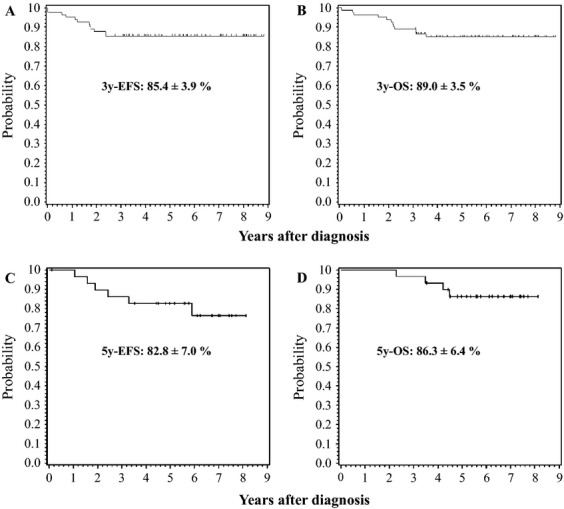
Probability of event-free survival (EFS) and overall survival (OS) in pediatric B-cell precursor acute lymphoblastic leukemia patients with TCF3-PBX1 fusion treated according to the JACLS ALL02 (n = 82, A and B) and CCLSG ALL2004 protocol (n = 30, C and D). (A and C) EFS, (B and D) OS. JACLS, Japan Association Childhood Leukemia Study; CCLSG, Children's Cancer and Leukemia Study Group.
Table 3.
Comparison of the characteristics of BCP-ALL patients with TCF3-PBX1 fusion according to relapse status in JACLS ALL02 and CCLSG ALL 2004 cohorts
| JACLS ALL02 | CCLSG ALL2004 | |||||
|---|---|---|---|---|---|---|
| Relapsed | Nonrelapsed | P-value | Relapsed | Nonrelapsed | P-value | |
| Number of patients | 9 | 73 | 5 | 25 | ||
| Gender (male/female) | 3/6 | 35/38 | 0.49 | 1/4 | 11/14 | 0.32 |
| Age (years) at diagnosis, median (range) | 7 (1−14) | 5 (1−15) | 0.81 | 11 (4–14) | 6 (1–14) | 0.05 |
| WBC count, cells/μL median (range) | 24,500 (9700−70,400) | 21,750 (2700−183,300) | 0.08 | 14,500 (9400–26,600) | 17,100 (3200–137,360) | 0.88 |
| NCI risk group, SR/HR | 4/5 | 45/28 | 0.53 | 1/4 | 12/13 | 0.25 |
| Chromosome | 0.12 | 0.95 | ||||
| Normal karyotype | 4 | 19 | 1 | 3 | ||
| t(1;19)(q23;p13) | 1 | 16 | 2 | 7 | ||
| der t(1;19)(q23;p13) | 2 | 23 | 1 | 12 | ||
| Unknown | 2 | 15 | 1 | 3 | ||
| SCT in 1st CR (n) | 1 | 1 | 0.21 | ND | ND | |
| Survival | <0.01 | <0.01 | ||||
| Alive, n (%) | 0 (0) | 70 (95.6) | 1 (25) | 25 (100) | ||
| Dead, n (%) | 9 (100) | 3 (4.4) | 4 (75) | 0 (0) | ||
JACLS, Japan Association of Childhood Leukemia Study; CCLSG, Children's Cancer and Leukemia Study Group; WBC, white blood cell; NCI, National Cancer Institute; SR, standard risk; HR, high risk. SCT, stem cell transplantation; CR, complete remission; ND, not determined.
In CCLSG cohort, five (16.7%) of the 30 patients experienced relapse. The site of relapse was the BM in four patients and the CNS combined with BM in one patient. Three of five patients experienced relapse during maintenance therapy. Four of the five patients eventually died. A comparison of the characteristics of patients according to the occurrence of relapse is shown in Table 3. The results of univariate analysis also showed that none of the conventional prognostic factors, including age at onset, initial WBC count, and NCI risk group were associated with poor OS in the 30 patients (Table S2).
IKZF1 deletion is identified in relapsed BCP-ALL patients with TCF3-PBX1 in the JACLS ALL02 cohort
The results of the MLPA analysis were summarized in Tables 4 and 5. Deletion of the IKZF1 gene was detected in 2 (3.8%) of 53 patients with available DNA sample of diagnostic leukemic blasts, which was a significantly lower frequency than that of pediatric BCP-ALL patients without TCF3-PBX1 fusion (3.8% vs. 10.4%, P < 0.001, Table 4). The JAK2 mutation was not identified in the two patients with IKZF1 deletion. Deletions of PAX5, CDKN2A, and CDKN2B were also less frequent in BCP-ALL patients with than in those without TCF3-PBX1 fusion. However, deletion of RB1 was detected in nine (17.0%) of 53 patients, which was a higher rate than that of patients without TCF3-PBX1 fusion (3.0%) (Table 4). In terms of the prognostic impact of these micro deletions, none were associated with an increase of relapse except the IKZF1 deletion (Table 5).
Table 4.
Summary of the results of MLPA analyses of TCF3-PBX1 positive and negative BCP-ALL patients in the JACLS ALL02 and CCLSG cohorts
| JACLS ALL02 cohort | CCLSG cohort | |||||
|---|---|---|---|---|---|---|
| TCF3-PBX1 (+) | TCF3-PBX1 (−) | P-value | TCF3-PBX1 (+) | TCF3-PBX1 (−) | P-value | |
| Number of patients | 53 | 163 | 22 | 155 | ||
| IKZF1 deletion (%) | 2 (3.8) | 17 (10.4) | <0.001 | 1 (4.5) | 21 (13.5) | 0.23 |
| CDKN2A deletion (%) | 10 (18.9) | 71 (43.6) | <0.001 | 6 (27.3) | 37 (23.9) | 0.73 |
| CDKN2B deletion (%) | 8 (15.1) | 61 (37.4) | <0.001 | 5 (22.7) | 40 (25.8) | 0.76 |
| PAX5 deletion (%) | 12 (22.6) | 47 (28.8) | 0.37 | 9 (40.9) | 28 (18.1) | 0.014 |
| ETV6 deletion (%) | 1 (1.9) | 46 (28.2) | <0.001 | 2 (9.1) | 40 (25.8) | 0.085 |
| RB1 deletion (%) | 9 (17.0) | 3 (1.8) | <0.001 | 4 (18.2) | 20 (12.9) | 0.50 |
| BTG1 deletion (%) | 1 (1.9) | 14 (8.6) | <0.001 | 1 (4.5) | 20 (12.9) | 0.26 |
| EBF1 deletion (%) | 2 (3.8) | 20 (12.3) | <0.001 | 2 (9.1) | 17 (10.9) | 0.79 |
JACLS, Japan Association of Childhood Leukemia Study; CCLSG, Children's Cancer and Leukemia Study Group.
Table 5.
Impact of genetic alterations on the outcomes of patients with BCP-ALL with TCF3-PBX1
| JACLS ALL02 cohort | CCLSG cohort | |||||
|---|---|---|---|---|---|---|
| Relapse | Nonrelapse | P-value | Relapse | Nonrelapse | P-value | |
| Number of patients | 8 | 45 | 4 | 18 | ||
| IKZF1 deletion (%) | 2 (25) | 0 | <0.01 | 1 (25) | 0 | 0.03 |
| CDKN2A deletion (%) | 1 (12.5) | 9 (20) | 0.62 | 2 (50) | 4 (22.2) | 0.26 |
| CDKN2B deletion (%) | 1 (12.5) | 7 (15.6) | 0.99 | 2 (50) | 3 (16.7) | 0.15 |
| PAX5 deletion (%) | 2 (25) | 10 (22.2) | 0.72 | 2 (50) | 7 (38.9) | 0.68 |
| ETV6 deletion (%) | 0 | 1 (2.2) | 0.67 | 0 | 2 (11.1) | 0.48 |
| RB1 deletion (%) | 2 (25) | 7 (15.6) | 0.51 | 0 | 4 (22.2) | 0.30 |
| BTG1 deletion (%) | 0 | 1 (12.5) | 0.67 | 1 (12.5) | 0 | 0.18 |
| EBF1 deletion (%) | 0 | 2 (25) | 0.54 | 0 | 2 (11.1) | 0.48 |
JACLS, Japan Association of Childhood Leukemia Study; CCLSG, Children's Cancer and Leukemia Study Group.
TP53 mutation has been associated with relapse in BCP-ALL patients with TCF3-PBX1 fusion 8. Therefore, we screened for mutations in TP53 in the diagnostic samples of eight cases experiencing relapse. In addition, TP53 mutation screening was performed in relapsed leukemic samples from four of the eight relapsed patients. However, TP53 mutations were not observed in these 12 samples.
IKZF1 deletion is also identified in relapsed patient with TCF3-PBX1 positive BCP-ALL in the validation cohort (CCLSG cohort)
To confirm the significance of the IKZF1 deletion in BCP-ALL with TCF3-PBX1, 22 diagnostic leukemic samples with TCF3-PBX1 fusion obtained from the patients registered in the CCLSG ALL2004 protocol were analyzed by MLPA. Although only 1 (4.5%) of 22 patients had the IKZF1 deletion, this patient experienced relapse and died of the disease (Tables 4 and 5).
Discussion
In this study, we showed that TCF3-PBX1 positive pediatric BCP-ALL patients treated according to the JACLS ALL02 and CCLSG ALL2004 protocol had favorable outcomes similar to those reported in western countries (Table 2) 2,4–6. A low frequency of CNS relapse (1 in 82 patients, 1.2%) was observed in JACLS ALL02 cohort, showing a more favorable result than those reported in the St. Jude and MRC cohorts (Table 2) 2,5. However, 9 (11%) of 82 in JACLS and 5 (16.7%) of 30 patients in CCLSG cohort experienced relapse and most of them died of disease progression or transplant-related complications. The clinical and biological features of the relapsed cases should be evaluated to further improve the prognosis of pediatric BCP-ALL with TCF3-PBX1 fusion. To the best of our knowledge, a reliable prognostic marker has not been identified in this subgroup 4,5,7. In this study, conventional prognostic markers, such as age at onset, initial WBC count, and NCI risk group, were not associated with poor prognosis, which was in agreement with previous studies. Furthermore, we found that the type of chromosomal abnormality had no prognostic impact 4,5. However, the pattern of relapse was unique in the present study, especially in JACLS cohort: eight of nine patients relapsed during the maintenance phase and one patient had primary induction failure, suggesting that leukemic blasts in these patients initially showed resistance to the chemotherapeutic agents used. However, BM status and assessment of MRD on days 15 and 33 did not identify those patients who experienced very early relapse, suggesting that a small fraction of leukemic cells contributed to relapse.
Few studies have investigated the association between genetic alterations and prognosis in BCP-ALL with TCF3-PBX1. Kawamura et al. identified point mutations of TP53 in two of 20 initial patients and in four relapsed patients with BCP-ALL expressing TCF3-PBX1 and suggested a possible relationship between TP53 mutation and disease progression in BCP-ALL patients with TCF3-PBX1 fusion 8. Therefore, in this study, we screened for TP53 mutations in TCF3-PBX1 positive BCP-ALL patients who experienced relapse. However, no mutations in TP53 (exons 5−9) were detected in four relapsed leukemia samples of eight relapsed patients. Furthermore, TP53 mutation analysis of the diagnostic samples of eight relapsed patients did not show any mutations. However, because the prognosis of BCP-ALL with TCF3-PBX1 fusion has improved dramatically in the last decades, TP53 mutations might not be associated with relapse in this subgroup.
IKZF1 deletion is known to be a strong prognostic factor associated with poor outcome in pediatric BCP-ALL 11,14–16. However, the significance of IKZF1 deletion in BCP-ALL with TCF3-PBX1 fusion has not been determined. Previous studies have shown that IKZF1 deletion is less frequent in BCP-ALL with recurrent chromosomal abnormalities 11,14–16, which is in agreement with our results showing that the frequency of IKZF1 deletion was lower in the TCF3-PBX1 fusion positive subgroup than in the other subgroups (Table 4). However, IKZF1 deletion was detected in 2 (25%) of eight evaluable relapsed patients, whereas it was not present in any of the patients maintaining continuous CR (Table 5). In addition, MLPA analysis of 22 diagnostic samples of BCP-ALL with TCF3-PBX1 in the independent CCLSG cohort identified one patient with IKZF1 deletion who eventually relapsed and died. Although our observation may be potentially interesting, further study employing larger cohort is warranted to determine the prognostic impact of IKZF1 deletion in this subgroup.
Ferreiros-Vidal et al. reported that Ikaros-regulated genes are highly represented in pre-Bcell receptor signaling, cell cycle regulation, and the somatic rearrangement of Ig genes, which are key to the differentiation of B-cell progenitors 17. These authors showed that inducible Ikaros expression in cycling pre-B cells was sufficient to drive transcriptional changes, such as repression of Myc, Cdk6, Ccnd2, Cdkn1a, and Cdkn1b, resembling the differentiation of cycling to resting pre-B cells in vivo. These findings suggested that haplo-insufficiency of IKZF1 might be associated with the differentiation block and accelerated cell cycle progression in BCP-ALL. In that study, 52% of TCF3 target genes were also bound by Ikaros, suggesting that these two transcriptional factors collaborate to regulate B-cell specification. These findings indicate that a severe impairment in the differentiation of cycling to resting pre-B cells in BCP-ALL with IKZF1 deletion and TCF3-PBX1 fusion may be associated with poor prognosis.
In this study, no additional genetic alterations were detected in ∼75% of relapsed patients with TCF3-PBX1. To identify additional genetic alterations related to poor prognosis, a comprehensive genomic analysis using matched pair samples of onset and relapse might be useful. Our group is therefore planning to perform exome analysis of relapsed and nonrelapsed patients with BCP-ALL expressing TCF3-PBX1.
In conclusion, the prognosis of pediatric BCP-ALL patients with TCF3-PBX1 fusion treated according to the JACLS ALL02 and CCLSG ALL2004 protocol was similar to that reported in studies performed in Western countries. Although concomitant IKZF1 deletion may account for ∼25% of treatment failure in this subgroup, further study of larger cohort is warranted to determine the prognostic impact of IKZF1 deletion in this subgroup.
Acknowledgments
The authors thank all the patients who participated in this study and their guardians. The authors also thank the staff of the OSCR data center for data management and all physicians who registered the patients in the JACLS ALL02 clinical trial. This work was supported by grants for Clinical Cancer Research and Research on Measures for Intractable Diseases from the Japanese Ministry of Health, Labor and Welfare and by grants-in-aid for scientific research from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. The primer list of TCF3-PBX1 and TP53 used in the current study.
Table S2. Univariate Cox model of event-free and overall survival of 112 patients with TCF3-PBX1
Figure S1. Comparison of minimal residual disease (MRD) between relapsed and nonrelapsed patients determined by quantitative RT-PCR of the TCF3-PBX1 transcript on days 15 and 33 of the induction phase in JACLS cohort: (A) day 15 and (B) day 33.
References
- 1.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 2.Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, et al. Prognostic effect of chromosomal abnormalities in childhood B cell precursor acute lymphoblastic leukaemia: results from the UK medical research council ALL97/99 randomized trial. Lancet Oncol. 2010;11:429–438. doi: 10.1016/S1470-2045(10)70066-8. [DOI] [PubMed] [Google Scholar]
- 3.Mellentin JD, Murre C, Donlon TA, McCaw PS, Smith SD, Carroll AJ, et al. The gene for enhancer binding proteins E12/E47 lies at the t(1;19) breakpoint in acute leukemias. Science. 1989;246:379–382. doi: 10.1126/science.2799390. [DOI] [PubMed] [Google Scholar]
- 4.Andersen MK, Autio K, Barbany G, Borgstrom G, Cavelier L, Golovleva I, et al. Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br. J. Haematol. 2011;155:235–243. doi: 10.1111/j.1365-2141.2011.08824.x. [DOI] [PubMed] [Google Scholar]
- 5.Jeha S, Pei D, Raimondi SC, Onciu M, Campana D, Cheng C, et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia. 2009;23:1406–1409. doi: 10.1038/leu.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felice MS, Gallego MS, Alonso CL, Alfaro EM, Guitter MR, Bernasconi AR, et al. Prognostic impact of t(1;19)/TCF3–PBX1 in childhood acutelymphoblastic leukemia in the context of Berlin–Frankfurt–Munster-based protocols. Leuk. Lymph. 2011;52:1215–1221. doi: 10.3109/10428194.2011.565436. [DOI] [PubMed] [Google Scholar]
- 7.Hunger SP, Fall MZ, Camitta BM, Carroll AJ, Link MP, Lauer SJ, et al. E2A-PBX1 chimeric transcript status at end of consolidation is not predictive of treatment outcome in childhood acute lymphoblastic leukemias with a t(1;19)(q23;p13): a pediatric oncology group study. Blood. 1998;91:1021–1028. [PubMed] [Google Scholar]
- 8.Kawamura M, Kikuchi A, Kobayashi S, Hanada R, Yamamoto K, Horibe K, et al. Mutations of the p53 and ras genes in childhood t(1;19)-acute lymphoblastic leukemia. Blood. 1995;85:2546–2552. [PubMed] [Google Scholar]
- 9.Suzuki N, Yumura-Yagi K, Yoshida M, Hara J, Nishimura S, Kudoh T, et al. Japan Association of Childhood Leukemia Study (JACLS). Outcome of childhood acute lymphoblastic leukemia with induction failure treated by the Japan Association of Childhood Leukemia Study (JACLS) ALL F-protocol. Pediatr. Blood Cancer. 2010;54:71–78. doi: 10.1002/pbc.22217. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa D, Hara J, Suenobu S, Takahashi Y, Sato A, Suzuki N, et al. Successful abolition of prophylactic cranial irradiation in children with non-T acute lymphoblastic leukemia (ALL) in the Japan Association of Childhood Leukemia Study (JACLS) ALL-02 trial. Blood (ASH Annual Meeting Abstracts) 2011;118:653. [Google Scholar]
- 11.Asai D, Imamura T, Suenobu S, Saito A, Hasegawa D, Deguchi T, et al. IKZF1 deletion is associated with a poor outcome in pediatric B cell precursor acute lymphoblastic leukemia in Japan. Cancer Med. 2013;2:412–419. doi: 10.1002/cam4.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyakuna N, Shimomura Y, Watanabe A, Taga T, Kikuta A, Matsushita A, et al. for the Japanese Childhood Cancer and Leukemia Study Group (JCCLSG) Assessment of corticosteroid-induced osteonecrosis in children undergoing chemotherapy for acute lymphoblastic leukemia: a report from the Japanese Childhood Cancer and Leukemia Study Group. J. Pediatr. Hematol. Oncol. 2014;36:22–29. doi: 10.1097/MPH.0000000000000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwab CJ, Jones LR, Morrison H, Ryan SL, Yigittop H, Schouten JP, et al. Evaluation of multiplex ligation-dependent probe amplification as a method for the detection of copy number abnormalities in B-cell precursor acute lymphoblastic leukemia. Genes Chromosom. Cancer. 2010;49:1104–1113. doi: 10.1002/gcc.20818. [DOI] [PubMed] [Google Scholar]
- 14.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Children's oncology group. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuiper RP, Waanders E, van der Velden VH, van Reijmersdal SV, Venkatachalam R, Scheijen B, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24:1258–1264. doi: 10.1038/leu.2010.87. [DOI] [PubMed] [Google Scholar]
- 16.Chen IM, Harvey RC, Mulligham CG, Gastier-Foster J, Wharton W, Kang H, et al. Outcome modeling with CRLF2 IKZF1 JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group Study. Blood. 2012;119:3512–3522. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferreiros-Vidal I, Carroll T, Taylor B, Terry A, Liang Z, Bruno L, et al. Genome-wide identification of Ikaros targets elucidates its contribution to mouse B-cell lineage specification and pre-B-cell differentiation. Blood. 2013;121:1769–1782. doi: 10.1182/blood-2012-08-450114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The primer list of TCF3-PBX1 and TP53 used in the current study.
Table S2. Univariate Cox model of event-free and overall survival of 112 patients with TCF3-PBX1
Figure S1. Comparison of minimal residual disease (MRD) between relapsed and nonrelapsed patients determined by quantitative RT-PCR of the TCF3-PBX1 transcript on days 15 and 33 of the induction phase in JACLS cohort: (A) day 15 and (B) day 33.