

Abstract
Direct tissue imaging mass spectrometry (IMS) by matrix-assisted laser desorption ionization and time-of-flight (MALDI-TOF) mass spectrometry has become increasingly important in biology and medicine, because this technology can detect the relative abundance and spatial distribution of interesting proteins in tissues. Five thyroid cancer samples, along with normal tissue, were sliced and transferred onto conductive glass slides. After laser scanning by MALDI-TOF equipped with a smart beam laser, images were created for individual masses and proteins were classified at 200-µm spatial resolution. Based on the spatial distribution, region-specific proteins on a tumor lesion could be identified by protein extraction from tumor tissue and analysis using liquid chromatography with tandem mass spectrometry (LC-MS/MS). Using all the spectral data at each spot, various intensities of a specific peak were detected in the tumor and normal regions of the thyroid. Differences in the molecular weights of expressed proteins between tumor and normal regions were analyzed using unsupervised and supervised clustering. To verify the presence of discovered proteins through IMS, we identified ribosomal protein P2, which is specific for cancer. We have demonstrated the feasibility of IMS as a useful tool for the analysis of tissue sections, and identified the tumor-specific protein ribosomal protein P2.
Graphical Abstract
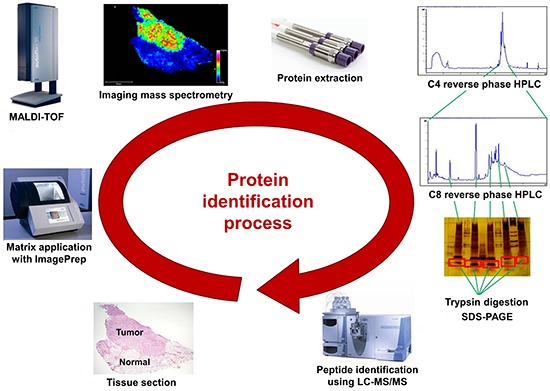
Keywords: Pathology; Proteins; Spectrometry, Mass, Matrix-Assisted Laser Desorption-Ionization; Thyroid Gland; Neoplasms
INTRODUCTION
Papillary thyroid carcinomas (PTC) are the most common form of thyroid cancer, accounting for nearly 85% of primary thyroid malignancies. Commonly, the solitary nodule is a palpably discrete swelling within an otherwise apparently normal thyroid gland, and is incidentally detected by radiological evaluation (1). From a clinical standpoint, the possibility of neoplastic disease is a major cause for concern in patients who present with thyroid nodules, because most conventional papillary carcinomas present as asymptomatic thyroid nodules. Recent studies have demonstrated 2 molecular mechanisms that function in the carcinogenesis of thyroid cancer: the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/Akt pathways (2, 3). Additionally, such pathways are related to genetic alterations such as rearrangements in the "rearranged during transfection" (RET) or neurotrophic tyrosine kinase receptor 1 (NTRK1) genes, and the activation of point mutations in the BRAF gene (4). In a previous study, protein expression patterns according to genetic variables have been demonstrated to correlate with the microscopic features, clinical manifestations, and prognostic characteristics of PTC (5). Although various protein alterations have been detected as diagnostic markers or prognostic predictors, the challenge remains to identify the expression of new molecules in tumors.
In biological research, protein expression profiling technology is a useful method to identify differential protein expression patterns and modifications. Analytical techniques with high sensitivity and increased throughput are required for the discovery of new biomarkers and new drugs. Recent progress in imaging mass spectrometry (IMS) has made it possible to identify several cellular components such as proteins, drugs, and other endogenous molecules directly on tissue sections (6, 7, 8, 9, 10). IMS uses matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) technology. The matrix is applied on cryosectioned tissue and a very small area of the matrix-applied tissue is analyzed using MALDI-MS. Differences between the analyzed areas are displayed by the imaging program. Each analyzed area of the tissue section, which can be as small as >200 µm, on a conductive surface such as gold-plated or indium tin oxide (ITO)-coated slide, is analyzed spot by spot using MALDI-MS after the application of the matrix. Using all the spectral data from each spot, the magnitudes of specific peaks on each spot can be displayed in terms of color intensity. In this way, spatial information on the tissue can be obtained. Based on the spatial distribution, region-specific proteins on a tissue can be identified by extracting proteins from the tissue, digesting them with trypsin, and analyzing the fragments using liquid chromatography with tandem MS (LC-MS/MS). This technology using IMS has broad applications in the detection of new proteins in various fields. Compared with conventional imaging methods, the advantages of IMS are that it does not require the additional use of a specific antibody against the protein, and that it integrates histopathology and protein expression (11). For this reason, IMS provides superior information regarding distinct molecular arrangements in tissue sections.
In the present study, we attempted to analyze thyroid samples, including those from PTC, using IMS. Before analysis using IMS, the diagnosis was confirmed by microscopic evaluation, immunohistochemical (IHC) staining of markers such as CK19, galectin-3, and RET, and pyrosequencing of the BRAF mutation, and then 5 homogenous samples of PTC were selected. After comparison of the differential molecular weight distribution between normal and tumor tissues using IMS, a protein of a specific molecular weight was identified by sequencing combined with LC-MS/MS, and then the presence of this protein was reconfirmed by western blot analysis.
MATERIALS AND METHODS
Patient selection
This study investigated 5 patients who had been diagnosed with papillary carcinoma in the thyroid and had undergone total thyroidectomy (Fig. 1). Four of the patients were female and one was male. Patient ages ranged from 48-68 yr (mean age, 54.6 yr). Perithyroidal invasions of the tumor were observed in all patients, lymph node metastasis in 3 patients, and association with nodular hyperplasia in 2 patients. Tumor sizes ranged from 0.6-1.5 cm (average size, 0.96 cm). To prepare tumor samples with identical molecular characteristics, the same configuration (BRAF mutation-positive, CK19 and galectin-3, and RET-negative), as confirmed by pyrosequencing and IHC staining, was selected.
Fig. 1.

Gross and microscopic findings of the thyroid. (A) The right thyroid showed a papillary carcinoma (black arrow) and nodular hyperplasia (black dash line arrow). Immunohistochemistry was positive in CK19 (B) and Galectin-3 (C).
Tissue sections
All samples were immersed in liquid nitrogen immediately after excision. The frozen tissue was then sectioned into 10-µm thick slices at -20℃ using a cryo-microtome (CM-1850; Leica Microsystems Nussloch GmbH, Nussloch, Germany). The optimum cutting temperature (OCT) polymer can cause deterioration of IMS signals; therefore, special care should be taken to avoid OCT contamination in sections where it has been used to stabilize the tissue. The optimal procedure involves the use of OCT to adhere the tissue to the sample disc, without allowing it to come into contact with the sliced tissue. The frozen tissue sections were then thaw-mounted on ITO-coated glass slides (Bruker Daltonics Inc. Bremen, Germany). The prepared slides were dried in desiccators for about 20 min and then stored at -80℃ until use.
Sample preparation for imaging mass spectrometry
Sinapinic acid (SA; Bruker Daltonics Inc.) was used as the protein MALDI matrix and prepared as a 10 mg/mL solution in a 50:50 solution of acetonitrile and 0.5% trifluoroacetic acid (TFA). An ImagePrep instrument (Bruker Daltonics Inc.) was used to spray a total of 3 mL of matrix solution on each tissue section. The optimal parameters (dry time, incubation time, and thickness) of the ImagePrep instrument were set to obtain a homogeneous matrix crystal on the tissue sections. After matrix application, the homogeneity of the matrix on the tissue was checked using the imaging function of Chip-1000 (Shimadzu Biotech, Kyoto, Japan). The ITO slide containing the tissue section was mounted on an MTP slide adapter (Bruker Daltonics Inc.), which was directly transferred to the MALDI mass spectrometer.
Matrix-assisted laser desorption ionization and time-of-flight mass spectrometry
IMS spectra were acquired in both positive- and negative-ion reflector modes with the aid of an Autoflex III MALDI-TOF mass spectrometer equipped with smart beam laser technology (Nd:YAG, 355 nm; Bruker Daltonics Inc.). MS data were acquired by averaging signals from 500 consecutive laser shots with a frequency of 200 Hz. All experiments conducted in this study were independently repeated 3 times. The spatial resolution of each of the mass images shown is 200 µm. Prior to each data acquisition, external calibration was conducted using a mixed peptide calibration standard with a m/z range of 800-3,200. All obtained spectra were baseline-subtracted.
Image analysis
Data analysis software such as FlexImaging ver. 2.0, FlexAnalysis ver. 3.0 and ClinProTools ver. 2.1 from Bruker Daltonics (Billerica, MA, USA) was employed to obtain the density maps of the species of interest and to check differences in the intensity of each spectrum. We manually analyzed normal and cancer regions according to supervised classification. Additionally, unsupervised classification was performed with Principal Component Analysis (PCA) using the ClinProTools software, and the results of this analysis were displayed using the FlexImaging software.
Tissue lysis and high performance liquid chromatography separation
After tissue sectioning for IMS, the remaining tissue masses were separated into normal and cancer regions and subjected to further protein identification. To remove any remaining OCT and blood, the tissue sample was washed with PBS. The T-PER tissue protein extraction reagent (Pierce/Thermo Fisher Scientific Inc. Rockford, IL, USA), with added protease and phosphatase inhibitors for preventing protein degradation, was used as the lysis buffer. The suspended tissue samples were Dounce-homogenized at 200 rpm for 2 min on ice followed by sonication (5 sec×2). The final tissue lysates were centrifuged at 10,000×g for 5 min at 4℃ to remove intact cells and cell debris.
For the separation of target proteins from tissue lysates, sequential chromatography using C4 and C8 columns was performed. For the C4 column, the initial protein amount was 3 mg, and this was fractionated into 60 fractions. The fractions that contained proteins with the target m/z values were pooled and separated further using a C8 column. The resulting fractions were verified by MALDI-MS, and then subjected to protein identification using LC-MS/MS.
In-gel digestion and mass spectrometry analysis
Fractions of interest were digested by a trypsin in-gel digestion procedure following separation by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Bands of interest were manually cut out, and the gel pieces were destained and washed prior to in-gel digestion. In-gel digestion was carried out with 250 ng/µL sequencing grade modified trypsin (Promega, Madison, WI, USA) in 50 mM NH4HCO4 buffer (pH 8.0) at 37℃ overnight. The digested peptides were extracted using 5% formic acid in acetonitrile and cleared by centrifugation at 1,000×g for 5 min. The supernatant was dried in a SpeedVac, and the resulting tryptic peptides were analyzed by LC-MS/MS with a fused-silica microcapillary C18 column (75 µm internal diameter; 100 mm length). LC was performed with the following linear gradient conditions: 0 min 3% B, 15 min 8% B, 23 min 16% B, 50 min 48% B, and 52 min 90% B (buffer A: 0.1% formic acid in H2O; buffer B: 0.1% formic acid in acetonitrile). The column was eluted with 90% solvent B for 10 min at the end of each run, and then re-equilibrated with the initial solvent condition (3% solvent B) for the next run. The flow rate was 250 nL/min. The separated peptides were subsequently analyzed by linear ion trap mass spectrometry (LIT-MS) on an LTQ spectrometer (Thermo Fisher Scientific Inc., San Jose, CA, USA). The electrospray voltage was set at 2.0 kV, and the threshold for switching from MS to MS/MS was 500. The normalized collision energy for MS/MS was 35% of the main radio frequency amplitude, and the duration of activation was 30 ms. All spectra were acquired in data-dependent mode. Each full MS scan was followed by nine MS/MS scans, corresponding to the 9 most intense peaks in the full MS scan. The repeat count of each peak for dynamic exclusion was 1, and the repeat duration was 30 sec. The dynamic exclusion duration was set at 180 sec and the exclusion mass width was ±1.5 Da. The list size of dynamic exclusion was 50.
Database analysis
The acquired LC-ESI-MS/MS fragment spectra were analyzed in the BioWorksBrowser™ (version Rev. 3.3.1 SP1, Thermo Fisher Scientific Inc., CA, USA) with the SEQUEST search engines against the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) non-redundant human database.
The SEQUEST criteria for identifying peptides included the following: Xcorr values greater than 1.8, 2.2, and 3.5 for +1, +2, and +3 charge state peptides, respectively, a delta Cn value >0.1, a fixed modification of carbarmidomethylation at cysteine (+58 Da), and a variable modification of oxidation at methionine (+16 Da).
Ethics statement
This study protocol was approved by the institutional review board of the Konkuk University Hospital (IRB No. KUH 1210010). Informed consent was confirmed by the board.
RESULTS
Tissue profiling by matrix-assisted laser desorption/ionization imaging mass spectrometry
Hematoxylin and eosin (HE)-stained slides allowed the visualization of tumor lesions as dark pinkish areas and normal tissue as light pinkish areas (Fig. 2). We obtained spectra from tumor and normal areas of the thyroid tissue during the scan (Supplementary Fig. 1). These spectra clearly showed multiple peaks, representing patterns of protein expression in tumor and normal tissue. For the evaluation of a particular protein expression pattern, these spectra were analyzed using unsupervised and supervised clustering analyses, and significant m/z values were acquired that allowed the classification of tissues as tumor or normal samples (Supplementary Table 1).
Fig. 2.
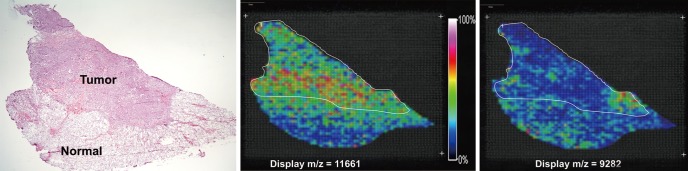
Selected molecular ion images from a papillary thyroid carcinoma section. The thyroid slide stained hematoxylin and eosin revealed papillary carcinoma. Imaging mass spectrometry based on Principal Component Analysis, molecular ions corresponding to 11,661 m/z and 9,282 m/z.
Spectrometric images of molecules with different m/z values on frozen sections revealed various imaging patterns. Images reconstructed from supervised-classification analysis and PCA of the acquired datasets clearly differentiated tumor and normal regions. The classification and PCA data revealed that the molecular distributions match the HE-stained images of excised thyroid tissue samples. Of five cases, 4 tissue samples demonstrated that peaks corresponding to m/z values of 11,651-11,667 exhibited higher intensities in tumor lesions, whereas the peak with an m/z value of 9,268-9,284 consistently showed higher intensities in normal thyroid. Color was used to represent ions, and color saturation was a function of the relative intensity (see color reference bar).
Protein identification of tumor tissues
In the above sections, we have described the usefulness of IMS for protein profiling in tumor and normal thyroid tissue samples. To identify differentially expressed proteins, we extracted proteins via lysis of surgically excised tissue specimens from 5 patients. For direct MALDI-TOF MS analysis, proteins extracted from tumor and normal tissue samples were separated by chromatography on C4 and C8 columns. The fractions contained a protein with an m/z value of 11,661, consistent with one of the significant m/z values obtained from the above supervised and unsupervised clustering analyses of MALDI-IMS data (Supplementary Fig. 2).
For further evaluation, the pooled fractions were digested by a tryptic in-gel digestion procedure following SDS-PAGE. The resulting tryptic peptides from the pooled fractions were sequenced using LC-MS/MS. LC-MS/MS results were analyzed using the BioWorksBrowser™ (version Rev. 3.3.1 SP1, Thermo Fisher Scientific Inc., CA, USA) for the identification of peptide sequences (Fig. 3).
Fig. 3.

Protein identification (include mass spectrum for protein in HPLC fractions). Identification of proteins of differential expression was carried out by digesting of separated proteins with trypsin and sequencing by LC-MS/MS. MS/MS spectra showing a peptide sequences. YVASYLLAALGGNSSPSAK of fraction 50 (pooled fraction 4): Xc value is 2.420, ΔCn value is 0.261 (A), NIEDVIAQGIGK of fraction 50 (pooled fraction 4): Xc value is 4.347, ΔCn value is 0.394 (B), LASVPAGGAVAVSAAPGSAAPAAGSAPAAAEEK of fraction 50 (pooled fraction 4): Xc value is 4.308 (C), ΔCn value is 0.608 and tolerances of m/z of the peptide were ± 2 and ± 1 for full mass and MS/MS fragmentation, respectively.
Peptide profiles analyzed by BioWorksBrowser™ software were matched to 64 amino acid sequences (55.7%) within 3 unique peptides of ribosomal protein P2 comprised of 115 amino acids. The correspondence between the identified protein and ribosomal protein P2 was compatible with high confidence levels. The 3 unique matching peptide sequences had m/z values of 1,870.11 (amino acids 3-21; sequence, R.YVASYLLAALGGNSSPSAK. D), 1,257.43 (amino acids 50-61; sequence, K.NIEDVIAQGIGK.L), and 2,776.08 (amino acids 62-94; sequence, K.LASVPAGGAVAVSAAPGSAAPAAGSAPAAAEEK.K) (Supplementary Fig. 3).
In addition, the MALDI-IMS data were re-analyzed to investigate whether the identified ribosomal protein P2, with an m/z value of 11,657, showed differential distributions in the tumor lesions of another thyroid sample. The tumor tissue showed high concentration of detecting protein with an m/z value 11,661, corresponding to ribosomal protein P2 as identified by LC-MS/MS (Fig. 4). Additionally, the molecular distribution matched the HE-stained image of the surgically excised thyroid tissue. Therefore, IMS showed that the localization of the protein was consistent with the location of the tumor lesion.
Fig. 4.

In imaging mass spectrometry, the distribution of detecting protein (11,661 m/z) displayed high concentration and corresponded with concentration of ribosomal protein p2 (11,657 m/z) in tumor lesion (Δmass: 4 m/z).
DISCUSSION
One of the most commonly used strategies in proteomics involves the identification of new proteins that are differentially expressed between normal and tumor tissues. The aims of cancer proteomics are to define a more precise molecular classification of tumors than is accomplished by conventional classification, and to discover meaningful biomarkers for diagnosis, development of therapeutic strategies, and prognosis. Recently, IMS has been highlighted as a promising tool for the visualization and identification of biomolecules in tissue sections (12). In addition to conventional methods such as genomics or proteomics, IMS has been considered to be of high importance for the discovery of new molecules. In the present study, we demonstrated the classification of tissue samples into tumor and normal tissues by the analysis of differential protein molecular weight distributions in human PTC samples. Among the differentially expressed peaks, ribosomal protein P2 was identified by LC-MS/MS as a protein that is differentially expressed in the tumor area. Consequently, these findings suggest that IMS may contribute to the identification of various proteins in future studies.
Profiling and imaging by IMS has been previously applied to tissue samples from various diseases, including lung, brain, breast, ovary, stomach and thyroid cancer (13, 14, 15, 16, 17, 18). Molecular protein signatures represent a unique data set with which to classify and correlate clinically relevant information and outcomes with changing molecular events during the progression and treatment of disease (19). The approach taken by most published studies are comparative proteomic analysis whereby mass spectral features (m/z peaks) are correlated with a variety of clinical data, such as therapeutic regimen and overall outcome. Some studies have demonstrated the identification of specific molecular changes associated with progression of disease. For instance, molecular signatures from glioma, ovarian cancer (20, 21) and non-small-cell lung cancer can be used to classify these lesions according to histological grade and to predict the patients' prognosis (13, 14). However, such significant molecular data have clinical limitations in predicting diagnosis and survival, because of the heterogeneous distributions of different proteins in spite of the fact that each tumor sample has the same origin.
Although clinical limitations still exist, IMS may be helpful in the identification of unknown biological variables in various tumors. Therefore, this technique will provide a common disease-wide approach that can be applied to search specific disease entities. Previous data suggest that IMS might be superior to immunohistochemical staining in identifying the site of origin for various tumors (22). Furthermore, the particular protein profiles identified by IMS could be useful to improve the selection of drug agents during the development of a therapeutic strategy.
Our IMS analyses of PTC sections reveal that the distributions of differentially expressed proteins show a separation between normal and tumor tissues based on discrete molecular weight. In particular, recently developed analytical methods, such as supervised clustering using manual analysis and unsupervised clustering using PCA, were conducted to improve the separation of distributional prototypes and to identify specific proteins in thyroid tissue. Subsequently, the presence of ribosomal protein P2 was identified by LC-MS/MS. A published study reports that ribosomal protein P2 is associated with tumor growth and could be a potential molecular target for antisense therapy of human malignancies, but its function in PTC is still unknown (23).
This study has several limitations. First, the small number of thyroid samples results in a limited ability to demonstrate an association between the detected protein and PTC. Second, in matching the m/z value of expected protein to that of ribosomal protein P2, the m/z value might have been altered due to technological limitations such as insufficient mass resolution in the higher m/z region, although our m/z value was within the allowable error limit. Such technological limitations can make it difficult to ascertain our conclusions. Therefore, progressive technological improvement is essential to improve accuracy.
Our study successfully showed the feasibility of using IMS as a useful tool for the analysis of tissue sections. This method may provide a new direction in the study of cancer, by identifying tumor-specific markers, and thereby leading to elucidation of the mechanism of tumorigenesis and the identification of therapeutic targets. Additionally, protein profiles obtained from IMS may allow the accurate prediction of tumor behavior, diagnosis, and prognosis, and uncover the etiologies underlying idiopathic disease.
Footnotes
Funding: The research was supported by grants from the National Research Foundation of Korea (Grant No., 2009-0072346) and the Converging Research Center Program (2011K000898) through the Ministry of Education, Science and Technology, Korea.
The authors have no conflict of interest to disclose.
Supplementary Material
Matrix-assisted laser desorption/ionization mass spectra taken a different locations within a papillary carcinoma. Over 100 different peaks could be detected with some of them having a distinct spatial distribution in the tissue. top, tumor area; bottom, normal area.
Matrix-assisted laser desorption/ionization mass spectrometry protein profiles from the HPLC fractions (fraction 50, pooled fraction 4). The peptide of 11,661 m/z (red arrow) was frequently found in tumor rather than in normal tissue.
Peptide profiles analyzed by BioWorksBrowserTM software were matched to 55.7% of amino acid sequences within 3 unique peptides (red circle) of ribosomal protein P2.
Comparisons between tumor and normal tissues according to supervised and unsupervised cluster analyses
References
- 1.Mazzaferri EL. Management of a solitary thyroid nodule. N Engl J Med. 1993;328:553–559. doi: 10.1056/NEJM199302253280807. [DOI] [PubMed] [Google Scholar]
- 2.Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6:292–306. doi: 10.1038/nrc1836. [DOI] [PubMed] [Google Scholar]
- 3.Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J Clin Endocrinol Metab. 2008;93:278–284. doi: 10.1210/jc.2007-1076. [DOI] [PubMed] [Google Scholar]
- 4.Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28:742–762. doi: 10.1210/er.2007-0007. [DOI] [PubMed] [Google Scholar]
- 5.Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, Biddinger PW, Nikiforov YE. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol. 2006;30:216–222. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- 6.Chaurand P, Schwartz SA, Reyzer ML, Caprioli RM. Imaging mass spectrometry: principles and potentials. Toxicol Pathol. 2005;33:92–101. doi: 10.1080/01926230590881862. [DOI] [PubMed] [Google Scholar]
- 7.Seeley EH, Caprioli RM. MALDI imaging mass spectrometry of human tissue: method challenges and clinical perspectives. Trends Biotechnol. 2011;29:136–143. doi: 10.1016/j.tibtech.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seeley EH, Caprioli RM. Imaging mass spectrometry: towards clinical diagnostics. Proteomics Clin Appl. 2008;2:1435–1443. doi: 10.1002/prca.200800013. [DOI] [PubMed] [Google Scholar]
- 9.Ko KH, Kwon CI, Park SH, Han NY, Lee HK, Kim EH, Hahm KB. Application of matrix-assisted laser desorption/ionization time-of-flight imaging mass spectrometry (MALDI-TOF IMS) for premalignant gastrointestinal lesions. Clin Endosc. 2013;46:611–619. doi: 10.5946/ce.2013.46.6.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko KH, Han NY, Kwon CI, Lee HK, Park JM, Kim EH, Hahm KB. Recent advances in molecular imaging of premalignant gastrointestinal lesions and future application for early detection of barrett esophagus. Clin Endosc. 2014;47:7–14. doi: 10.5946/ce.2014.47.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimma S, Sugiura Y, Hayasaka T, Hoshikawa Y, Noda T, Setou M. MALDI-based imaging mass spectrometry revealed abnormal distribution of phospholipids in colon cancer liver metastasis. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;855:98–103. doi: 10.1016/j.jchromb.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 12.Shanta SR, Zhou LH, Park YS, Kim YH, Kim Y, Kim KP. Binary matrix for MALDI imaging mass spectrometry of phospholipids in both ion modes. Anal Chem. 2011;83:1252–1259. doi: 10.1021/ac1029659. [DOI] [PubMed] [Google Scholar]
- 13.Yanagisawa K, Shyr Y, Xu BJ, Massion PP, Larsen PH, White BC, Roberts JR, Edgerton M, Gonzalez A, Nadaf S, et al. Proteomic patterns of tumour subsets in non-small-cell lung cancer. Lancet. 2003;362:433–439. doi: 10.1016/S0140-6736(03)14068-8. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, Johnson MD, Caprioli RM. Proteomic-based prognosis of brain tumor patients using direct-tissue matrix-assisted laser desorption ionization mass spectrometry. Cancer Res. 2005;65:7674–7681. doi: 10.1158/0008-5472.CAN-04-3016. [DOI] [PubMed] [Google Scholar]
- 15.Cornett DS, Mobley JA, Dias EC, Andersson M, Arteaga CL, Sanders ME, Caprioli RM. A novel histology-directed strategy for MALDI-MS tissue profiling that improves throughput and cellular specificity in human breast cancer. Mol Cell Proteomics. 2006;5:1975–1983. doi: 10.1074/mcp.M600119-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Lemaire R, Menguellet SA, Stauber J, Marchaudon V, Lucot JP, Collinet P, Farine MO, Vinatier D, Day R, Ducoroy P, et al. Specific MALDI imaging and profiling for biomarker hunting and validation: fragment of the 11S proteasome activator complex, Reg alpha fragment, is a new potential ovary cancer biomarker. J Proteome Res. 2007;6:4127–4134. doi: 10.1021/pr0702722. [DOI] [PubMed] [Google Scholar]
- 17.Balluff B, Rauser S, Meding S, Elsner M, Schöne C, Feuchtinger A, Schuhmacher C, Novotny A, Jütting U, Maccarrone G, et al. MALDI imaging identifies prognostic seven-protein signature of novel tissue markers in intestinal-type gastric cancer. Am J Pathol. 2011;179:2720–2729. doi: 10.1016/j.ajpath.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meding S, Nitsche U, Balluff B, Elsner M, Rauser S, Schöne C, Nipp M, Maak M, Feith M, Ebert MP, et al. Tumor classification of six common cancer types based on proteomic profiling by MALDI imaging. J Proteome Res. 2012;11:1996–2003. doi: 10.1021/pr200784p. [DOI] [PubMed] [Google Scholar]
- 19.Walch A, Rauser S, Deininger SO, Höfler H. MALDI imaging mass spectrometry for direct tissue analysis: a new frontier for molecular histology. Histochem Cell Biol. 2008;130:421–434. doi: 10.1007/s00418-008-0469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang S, Lee A, Park YS, Lee SC, Park SY, Han SY, Kim KP, Kim YH, Yoo CW, Kim HK. Alteration in lipid and protein profiles of ovarian cancer: similarity to breast cancer. Int J Gynecol Cancer. 2011;21:1566–1572. doi: 10.1097/IGC.0b013e318226c5f5. [DOI] [PubMed] [Google Scholar]
- 21.Kang HS, Lee SC, Park YS, Jeon YE, Lee JH, Jung SY, Park IH, Jang SH, Park HM, Yoo CW, et al. Protein and lipid MALDI profiles classify breast cancers according to the intrinsic subtype. BMC Cancer. 2011;11:465. doi: 10.1186/1471-2407-11-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaurand P, Sanders ME, Jensen RA, Caprioli RM. Proteomics in diagnostic pathology: profiling and imaging proteins directly in tissue sections. Am J Pathol. 2004;165:1057–1068. doi: 10.1016/S0002-9440(10)63367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner-Thorpe J, Ito H, Ashley SW, Whang EE. Ribosomal protein P2: a potential molecular target for antisense therapy of human malignancies. Anticancer Res. 2003;23:4549–4560. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Matrix-assisted laser desorption/ionization mass spectra taken a different locations within a papillary carcinoma. Over 100 different peaks could be detected with some of them having a distinct spatial distribution in the tissue. top, tumor area; bottom, normal area.
Matrix-assisted laser desorption/ionization mass spectrometry protein profiles from the HPLC fractions (fraction 50, pooled fraction 4). The peptide of 11,661 m/z (red arrow) was frequently found in tumor rather than in normal tissue.
Peptide profiles analyzed by BioWorksBrowserTM software were matched to 55.7% of amino acid sequences within 3 unique peptides (red circle) of ribosomal protein P2.
Comparisons between tumor and normal tissues according to supervised and unsupervised cluster analyses