

Abstract
The inflammatory response is a primary mechanism in the pathogenesis of ventilator-induced lung injury. Autophagy is an essential, homeostatic process by which cells break down their own components. We explored the role of autophagy in the mechanisms of mechanical ventilation-induced lung inflammatory injury. Mice were subjected to low (7 ml/kg) or high (28 ml/kg) tidal volume ventilation for 2 h. Bone marrow-derived macrophages transfected with a scrambled or autophagy-related protein 5 small interfering RNA were administered to alveolar macrophage-depleted mice via a jugular venous cannula 30 min before the start of the ventilation protocol. In some experiments, mice were ventilated in the absence and presence of autophagy inhibitors 3-methyladenine (15 mg/kg ip) or trichostatin A (1 mg/kg ip). Mechanical ventilation with a high tidal volume caused rapid (within minutes) activation of autophagy in the lung. Conventional transmission electron microscopic examination of lung sections showed that mechanical ventilation-induced autophagy activation mainly occurred in lung macrophages. Autophagy activation in the lungs during mechanical ventilation was dramatically attenuated in alveolar macrophage-depleted mice. Selective silencing of autophagy-related protein 5 in lung macrophages abolished mechanical ventilation-induced nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome activation and lung inflammatory injury. Pharmacological inhibition of autophagy also significantly attenuated the inflammatory responses caused by lung hyperinflation. The activation of autophagy in macrophages mediates early lung inflammation during mechanical ventilation via NLRP3 inflammasome signaling. Inhibition of autophagy activation in lung macrophages may therefore provide a novel and promising strategy for the prevention and treatment of ventilator-induced lung injury.
Keywords: lung injury, autophagy, mechanical ventilation, inflammasome, macrophage
mechanical ventilation not only saves the lives of patients with respiratory failure but also initiates and exacerbates lung injury known as ventilator-induced lung injury (VILI), which in turn contributes to patient morbidity and mortality (11). VILI is characterized by an influx of inflammatory cells, increased pulmonary vascular permeability, and endothelial and epithelial cell death. The release of proinflammatory cytokines by lung cells in response to mechanical stretch is a crucial event for initiation and/or potentiation of VILI and may cause systemic inflammatory response and multiple system organ failure (4, 22, 35). The alveolar macrophages (AMs) (5, 7, 26), neutrophils (42), alveolar epithelium (35), and endothelium (10) are all involved in the production of proinflammatory cytokines during mechanical ventilation.
Autophagy is an essential “self-eating” process important for cell survival and homoeostasis via degradation of unnecessary or dysfunctional cellular components as well as invading microorganisms (17, 24, 27). Following the induction of autophagy, omegasomes are derived from endoplasmic reticulum, followed by the formation and elongation of isolation membranes, or phagophores. The cytoplasmic cargo is then sequestered, and the double-membrane autophagosome fuses with a lysosome to form an autolysosome for degradation or recycling of cytoplasmic components. Autophagy is orchestrated by autophagy gene (Atg)-related protein complexes, which are involved in autophagy induction and sequestration. Class III phosphatidylinositol 3-kinase (PI3-kinase) complex and ATG proteins are required for the formation of isolation membrane. Two ubiquitin-like protein conjugation pathways cause the elongation of the isolation membrane. The cytosolic form of microtubule-associated protein 1A/1B-light chain 3 (LC3-I) is conjugated to phosphatidylethanolamine to form LC3-II, which is recruited to autophagosomal membranes, the process of which is essential for the autophagosome formation (24, 27). Under physiological conditions, autophagy occurs in most tissues and contributes to the routine turnover of cytoplasmic components. The autophagic/lysosomal pathway is a critical mechanism to maintain cellular homeostasis and may be considered as a cell survival program (17, 24). However, under pathological conditions, autophagy can be induced to protect the cell from injurious stimuli or, alternatively, to contribute to irreversible cell injury and autophagic death (9, 17, 27).
In addition to its role in starvation adaptation, development, anti-aging, cell death, and tumor suppression, autophagy is emerging as a process of high importance for the regulation of innate immunity and inflammation (17). Recent studies indicated that autophagy regulates the release of proinflammatory cytokines including IL-1β that is among the best markers of ventilator-induced lung inflammation (3). The role of autophagy activation in VILI has recently been shown in autophagin-1/ATG4B-deficient mice. Activation of autophagy following mechanical ventilation mediated lung inflammation and injury via NF-κB signaling pathway (21). In the present study, our findings extend these observations by indicating that autophagy activation specifically in lung macrophages mediated lung inflammatory injury via nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome activation during mechanical ventilation. Inhibition of autophagy activation in lung macrophages may therefore provide a novel and promising strategy for the prevention and treatment of VILI.
MATERIALS AND METHODS
Animals
Wild-type C57BL/6J and gp91phox null (gp91phox−/−) male mice were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in microisolator cages under specific pathogen-free conditions, fed with autoclaved food, and used in experiments at 8–12 wk of age. Mice were handled under a protocol approved by the University of Illinois Institutional Animal Care and Use Committee.
Depletion of AMs in Mice
Mouse AMs were depleted as previously described (40). Briefly, clodronate liposomes were made with phosphatidylserine, phosphatidylcholine, and cholesterol at a molar ratio of 1:6:4 in chloroform. The chloroform is removed by rotary evaporation (100 rpm) at 40°C. The clodronate liposome solution was filtered through 200-nm filters and then delivered by nebulization to anesthetized mice (ketamine 90 mg/kg ip).
Isolation and Culture of Mouse BMDMs
Bone marrow-derived macrophages (BMDMs) were generated from femurs of C57/BL6J mice as described previously (41). Briefly, bone marrow was flushed from femurs with PBS (without Ca2+ and Mg2+). Wash medium (2–5 ml) was used in each femur. Collected bone marrow cell suspensions were centrifuged for 5 min at 500 g at room temperature. Cell pellets were resuspended in macrophage complete medium (DMEM with 10% FBS, 20% L-929 cells conditioned medium, 10 mM l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin). Cells (4 × 105) were then added to each sterile plastic petri dish in 10 ml macrophage complete medium and incubated at 37°C and 5% CO2. On day 3, 5 ml of macrophage complete medium was replaced to each dish. After 7 days in culture, the adherent cells were ∼95% pure macrophages as evidenced by the expression of macrophage surface markers F40/8 (Santa Cruz), and these cells were used in subsequent experiments.
ATG5 Knockdown in BMDMs
ATG5 small interfering (si)RNA, which is a pool of three target-specific 20–25 nt siRNAs (Dharmacon) at a concentration of 50 nM, was added to 2×106 BMDMs in six-well plates to deplete ATG5 following the manufacturer's instruction. Successful downregulation of ATG5 in BMDMs in all experiments was confirmed by Western blot analysis. All experiments were performed 48 h posttransfection.
Experimental Protocols
A well-established in vivo mouse model of VILI was utilized as described previously (12, 19, 40). Briefly, mice were anesthetized with an intraperitoneal injection of a ketamine/xylazine mixture (75 mg/kg) and inhalation of 1% isoflurane for maintenance. Mice were tracheotomized and subjected to mechanical ventilation with a low tidal volume of 7 ml/kg and a frequency of 120 breath/min or a high tidal volume of 28 ml/kg and a frequency of 60 breath/min, by use of a Harvard Apparatus ventilator (MiniVent, Harvard Biosciences; Holliston, MA). All experiments were conducted with room air and 0 cmH2O end-expiratory pressure. The dead space (∼5 ml) was added to the ventilator circuit to maintain arterial Pco2 between 35 and 45 Torr (4.7–6.0 kPa). Body temperature was maintained between 37 and 38°C, by use of a heating lamp. In some experiments, saline or BMDMs transfected with a scrambled siRNA or ATG5 siRNA (2 × 106 cells, 200 μl total volume each) were administered to AM-depleted mice via a jugular venous cannula 30 min before mechanical ventilation. In other experiments, animals were pretreated with autophagy inhibitors 3-methyladenine (3-MA, 15 mg/kg ip) or trichostatin A (TSA, 1 mg/kg ip) 1 h before exposure to mechanical ventilation. After 2 h of mechanical ventilation, mice were euthanized and lung injury was assessed by analysis of bronchoalveolar lavage (BAL) fluid, extravascular lung water measurement, and biochemical analysis of lung tissue.
Isolation of Macrophages from Whole Mouse Lungs
AMs and interstitial macrophages were isolated from mouse lungs following mechanical ventilation.
Isolation of AMs.
AMs were harvested as previously described (8, 40). In brief, BAL was performed by intratracheal injection of 1 ml of ice-cold Mg2+- and Ca2+-free PBS containing 0.6 mM EDTA, followed by gentle aspiration. The lavage procedure was repeated 10 times. After washing the lavage cells, we made differential cell counts. Lavage fluid from mice contained 98% AMs. AMs were >96% viable as measured by Trypan blue exclusion.
Isolation of lung interstitial macrophages.
The lung after BAL was used for preparation of interstitial macrophages (8). The lung vascular bed was perfused with ice-cold PBS-EDTA until the tissue turned white, followed by perfusion with RPMI 1640 medium containing 50 U/ml collagenase (Worthington Type IV, Copper Biomedical, Malvern, PA) (enzyme solution). The lungs were excised and sliced into little pieces. The lung tissue fragments were incubated in a 15-ml enzyme solution for 60 min at 37°C in a shaking water bath. The solution was passed through a thin layer of premoistened nylon wool (70 μm) to remove cell debris. The lung-digested cells were then washed twice with RPMI 1640 medium and finally layered on top of a two-step Percoll gradient (20–50%). After centrifugation at 300 g at 4°C, the interphase between the 20 and 50% Percoll layers was harvested, washed again, and resuspended in RPMI 1640 medium. The suspension contained 96% interstitial macrophage. Interstitial macrophage were >95% viable as measured by Trypan blue exclusion.
Western Blotting and Immunoprecipitation
Western blot analysis of lysates of mouse resident alveolar macrophages and lung homogenates was performed as previously described (19, 36, 40). Briefly, fresh lung tissues were lysed on ice in the buffer containing 1 mM PMSF, 0.2 U/ml aprotinin, and 1 mM sodium orthovanadate. Protein concentrations of the samples were determined by use of the bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Total tissue lysate proteins (20 μg) were loaded for 16% SDS-PAGE, and the separated proteins were transferred onto nitrocellulose membranes by electroblotting. The membranes were incubated with primary antibodies overnight, washed, and then incubated with goat anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (1:3,000∼5,000) for 60 min. Protein bands were detected with the ECL SuperSignal reagent (Pierce). Relative band densities of the various proteins were measured from scanned films with NIH ImageJ Software.
Immunoprecipitation analysis was performed as described previously (32, 40). Lung tissue lysates were precleared by using 1 mg control IgG together with protein A/G PLUS-agarose beads and were then incubated overnight at 4°C with anti-NLRP3, anti-ASC, or anti-caspase-1 antibodies, followed by addition of 25 ml protein A/G PLUS-agarose beads. The resulting immunoprecipitates were dissolved in SDS-PAGE sample buffer for electrophoresis and immunoblot analysis.
Transmission Electron Microscopy
Lungs were excised and fixed with a fixative buffer containing 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M of phosphate-buffered solution and were stored at 4°C until embedding. Tissue samples were then postfixed in 1% phosphate-buffered osmium tetroxide and embedded in Spurr's resin. Ultrathin sections (0.1 μm) were stained with 1% uranyl acetate and 0.2% lead citrate and analyzed by transmission electron microscope (JEM-1220). The total areas of the autophagosome were calculated with Adobe Photoshop CS3 Extended software, and the percentage of the cell occupied by the autophagosome was calculated.
Lung Histology
For mice ventilated with normal or high tidal volume, the trachea was cannulated, and the lungs were fixed by instillation of 4% paraformaldehyde (Sigma-Aldrich) in PBS and held for 2 h under 25 cmH2O pressure. The lungs were removed and preserved in 10% neutral buffered formalin overnight. Formalin-fixed tissue was washed with PBS and dehydrated in 70% ethanol before paraffin embedding. Sections (4 μm thick) were stained with hematoxylin and eosin and examined by light microscopy.
Lung MPO Assay
MPO activity in the lung homogenates was determined by using a MPO assay kit (Invitrogen) according to manufacturer's protocol (38). Briefly, lung tissue (∼50 μg) was homogenized in 1 ml of cold sample buffer, sonicated three times for 15 s on ice, and centrifuged at 16,000 g for 30 min at 4°C. MPO activity was obtained in 100 μl of supernatants in duplicate by using development reagent over 25 s at 450 nm. Development reagent without sample was used as a control. MPO activity was expressed as change in absorbance per milligram of protein.
Determination of PMN Counts in BAL Fluid
At the end of experiments, BAL was performed by intratracheal injection of 1 ml of PBS followed by gentle aspiration. The lavage was repeated three times. The pooled BAL fluid was centrifuged at 400 g for 5 min and cell pellets were suspended in PBS. Cell suspensions were diluted to a final concentration of 1 × 105 cells/ml, and a 200-μl volume of resuspended cells was cytospun onto slides at 300 rpm for 5 min with a cytocentrifuge (Shandon Southern Instruments, Sewickley, PA). Slides were stained with Diff-Quick dye (Dade Behring, Newark, DE) and examined at a magnification of ×20 and ×40 by light microscopy. The percentage of polymorphonuclear neutrophils (PMNs) was determined after counting 300 cells in randomly selected fields. The total cell count in BAL fluid was manually measured with a hemocytometer.
Assessment of Pulmonary Vascular Permeability and Edema Formation
Protein concentration in BAL was determined with bicinchoninic acid method as an index of increased permeability of the alveolar-capillary barriers (12, 19, 38). Extravascular lung water (ELW) was used as an index of lung water content and edema (19). At the end of experiments, the lungs were removed, weighed, and homogenized after addition of 1 ml of distilled water. The lung homogenate was weighed and an aliquot was centrifuged (16,000 g, 10 min) for assay of hemoglobin concentration in the supernatant. Blood was collected through right ventricular puncture. A second aliquot of homogenate, supernatant, and blood was weighed and then desiccated in an oven (60°C for 48 h) for determination of ELW.
Cytokine Assay
Levels of IL-1β and IL-18 in BAL were measured by using commercial ELISA kits (R&D System and BD Biosciences) according to the manufacturer's instructions (19). Each value represents the means of triplicate determinations.
Drugs, Reagents, and Antibodies
TSA was purchased from Selleckchem and dissolved in DMSO. Peptide SS-31 was obtained from AnaSpec. LC3 polyclonal antibody was purchased from Cell Signaling. Anti-NLRP3, ASC, caspase-1, Pro-IL-1β, IL-1β, Pro-IL-18, and IL-18 antibodies were from Santa Cruz Biotechnology. Armenian hamster IgG isotype control was obtained from BioLegend and horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG antibodies from Santa Cruz Biotechnology.
All other chemicals and reagents used were obtained from Sigma Chemical (St. Louis, MO) unless otherwise stated.
Statistical Analysis
One-way ANOVA and Student-Newman-Keuls test for post hoc comparisons were used to determine differences between control and experimental groups. Student's t-test was performed for paired samples. Parameter changes between different groups over time were evaluated by a two-way ANOVA with repeated measures. Data are expressed as means ± SE, and differences between group means were considered significant at P < 0.05.
RESULTS
Mechanical Ventilation with a High Tidal Volume Activates Autophagy
To elucidate the role of autophagy in the mechanism of VILI, we first determined whether mechanical ventilation causes autophagy activation in the lung. LC3 is a soluble protein with a molecular mass of ∼16 kDa that is distributed ubiquitously in mammalian cells. Upon induction of autophagy, newly synthesized LC3 is cleaved by Atg4B to generate LC3-I. Cytosolic LC3-I is further converted to membrane-bound LC3-II (lapidated LC3) by the addition of phosphatidylethanolamine to the COOH-terminal glycine residue of LC3-I (13). LC3-II is stably associated to the autophagosomal membrane and is therefore considered a specific autophagy marker (13). We observed that LC3-II level in the lung remarkably increased within 15 min following mechanical ventilation with a high tidal volume (28 ml/kg) and maintained a maximal level for at least 2 h (Fig. 1A). LC3-II level was not altered during 2 h of low tidal volume ventilation (7 ml/kg). Mechanical ventilation also increased LC3-II protein expression in a tidal volume-dependent manner (Fig. 1B). These results indicate that mechanical stretch at a high tidal volume induces autophagy activation in the lung.
Fig. 1.
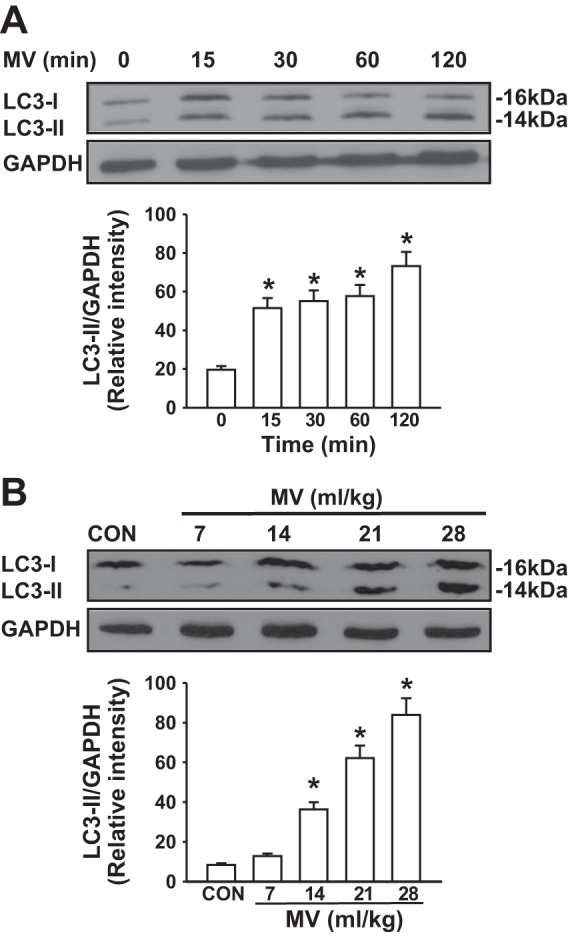
Mechanical ventilation (MV) induced autophagy activation in mouse lungs. Western blot analysis of lung homogenates was performed to determine the level of LC3-II (autophagy marker). A: mechanical ventilation induced autophagy activation. Mice were ventilated with a high tidal volume (28 ml/kg) for the indicated time (n = 3 for each time point). Top, representative Western blots for LC3-I and LC3-II; bottom, bar graph showing the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. B: mechanical ventilation induced autophagy activation in a tidal volume-dependent manner. Mice were ventilated with different tidal volumes (7–28 ml/kg) for 2 h (n = 3 for each tidal volume). Top, representative Western blots for LC3-II; bottom, bar graph showing the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. *P <0.05 vs. nonventilated group. CON, control.
Mitochondrial ROS Mediates Autophagy Activation in Response to Mechanical Ventilation
Signal transduction by reactive oxygen species (ROS) plays a critical role in the modulation of autophagy (30). Mechanical ventilation induces ROS generation via both mitochondrial and NADPH oxidase signaling pathways (40). To this end, the role of ROS in mechanical ventilation-induced autophagy activation in the lung was assessed. We observed that mitochondrial ROS specific inhibitor SS-31 resulted in nearly complete inhibition of LC3-II generation attributable to mechanical ventilation (Fig. 2). To determine whether NADPH-derived ROS are also involved in autophagy activation following mechanical ventilation, we took advantage of gp91phox-deficient mice that have been used previously to study the role of gp91phox and NADPH oxidase as an oxygen sensor in vivo (40). Deletion of gp91phox did not inhibit mechanical ventilation-induced autophagy activation in the lung (Fig. 2). Collectively, these results indicate that mitochondrial ROS mediates mechanical ventilation-induced autophagy activation in the lung.
Fig. 2.
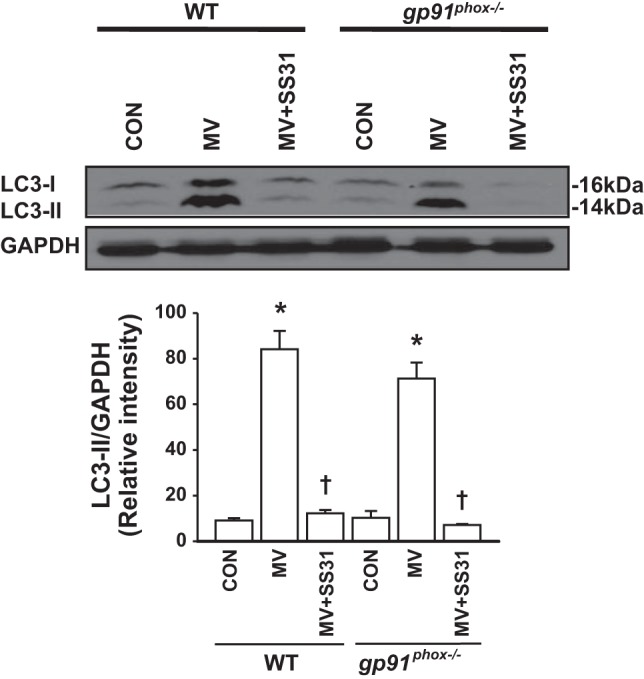
Mitochondrial reactive oxygen species (ROS) mediates autophagy activation in response to mechanical ventilation. Wild-type (WT) and gp91phox−/− mice were pretreated with the mitochondria-targeted antioxidant SS-31, a scavenger for mitochondrial ROS, and then ventilated with a high tidal volume (MV) for 2 h. At the end of experiments, lung tissues were recovered. Top, representative Western blots for LC3-I and LC3-II; bottom, bar graph showing the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. *P < 0.05 vs. the control (nonventilated and nontreated) group, †P < 0.05 vs. corresponding MV alone group.
Mechanical Ventilation Stimulates Autophagy Activation in Lung Macrophages
To further identify which specific cell type(s) plays a principal role in mechanical stretch-induced autophagy activation in the lung, we examined newly formed autophagosomes using transmission electron microscopy (TEM) on lung sections. Based on the typical autophagosomes with double membranes and cellular contents, vacuole-containing autophagosomes were identified by TEM. We found that autophagosomes in lung macrophages were significantly increased following mechanical ventilation with a high tidal volume compared with noninjurious ventilation (Fig. 3). There were also a small number of autophagosomes in lung endothelium. Surprisingly, we did not detect autophagosomes in epithelial cells including type I and type II cells. Western blot analysis of LC3-II expression in isolated macrophages from mouse lungs showed that mechanical stretch induced autophagy activation in lung macrophages (Fig. 4A), consistent with our morphological evidence obtained by TEM. To further verify whether autophagy activation in macrophages is predominant in the lung during mechanical ventilation, we depleted alveolar macrophages using a liposomal clodronate technique (40). Lavageable alveolar macrophage count was reduced by 75% at day 4 with a 10-ml aerosolized dose of 20 mg/ml clodronate liposome solution. Depletion of alveolar macrophages resulted in significantly reduced autophagy activation in the lung (Fig. 4B). Accordingly, these findings strongly corroborate the view that mechanical stretch-induced autophagy activation mainly occurs in lung macrophages.
Fig. 3.

Transmission electron microscopy (TEM) images of lung ventilated at a high tidal volume (MV). A: representative images of lung macrophages (MΦ), endothelial cell (Endo), and type I and II epithelial cell (Epi). Arrows indicate autophagosomes. B: the bar graph indicates the percentage of the area in a cell that is occupied by autophagosomes. Data are from 3 experiments. Bar graph shows the average area ± SE of the cell that contains autophagosomes from 50 fields. *P < 0.05 vs. the control (nonventilated) group.
Fig. 4.
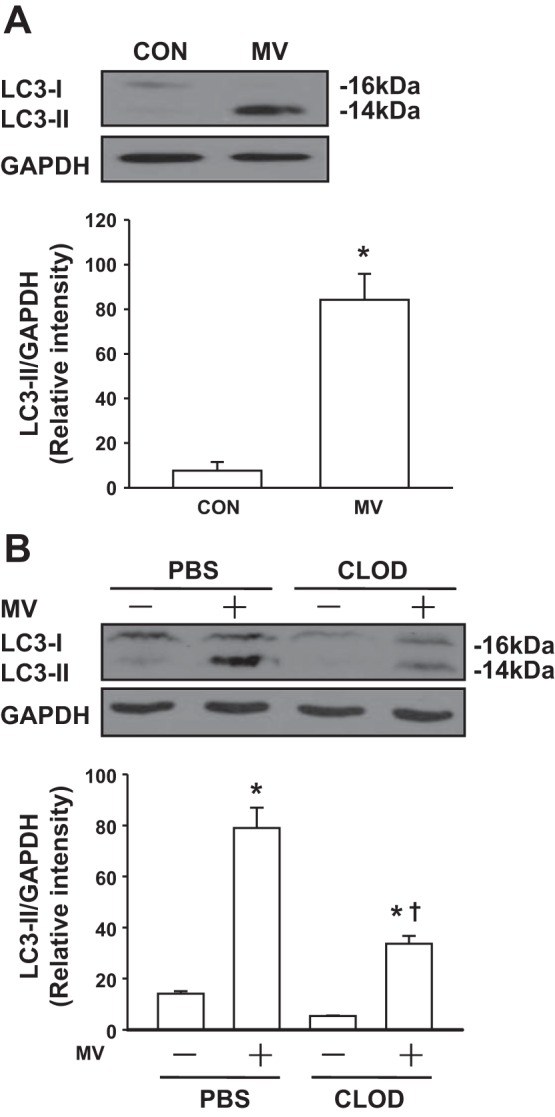
Mechanical ventilation induces autophagy in lung macrophages. A: mechanical ventilation activates autophagy in lung macrophages. Mice were ventilated with a low (7 ml/kg) or high tidal volume (28 ml/kg) for 2 h (n = 3 for each group). Lung macrophages were then isolated from the whole lung homogenates. Lysates from macrophages were analyzed by Western blot. Top, representative Western blots for LC3-I and LC3-II; bottom, bar graph showing the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. B: effects of alveolar macrophage (AM) depletion on mechanical ventilation-induced autophagy activation in the lung. Mice were depleted of AMs with clodronate liposomes (CLOD) or PBS (control) and then ventilated with a high tidal volume (28 ml/kg) for 2 h (n = 3 for each group). Top, representative Western blots for LC3-I and LC3-II; bottom, bar graph showing the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. *P < 0.05 vs. the control (nonventilated and nontreated) group, †P < 0.05 vs. corresponding MV group.
Genetic Inhibition of Autophagy Activation in Lung Macrophages Reduces Mechanical Ventilation-Induced Lung Inflammatory Injury
To further elucidate the role of autophagy activation in macrophages in lung inflammatory injury during mechanical ventilation, we observed the effects of injurious ventilation in AM-depleted mice receiving autophagy-deficient BMDMs. ATG5 protein expression following mechanical ventilation with a high tidal volume was dramatically increased in the lung tissues and lung macrophages (Fig. 5A). Therefore, BMDMs transfected with scrambled or ATG5 siRNAs were utilized in this study. ATG5 expression was depleted by ∼90% in BMDMs transfected with a specific ATG5 siRNA (Fig. 5B). Frozen sections of lung tissue showed that in vivo injection of BMDMs treated with scrambled siRNA or ATG5 siRNA resulted in comparable reconstitution of macrophages in the lung (Fig. 5C). Moreover, Western blot analysis of lung homogenates demonstrated that LC3-II level following mechanical ventilation with a tidal volume was markedly increased in AM-depleted lungs receiving BMDMs transfected with scrambled siRNA compared with normal lungs. However, the increase in LC3-II expression induced by mechanical ventilation was dramatically attenuated in AM-depleted lungs receiving BMDMs transfected with ATG5 siRNA (Fig. 5D). The PMN and total cell counts, and protein content in BAL fluid as well as ELW, dramatically increased following mechanical ventilation in control mice (no AM depletion) and AM-depleted mice receiving scrambled siRNA-transfected BMDMs, respectively. AM-depleted mice receiving ATG5 siRNA-transfected BMDMs showed a significant decrease in the PMN and total cell counts (Fig. 5, E and F) and protein content (Fig. 5G) in BAL fluid, and a reduction of ELW (Fig. 5H) compared with mice receiving scrambled siRNA-transfected BMDMs. Mice depleted of AMs prior to mechanical ventilation showed less PMN infiltration and lung edema formation. Histological analysis of lung tissue sections stained with hematoxylin and eosin revealed that, in contrast to normal lungs, mechanical ventilation with a high tidal volume induced neutrophil infiltration in the lung parenchyma and accumulation of protein-rich fluid in alveolar space in control mice (no AM depletion) and AM-depleted mice receiving scrambled siRNA-transfected BMDMs. Consistent with results of BAL analysis and ELW assay, these responses caused by injurious ventilation were markedly attenuated in AM-depleted mice receiving ATG5 siRNA-transfected BMDMs (Fig. 5I).
Fig. 5.

Repletion of bone marrow-derived macrophages (BMDMs) with decreased ATG5 aggravates lung inflammatory injury in AM-depleted mice during mechanical ventilation. A: effects of mechanical ventilation on ATG5 protein expression in lung tissues and lung macrophages (MΦ). Mice were ventilated with a low (7 ml/kg) or high tidal volume (28 ml/kg) for 2 h. Lung macrophages were then isolated from the whole lung homogenates. Lysates from lung homogenates and macrophages were analyzed by Western blot. Data are representative of 3 independent experiments. B: silencing of ATG5 in BMDMs with a specific siRNA. BMDMs isolated from donor mice were transfected with a scrambled siRNA (siSc) or ATG5 siRNA (siAtg5). After 48 h, the efficiency of transfection was evaluated by Western blot. Representative data of Western blot showed the depletion of ATG5 protein expression from BMDM cell lysates. C: effects of in vivo injection of BMDMs treated with scrambled siRNA or ATG5 siRNA on reconstitution of macrophages in the lung. BMDMs were transfected with scrambled siRNA or ATG5 siRNA. After 48 h, BMDMs transfected with a scrambled siRNA or ATG5 siRNA (2 × 106 cells, 200 μl total volume each) were labeled with PKH26 (red) and then given to AM-depleted mice via a jugular venous cannula. After 2 h, lung frozen sections (8 μm) were performed immediately. Photomicrographs were obtained with an Nikon Eclipse E400 microscope with a ×20 objective. D: effects of repletion of BMDMs with decreased ATG5 on mechanical ventilation (28 ml/kg)-induced autophagy activation in the lung. Mouse AMs were depleted with clodronate liposomes (CLOD) as described in materials and methods. BMDMs transfected with siSc or siAtg5 were intravenously injected into AM-depleted mice. Left, representative Western blots for LC3-I and LC3-II; right, bar graph showing the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. E: neutrophils from bronchoalveolar lavage (BAL) were enumerated to evaluate lung air space inflammation. F: total cell count from BAL fluid. G: pulmonary vascular protein permeability as determined by protein concentration of BAL fluid. H: pulmonary edema formation measured by extravascular lung water (ELW). I: histological analysis of lung tissue by hematoxylin and eosin staining (×40 magnification). *P < 0.05 compared with control group (nonventilated normal lungs); †P < 0.05 compared with MV alone group; ‡P < 0.05 compared with siSc group; n = 6/each group.
Genetic Inhibition of Autophagy in Lung Macrophages Dampens NLRP3 Inflammasome Signaling Following Mechanical Ventilation
It is recognized that NLRP3 inflammasome in AMs mediates lung inflammatory response during mechanical ventilation (40). We thus determine the effects of autophagy activation on NLRP3 inflammasome signaling pathway. As expected, mechanical ventilation induced NLRP3 inflammasome activation as evidenced by enhanced assembly of NLRP3, adaptor molecule apoptosis-associated specklike protein containing a caspase activation and recruitment domain (ASC), and caspase-1 in control mice (no AM depletion) and AM-depleted lungs receiving BMDMs transfected with scrambled siRNA, whereas NLRP3 inflammasome activation following mechanical ventilation was markedly inhibited in AM-depleted lungs receiving BMDMs transfected with ATG5 siRNA (Fig. 6, A and B). Western blot analysis showed that mechanical ventilation induced proteolytic cleavage of Pro-IL-1β and Pro-IL-18 and resulted in the release of mature 17-kDa IL-1β and 18-kDa IL-18 into the media in control mice (no AM depletion) and AM-depleted lungs receiving BMDMs transfected with scrambled siRNA (Fig. 7, A–C). IL-1β and IL-18 concentrations in BAL fluid of these lungs were also increased after mechanical ventilation (Fig. 7, D and E). However, AM-depleted mice receiving ATG5-silenced BMDMs demonstrated the lower levels of IL-1β and IL-18 following mechanical ventilation (Fig. 7, A–E). These findings suggest that mechanical ventilation with a high tidal volume activates pulmonary NLRP3 inflammasomes via autophagy signaling.
Fig. 6.

Autophagy mediates NLRP3 inflammasome activation during mechanical ventilation. BMDMs (MΦ) isolated from donor mice were transfected with a scrambled siRNA (siSc) or ATG5 siRNA (siAtg5). After 48 h, the efficiency of transfection was evaluated by Western blot. Mouse AMs were depleted with CLOD as described in materials and methods. BMDMs transfected with a siSc or siAtg5 were intravenously injected into AM-depleted mice. Mice were ventilated with a high tidal volume (MV, 28 ml/kg) for 2 h. At the end of experiments, lung tissues were recovered. A: effects of mechanical ventilation on NLRP3 inflammasome activation in the lung. The assembly of NLRP3 inflammasome was detected using immunoprecipitation (IP) with anti-NLRP3 antibody followed by immunoblotting (IB) for ASC, NLRP3, and caspase-1 cleavage product p10 fragments (Casp-1). B: relative densities of the bands of ASC proteins. Bar graph shows the relative abundance of ASC protein (normalized to that of NLRP3) from 3 experiments. C: relative density of the bands of Casp-1. Bar graph shows the relative abundance of Casp-1 protein (normalized to that of NLRP3) from 3 experiments. *P < 0.05 vs. the control (nonventilated and nontreated) group, †P < 0.05 vs. MV alone group, ‡P < 0.05 compared with siSc group.
Fig. 7.

Autophagy mediates the release of active forms of IL-1β and IL-18 from mouse lungs during mechanical ventilation. BMDMs (MΦ) isolated from donor mice were transfected with a scrambled siRNA (siSc) or ATG5 siRNA (siAtg5). After 48 h, the efficiency of transfection was evaluated by Western blot. Mouse AMs were depleted with clodronate liposomes (CLOD) as described in materials and methods. BMDMs transfected with a siSc or siAtg5 were intravenously injected into AM-depleted mice. A: effects of mechanical ventilation on the release of IL-1β and IL-18 in lung tissue. Following mechanical ventilation, the release of mature IL-1β and IL-18 was measured in the lung homogenates by Western blot analysis. The levels of Pro-IL-1β and Pro-IL-18 were determined in lung homogenates after mechanical ventilation. B: relative densities of the bands of IL-1β (p17) protein expression. Bar graph shows the relative abundance of IL-1β protein (normalized to that of GAPDH) from 3 experiments. C: relative densities of the bands of IL-18 protein expression. Bar graph shows the relative abundance of IL-18 protein (normalized to that of GAPDH) from 3 experiments. D: the levels of IL-1β in BAL fluid were measured by ELISA (n = 6). E: the levels of IL-18 in BAL fluid were measured by ELISA (n = 6). *P < 0.05 vs. the control (nonventilated and nontreated) group, †P < 0.05 vs. MV alone group, ‡P < 0.05 vs. siSc group.
Inhibition of Autophagy Dramatically Attenuates Mechanical Ventilation-Induced Lung Inflammation and Injury
In view of mechanical ventilation-induced autophagy activation, we next evaluated the effect of a selective autophagy inhibitor on VILI. 3-MA blocks autophagosome formation to inhibit autophagy activation (31). As shown in Fig. 8A, 3-MA completely inhibited autophagy activity at a concentration of 10 mg/kg. The effects of autophagy inhibitor on the integrity of the alveolar-capillary barrier and pulmonary edema formation were examined by measuring the concentration of total protein in BAL fluid and ELW, respectively. Mechanical ventilation with a high tidal volume induced an increase in transalveolar protein permeability and lung edema formation compared with low tidal volume ventilation, consistent with our previous findings (19). Mice treated with 3-MA showed lower protein permeability and less lung edema than vehicle-treated mice after 2 h of high tidal volume ventilation (Fig. 8, B and C). As PMN adhesion to pulmonary vascular endothelial cells and migration into the alveolar space are critical for induction of lung inflammation (19), we investigated the role of autophagy in the mechanism of PMN infiltration into the lung. As expected, mechanical ventilation with a high tidal volume caused a marked increase in lung MPO level (Fig. 8D) and PMN counts in BAL fluid in vehicle-treated mice (Fig. 8E). PMN infiltration into the lung was significantly reduced in mice pretreated with 3-MA (Fig. 8, D and E). Autophagy inhibitor had no effect on pulmonary vascular permeability, edema formation, or PMN infiltration in mice ventilated with a low tidal volume.
Fig. 8.

Autophagy inhibitor 3-methyladenine (3-MA) attenuated mechanical ventilation-induced lung injury in mice. Mice were pretreated with autophagy inhibitor 3-MA 1 h before exposure to high tidal volume mechanical ventilation (MV) for 2 h. Low tidal volume ventilation was used in the control group. A: effects of 3-MA on LC3-II protein expression in the lung following mechanical ventilation. Left, representative Western blots for LC3-I and LC3-II; right, protein quantification by densitometry. Bar graph shows the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. B: protein concentrations in BAL fluid. C: ELW. D: polymorphonuclear neutrophil (PMN) counts in BAL fluid. E: lung MPO activity. *P < 0.05 vs. control (CON) group. †P <0.05 vs. MV group; n = 6 per groups.
TSA is a classic histone deacetylases inhibitor and an anticancer drug in clinical settings. Recently, TSA has been reported to block autophagy in the heart (1) so we tested the effect of TSA on autophagy activation and lung inflammatory injury during mechanical ventilation. We observed that TSA pretreatment inhibited mechanical ventilation-induced autophagy activation. Pretreatment of mice with TSA for 1 h completely blocked mechanical stretch-induced autophagy activation in the lung (Fig. 9A). TSA alone had no effect on the levels of LC3-II. Consistent with findings from 3-MA, mice treated with TSA showed lower protein permeability (Fig. 9B), less lung edema (Fig. 9C), decreased MPO level (Fig. 9D) and PMN counts in BAL fluid (Fig. 9E) than vehicle-treated mice after 2 h of high tidal volume ventilation. TSA alone had no effect on pulmonary vascular permeability, edema formation, or PMN infiltration in mice ventilated with a low tidal volume (Fig. 9, B–E). Collectively, these results demonstrate that selective pharmacological inhibition of autophagy activation has a protective effect on VILI in this experimental model.
Fig. 9.

Autophagy inhibitor trichostatin A (TSA) attenuated mechanical ventilation-induced lung injury in mice. Mice were pretreated with autophagy inhibitor TSA 1 h before exposure to high tidal volume mechanical ventilation (MV) for 2 h. Low tidal volume ventilation was used in the control group. A: effects of pretreatment with TSA on LC3-II protein expression in the lung following mechanical ventilation. Left, representative Western blots for LC3-I and LC3-II; right, protein quantification by densitometry. Bar graph shows the relative abundance of LC3-II protein (normalized to that of GAPDH) from 3 experiments. B: protein concentrations in BAL fluid. C: ELW. D: lung MPO activity. E: PMN counts in BAL fluid. *P < 0.05 vs. CON group. †P <0.05 vs. MV group; n = 6 per groups.
DISCUSSION
In the present study, we identified a crucial role for excessive autophagy activation in lung macrophages that contributed to the molecular pathogenesis of VILI. Mechanical ventilation with a high tidal volume induced a rapid activation of autophagy in lung macrophages. Genetic inhibition of autophagy in the lung macrophages abolished mechanical ventilation-induced NLRP3 inflammasome activation, the release of proinflammatory cytokines IL-1β and IL-18, and lung inflammation and injury. Pharmacological blockade of autophagy activation in the lung also remarkably attenuated lung inflammatory injury during mechanical ventilation. Our findings therefore support the notion that mechanical ventilation induces lung inflammation and injury via autophagy signaling-mediated NLRP3 inflammasome activation and subsequent release of proinflammatory cytokines in lung macrophages.
One of the main findings of this study is the identification of lung macrophages as a key target for autophagy activation induced by mechanical stretch. This conclusion is based on the following findings: First, LC3 II protein expression was significantly increased in lung macrophages isolated from the whole lungs after mechanical ventilation. Depletion of AMs caused a dramatic decrease in mechanical ventilation-induced autophagy activation in the lung. Furthermore, TEM reveals robust autophagosomes in lung macrophages, but few autophagosomes in lung endothelium and none in epithelium in our model system. Finally, administration of ATG5 silencing BMDMs into AM-depleted mice dramatically attenuated mechanical ventilation-induced inflammation and lung injury. These results suggest that the lung macrophage is a critical autophagy sensor in response to mechanical stretch. In addition to macrophages, hyperinflation of the lung generates mechanical stress on many other cell types. In a previous study, the monocyte/macrophage, epithelial, endothelial, and fibroblastic lineages were exposed to a pressure/stretching strain resembling that of mechanical ventilation. Among various lung cell types, lung macrophages have been shown to be the main cellular source for IL-8 (5). Our recent study also demonstrated that mechanical stretch activated NLRP3 inflammasome in alveolar macrophages leading to release of IL-1β and IL-18 (40). Collectively, our findings suggest that lung macrophages are the most sensitive sensor of autophagy that may initiate lung inflammation during mechanical ventilation.
The role of autophagy in the mechanisms of acute lung injury has been controversial with studies that have suggested both protective and injurious aspects of autophagy (23). Activation of autophagy has been shown to play a protective role against hyperoxia-induced cell death by potentially interacting with and inhibiting apoptotic pathways in lung epithelial cells (34). Likewise, carbon monoxide attenuated hyperoxia-induced human alveolar epithelial cell death via activation of autophagy (16). In contrast, inhibition of autophagy ameliorates acute lung injury in mice caused by infection with live H5N1 virus (33) and by mechanical ventilation (21).
Whether autophagy is protective or detrimental in lung inflammation and injury seems to depend on the extent of its activation, specific stimuli, and specific cell type (23, 33, 34, 37). Inhibition of autophagy has been shown to promote or prevent apoptosis in the same cell dependent on the nature of the death stimulus and compensatory changes in other forms of autophagy (37). Our data clearly demonstrated that selective silencing of ATG5 in lung macrophages abolished mechanical ventilation-induced lung inflammatory injury. Pretreatment of mice with two different pharmacological inhibitors of autophagy prevented VILI. These results suggest that autophagy activation in lung macrophages mediated mechanical ventilation-induced lung injury and inflammation. Our findings were supported by a recent study using Atg4b knockout mice that showed that inhibition of autophagy activation in the whole lung attenuated VILI (21).
The detailed biochemical mechanisms by which mechanical stretch activates autophagy activation in lung macrophages await further characterization. ROS generated from mitochondria and NADPH oxidases have been demonstrated to be essential to autophagy activation (2, 29, 30). Increased ROS induce autophagy activation via oxidative modification of proteins that is involved in either the autophagy machinery or autophagic cell signaling (20). Hydrogen peroxide has been shown to oxidize and subsequently inactivate Atg4 during starvation. Atg4 is responsible for recycling of LC3 by cleaving phosphatidylethanolamine from LC3-II and inhibition of Atg4 leads to accumulation of LC3-II and increased autophagosome formation (30). ROS may also activate autophagy by indirect mechanisms causing damage to organelles such as mitochondria that induces mitochondrial autophagy (15). ROS interaction with aconitase can result in iron release and promotion of lipid peroxidation, which induces accumulation of LC3-II (30). The results in this study showed that inhibition of mitochondrial ROS generation resulted in nearly total inhibition of mechanical ventilation-induced activation of autophagy. However, depletion of gp91phox had no effect on autophagy activation in the lung during mechanical ventilation. Our previous study demonstrated that gp91phox deletion did not alter mitochondrial ROS production induced by mechanical stretch (40). These results suggest that mitochondria are a major source of ROS, which promotes activation of autophagy in the lung during mechanical ventilation. Findings from our laboratory (40) and others (39) have demonstrated that mechanical ventilation causes massive generation of mitochondrial ROS, although the mechanisms stimulating mitochondrial ROS following mechanical stretch are not clear. It is likely that mitochondrial distortion caused by mechanical stretch may result in the release of ROS (14). Our data thus indicate that mitochondrial ROS induced by mechanical ventilation play a key role in activation of autophagy in lung macrophages.
Numerous studies have indicated that autophagy plays an inhibitory role in inflammasome activation and subsequent release of IL-1β and IL-18 (25, 28, 29, 43). Blockade of autophagy by genetic ablation of the autophagy regulator Atg16L1 or Atg7 enables LPS-dependent inflammasome activation, suggesting that autophagy normally counters inflammasome activation by LPS (29). The mechanisms by which autophagy inhibits inflammasome activation may be associated with autophagy-dependent mitochondrial homeostasis that normally suppresses release of ROS (43) and mitochondrial DNA (17), both of which can activate inflammasome. However, our results indicate that induction of autophagy enhances assembly of NLRP3 inflammasome-activating components and the expression of the proinflammatory cytokines during mechanical ventilation. Supporting this view, it has been observed that autophagy activation contributes to an NLRP3-dependent proinflammatory response in the pancreatic INS-1(823/13) cell line (18). Excessive autophagy activation may enhance lysosomal destabilization that releases cathepsin B, which triggers NLRP3 inflammasome activation (18). Furthermore, stimulation of autophagy by starvation promoted NLRP3 inflammasome activation and IL-1β secretion in an Atg5-dependent manner in BMDMs (6). Taken together, our findings indicate a fundamental role of autophagy in lung macrophages as an upstream signaling of NLRP3 inflammasome activation during mechanical ventilation.
In summary, this is the first study to demonstrate the essential role of autophagy activation in lung macrophages in the molecular pathogenesis of VILI. Mechanical stretch stimulates mitochondrial ROS production in lung macrophages, which in turn activates autophagy and signals assembly of ASC, NLRP3, and caspase-1 to activate NLRP3 inflammasome, leading to the processing and maturation of Pro-IL-1β into the active IL-1β variant (Fig. 10). Our findings provide new insights into the release mechanisms of proinflammatory cytokines regulated by autophagy, showing that excessive autophagy activation in lung macrophages may initiates and exaggerates lung inflammation and injury during mechanical ventilation. The results thus point to potential therapeutic approaches to inhibiting autophagy activation in lung macrophages for treatment of VILI.
Fig. 10.
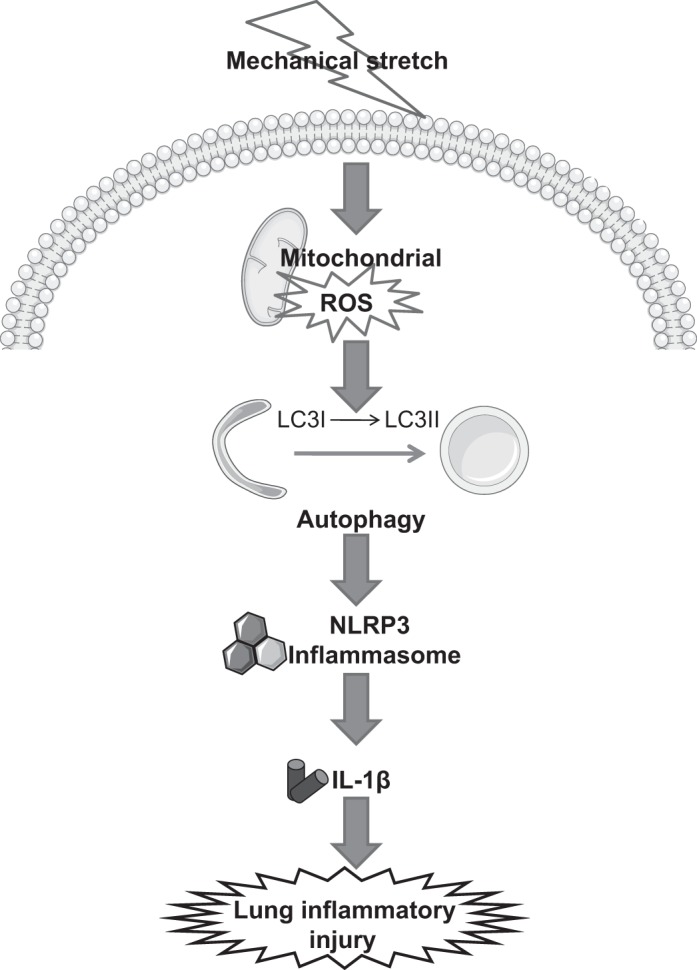
Model of macrophage autophagy-mediated inflammasome activation and lung inflammatory injury by mechanical ventilation.
GRANTS
This work was supported by NIH NHLBI grant 5R01HL104092 (G. Hu) and Natural Science Foundation of China 81070058 (G. Hu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Y.Z. and G.H. conception and design of research; Y.Z. performed experiments; Y.Z. prepared figures; G.L., R.O.D., D.E.S., and G.H. edited and revised manuscript; G.H. analyzed data; G.H. interpreted results of experiments; G.H. drafted manuscript; G.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Maricela Castellon (Department of Anesthesiology, University of Illinois College of Medicine) for technical assistance.
REFERENCES
- 1. Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA 108: 4123–4128, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 120: 4155–4166, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Conway Morris A, Kefala K, Wilkinson TS, Moncayo-Nieto OL, Dhaliwal K, Farrell L, Walsh TS, Mackenzie SJ, Swann DG, Andrews PJ, Anderson N, Govan JR, Laurenson IF, Reid H, Davidson DJ, Haslett C, Sallenave JM, Simpson AJ. Diagnostic importance of pulmonary interleukin-1β and interleukin-8 in ventilator-associated pneumonia. Thorax 65: 201–207, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dreyfuss D, Ricard JD, Saumon G. On the physiologic and clinical relevance of lung-borne cytokines during ventilator-induced lung injury. Am J Respir Crit Care Med 167: 1467–1471, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Dunn I, Pugin J. Mechanical ventilation of various human lung cells in vitro: identification of the macrophage as the main producer of inflammatory mediators. Chest 116: 95, 1999 [DOI] [PubMed] [Google Scholar]
- 6. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J 30: 4701–4711, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frank JA, Wray CM, McAuley DE, Schwendener R, Matthay MA. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 291: L1191–L1198, 2006 [DOI] [PubMed] [Google Scholar]
- 8. Franke-Ullmann G, Pförtner C, Walter P, Steinmüller C, Lohmann-Matthes ML, Kobzik L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J Immunol 157: 3097–3104, 1996 [PubMed] [Google Scholar]
- 9. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333: 1109–1112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grembowicz KP, Sprague D, McNeil PL. Temporary disruption of the plasma membrane is required for c-fos expression in response to mechanical stress. Mol Biol Cell 10: 1247–1257, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hess DR, Thompson BT, Slutsky AS. Update in acute respiratory distress syndrome and mechanical ventilation 2012. Am J Respir Crit Care Med 188: 285–292, 2013 [DOI] [PubMed] [Google Scholar]
- 12. Hu G, Malik AB, Minshall RD. Toll-like receptor 4 mediates neutrophil sequestration and lung injury induced by endotoxin and hyperinflation. Crit Care Med 38: 194–201, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knight MM, Bomzon Z, Kimmel E, Sharma AM, Lee DA, Bader DL. Chondrocyte deformation induces mitochondrial distortion and heterogeneous intracellular strain fields. Biomech Model Mechanobiol 5: 180–191, 2006 [DOI] [PubMed] [Google Scholar]
- 15. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441: 523–540, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim HP, Choi AM, Kim YS. Carbon monoxide activates autophagy via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol 45: 867–873, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 469: 323–335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li S, Du L, Zhang L, Hu Y, Xia W, Wu J, Zhu J, Chen L, Zhu F, Li C, Yang S. Cathepsin B contributes to autophagy-related 7 (Atg7)-induced nod-like receptor 3 (NLRP3)-dependent proinflammatory response and aggravates lipotoxicity in rat insulinoma cell line. J Biol Chem 288: 30094–30104, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu D, Yan Z, Minshall RD, Schwartz DE, Chen Y, Hu G. Activation of calpains mediates early lung neutrophilic inflammation in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 302: L370–L379, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lizama-Manibusan B, McLaughlin B. Redox modification of proteins as essential mediators of CNS autophagy and mitophagy. FEBS Lett 587: 2291–2298, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. López-Alonso I, Aguirre A, González-López A, Fernández AF, Amado-Rodríguez L, Astudillo A, Batalla-Solís E, Albaiceta GM. Impairment of autophagy decreases ventilator-induced lung injury by blockade of the NF-κB pathway. Am J Physiol Lung Cell Mol Physiol 304: L844–L852, 2013 [DOI] [PubMed] [Google Scholar]
- 22. McKechnie SR, Drummond GB. Cytokines, neurokines or both? Mixed mechanisms of mechanical lung injury. J Physiol 588: 1813–1814, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizumura K, Cloonan SM, Haspel JA, Choi AM. The emerging importance of autophagy in pulmonary diseases. Chest 142: 1289–1299, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat JL, Nicod LP, Chevrolet JC. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol Lung Cell Mol Physiol 275: L1040–L1050, 1998 [DOI] [PubMed] [Google Scholar]
- 27. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90: 1383–1435, 2010 [DOI] [PubMed] [Google Scholar]
- 28. Rodgers MA, Bowman JW, Liang Q, Jung JU. Regulation where autophagy intersects the inflammasome. Antioxid Redox Signal 20: 495–506, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456: 264–268, 2008 [DOI] [PubMed] [Google Scholar]
- 30. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of atg4. EMBO J 26: 1749–1760, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79: 1889–1892, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun Y, Hu G, Zhang X, Minshall RD. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res 105: 676–685, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun Y, Li C, Shu Y, Ju X, Zou Z, Wang H, Rao S, Guo F, Liu H, Nan W, Zhao Y, Yan Y, Tang J, Zhao C, Yang P, Liu K, Wang S, Lu H, Li X, Tan L, Gao R, Song J, Gao X, Tian X, Qin Y, Xu KF, Li D, Jin N, Jiang C. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci Signal 5: ra16, 2012 [DOI] [PubMed] [Google Scholar]
- 34. Tanaka A, Jin Y, Lee SJ, Zhang M, Kim HP, Stolz DB, Ryter SW, Choi AM. Hyperoxia induced LC3B interacts with the Fas apoptotic pathway in epithelial cell death. Am J Respir Cell Mol Biol 46: 507–514, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tremblay LN, Miatto D, Hamid Q, Govindarajan A, Slutsky AS. Injurious ventilation induces widespread pulmonary epithelial expression of tumor necrosis factor-α and interleukin-6 messenger RNA. Crit Care Med 30: 1693–1700, 2002 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y, Minshall RD, Schwartz DE, Hu G. Cyclic stretch induces alveolar epithelial barrier dysfunction via calpain-mediated degradation of p120-catenin. Am J Physiol Lung Cell Mol Physiol 301: L197–L206, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, Xiang Y, Cuervo AM, Czaja MJ. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem 283: 4766–4777, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang YL, Malik AB, Sun Y, Hu S, Reynolds AB, Minshall RD, Hu G. Innate immune function of the adherens junction protein p120-catenin in endothelial response to endotoxin. J Immunol 186: 3180–3187, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weinacker AB, Vaszar LT. Acute respiratory distress syndrome: physiology and new management strategies. Annu Rev Med 52: 221–237, 2001 [DOI] [PubMed] [Google Scholar]
- 40. Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, Hu G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J Immunol 190: 3590–3599, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, Rocchetti M, Mingozzi F, Foti M, Chirico G, Costa B, Zaza A, Ricciardi-Castagnoli P, Granucci F. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature 460: 264–268, 2009 [DOI] [PubMed] [Google Scholar]
- 42. Zhang H, Downey GP, Suter PM, Slutsky AS, Ranieri VM. Conventional mechanical ventilation is associated with bronchoalveolar lavage-induced activation of polymorphonuclear leukocytes: a possible mechanism to explain the systemic consequences of ventilator-induced lung injury in patients with ARDS. Anesthesiology 97: 1426–1433, 2002 [DOI] [PubMed] [Google Scholar]
- 43. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2011 [DOI] [PubMed] [Google Scholar]