

Abstract
The lomaiviticins are a family of cytotoxic marine natural products that have captured the attention of both synthetic and biological chemists due to their intricate molecular scaffolds and potent biological activities. Here we describe the identification of the gene cluster responsible for lomaiviticin biosynthesis in Salinispora pacifica strains DPJ-0016 and DPJ-0019 using a combination of molecular approaches and genome sequencing. The link between the lom gene cluster and lomaiviticin production was confirmed using bacterial genetics, and subsequent analysis and annotation of this cluster revealed the biosynthetic basis for the core polyketide scaffold. Additionally, we have used comparative genomics to identify candidate enzymes for several unusual tailoring events, including diazo formation and oxidative dimerization. These findings will allow further elucidation of the biosynthetic logic of lomaiviticin assembly and provide useful molecular tools for application in biocatalysis and synthetic biology.
Keywords: Natural product biosynthesis, Comparative genomics, Diazo group, Polyketide, Dimerization
1. Introduction
The lomaiviticins are a cytotoxic family of secondary metabolites produced by marine actinomycetes in the genus Salinispora (Figure 1A).1 These natural products, which are members of the angucycline family of aromatic polyketides, contain a distinctive diazotetrahydrobenzo[b]fluorene scaffold also found in the kinamycins (Figure 1B). Both classes of diazofluorene natural products exhibit potent cytotoxicity against human cancer cell lines, with lomaiviticin A displaying the greatest activity. Lomaiviticins A and B (structure not shown) were initially isolated from the halophilic actinomycete strain LL-37I366.2 Although LL-37I366 was originally proposed to be a new species in the genus Micromonospora, (“Micromonospora lomaivitiensis”), subsequent phylogenetic analyses showed that LL-37I366 is actually a strain of Salinispora pacifica (Figure S1). Lomaiviticin A was later re-identified in fermentation broths from a different S. pacifica strain, DPJ-0019. This organism also produces neolymphostins A–D, a family of alkaloids which are potent inhibitors of mTOR.3 Most recently, additional lomaiviticins (C–E) were isolated from DPJ-0019, an effort that led to elucidation of the complete absolute and relative stereochemistry of this natural product family.4
Figure 1.

The lomaiviticin and kinamycin natural product families possess unusual molecular architecture. A) Structures of the lomaiviticins isolated from Salinispora pacifica DPJ-0019. B) Structures of the kinamycins isolated from Streptomyces ambofaciens ATCC 23877. C) Structure of neolymphostin A, an additional bioactive metabolite from S. pacifica DPJ-0019. D) Transformations in lomaiviticin biosynthesis that may involve unusual enzymatic chemistry.
Though the lomaiviticins share their unusual diazofluorene pharmacophore with the kinamycins, this natural product family possesses additional, unique structural features that make it of particular interest to chemists (Figure 1C). Perhaps most notably, the lomaiviticins are C2-symmetric diazofluorene dimers, with the two halves of the molecule connected via a sterically congested C–C bond. They are also glycosidated with the unusual sugars N,N-dimethyl-L-pyrrolosamine and L-oleandrose. Finally, the lomaiviticins are more extensively oxidized than the kinamycins, possessing an additional A-ring hydroxyl group and a D-ring carbon at the ketone oxidation state.
The complex structural framework of the lomaiviticins has captured the attention of organic chemists, who have completed syntheses of both the lomaiviticin aglycone5 and core scaffold6 and continue to work toward a total synthesis. Despite their prominence as synthetic targets, the enzymatic chemistry involved in lomaiviticin assembly has not yet been extensively investigated. The unusual molecular architecture of these natural products implies that their biosynthesis will likely be a rich source of novel enzymatic transformations. Understanding the genetic and biochemical basis for lomaiviticin biosynthesis will not only enable the production of new lomaiviticin analogs through metabolic engineering or chemoenzymatic synthesis, but could also deliver useful tools for biocatalysis and metabolic engineering. An initial step toward gaining this knowledge has been achieved by Moore and coworkers, who used a bioactivity-guided genome mining strategy to locate a lomaiviticin (lom) gene cluster in Salinispora tropica CNB-440 and put forth a biosynthetic scheme for the assembly of lomaiviticin A. 7
Here we report our contemporaneous efforts that have led to the discovery of related lom gene clusters in S. pacifica DPJ-0016 and DPJ-0019. We have employed bacterial genetics to confirm the link between the lom gene cluster and lomaiviticin assembly, and we have also uncovered striking effects of growth media on lomaiviticin production by these two organisms. Annotation of the lom cluster and comparative genomic analysis have enabled us to formulate an alternate biosynthetic hypothesis for lomaiviticin assembly that implicates unusual logic for polyketide initiation and tailoring.8 Specifically, comparison of the lom cluster with a complete kinamycin biosynthetic gene cluster from Streptomyces ambofaciens ATCC 23877 and a partial biosynthetic gene cluster from Streptomyces murayamaensis has allowed us to identify candidate enzymes for both diazo assembly and dimerization that are also located in additional characterized and cryptic biosynthetic gene clusters from other organisms. These insights illustrate the power of comparative genomics for predicting biochemical function and further affirm that biosynthetic pathways producing architecturally unusual natural products are rich sources of new enzymatic chemistry and biosynthetic logic.
2. Results and discussion
2.1 Discovery of the lomaiviticin biosynthetic gene cluster
At the start of our studies, the genetic and biochemical basis for lomaiviticin biosynthesis was unknown. However, the structural similarities between these natural products and the kinamycins suggested substantial shared biosynthetic logic. Knowledge of kinamycin biosynthesis has been primarily gained through feeding experiments, which have revealed the polyketide origin of these metabolites and identified biosynthetic intermediates.9 Sequencing of the kinamycin biosynthetic gene cluster from Streptomyces murayamaensis by Gould and coworkers revealed both type II polyketide biosynthetic machinery and putative tailoring enzymes; however this cluster failed to produce diazo-containing metabolites when expressed in Streptomyces lividans ZX7, indicating that it was likely incomplete.10 More recently, duplicate kinamycin biosynthetic gene clusters were identified in the genome of S. ambofaciens ATCC 23877 but a detailed biosynthetic hypothesis has not yet been proposed.11 We anticipated that knowledge of genes involved in kinamycin assembly would facilitate identification of the lomaiviticin gene cluster and enable a more complete annotation of both diazofluorene biosynthetic pathways via comparative genomics.
We identified putative lomaiviticin biosynthetic gene clusters in two isolates of S. pacifica using complimentary approaches (Figure 2A). For S. pacifica DPJ-0016, we screened a cosmid library of genomic DNA using a probe for dTDP glucose 4,6-dehydratase, an enzyme involved in the biosynthesis of 6-deoxy sugars.12 This initial attempt identified two overlapping cosmids, 2C7 and 5E8b. Cosmid 2C7 (37.4 kb) encoded a type II polyketide β-ketoacyl synthase (KSα) and chain length factor (CLF), as well as several genes required for deoxy sugar biosynthesis. Cosmid 5E8b (40.9 kb) was found to contain homologs of several genes involved in type II PKS tailoring, including an aromatase/cyclase, anthrone oxidase, ketoreductase, and several monooxygenases. Further screening of the library with a ketoreductase probe generated from an open reading frame (ORF) in 5E8b revealed an additional overlapping cosmid, p730497 (23.9 kb), that encoded homologs of additional type II PKS tailoring genes and an NDP-hexose 2,3-dehydratase. These three overlapping cosmids covered a ~70 kb region of sequence that was designated as the lom gene cluster.
Figure 2.

Discovery of the lomaiviticin (lom) gene cluster and confirmation of function. A) The lom gene clusters from S. pacifica strains DPJ-0016 and DPJ-0019 and the deduced functions of gene products. Each arrow represents the direction of transcription of an open reading frame. See Table S3 for complete cluster annotation. B) HPLC analysis (500 nm) of crude fermentation extracts showing yield increases of lomaiviticins through medium optimization with strain DPJ-0019. C) Comparative HPLC analysis (500 nm) of crude fermentation extracts from S. pacifica DPJ-0016, DPJ-0019, and DPJ-0019-MT1 revealing loss of lomaiviticin production in the lom58 mutant, DPJ-0019-MT1.
In parallel, we located the lomaiviticin biosynthetic gene cluster in the neolymphostin producer S. pacifica DPJ-0019 using whole genome sequencing. Searching the assembled Illumina sequencing data for homologs of the type II PKS machinery from the kinamycin gene cluster revealed a cluster that contained all of the ORFs found in DPJ-0016, with the exception of a cassette of 7 genes (lom36–42). The absence of this gene cassette from DPJ-0019 suggests that these seven genes are not required for biosynthesis of the lomaiviticins. The DPJ-0019 lom cluster is also identical in content and organization to the recently disclosed lom cluster identified in S. tropica CNB-440.7
To establish optimal conditions for lomaiviticin production in S. pacifica DPJ-0016 and DPJ-0019, both strains were fermented in five different media, including the original production medium PCAA (production medium containing peptone and casamino acids).13 DPJ-0016 produced lomaiviticin C at 18 mg/L in PCAA and 16 mg/L in PYE but did not produce any detectable lomaiviticins in the other three media tested despite excellent growth. In contrast, DPJ-0019 produced lomaiviticins in all five media, including lomaiviticins C–E at titers of 129, 95, and 31 mg/L, respectively, in SPYESS (Figure 2B and Table S2). Notably, lomaiviticin production varies dramatically between the two strains depending on the nitrogen and carbon sources supplied during fermentation; this result highlights the important role played by medium in modulating expression of secondary metabolic pathways, even amongst strains of the same species.
To validate that the lom gene cluster in DPJ-0019 was indeed responsible for lomaiviticin biosynthesis, an insertional inactivation was generated with an apramycin resistance marker (aminoglycoside-(3)-acetyltransferase IV gene (aac3IV)) to disrupt the function of the putative KSα, lom58. Mutants were confirmed by colony PCR (experimental section, Figure S2). A successful double crossover mutant, DPJ-0019-lom58::acc(3)IV-MT1 (DPJ-0019-MT1), was confirmed by PCR and fermented in PCAA in parallel with the DPJ-0019 parent strain and DPJ-0016. Both DPJ-0016 and DPJ-0019 produced lomaiviticin C as the main component at 18 mg/L and 9 mg/L, respectively. In contrast, DPJ-0019-MT1 did not produce any detectable lomaiviticins, confirming the role of lom58 and the lom gene cluster in natural product production (Figure 2C).
In addition to lomaiviticins, DPJ-0019 also produces the potent mTOR inhibitor neolymphostin A; however, DPJ-0019 does not produce any detectable neolymphostins in SPYESS medium. Elimination of soy peptone as a nitrogen source from SPYESS and increasing the amount of yeast extract three-fold (medium YESS) favors production of the neolymphostins in DPJ-0019.3 To determine if the abolition of lomaiviticin production in the lom58 mutant DPJ-0019-MT1 would positively affect the yield of neolymphostins, DPJ-0019-MT1 was fermented in parallel with the DPJ-0019 parent strain in YESS. DPJ-0019-MT1 produced 12.5 mg/L of neolymphostin A, a three and a half fold increase in production compared to the parent strain (3.5 mg/L) (Figure S3). The parent strain still produced many lomaiviticins in YESS medium in good yields, including lomaiviticins C, D and E at 53 mg/L, 32 mg/L and 5 mg/L, respectively. This result suggests that the central metabolites involved in lomaiviticin production can be redirected to produce neolymphostins if lomaiviticin biosynthesis is interrupted. Therefore, eliminating lomaiviticin production in DPJ-0019 can also facilitate the isolation and purification of neolymphsotin A from fermentations.
2.2 Annotation of the lomaiviticin gene cluster and generation of a biosynthetic hypothesis
With evidence from the insertion mutant DPJ-0019-MT1 confirming the connection between lom58 and lomaiviticin production, we performed a detailed bioinformatic analysis of the lom cluster and generated a biosynthetic hypothesis. The lom gene cluster in DPJ-0019 encodes a total of 59 ORFs, including genes involved in the biosynthesis of the angucycline polyketide core,14 construction of L-oleandrose and pyrrolosamine sugars, and tailoring of the core scaffold (Table S3). Our cluster annotation and formulation of a biosynthetic hypothesis (Scheme 1) were aided by comparing the ORFs present in the lom cluster to those found in other characterized gene clusters, most notably the jad cluster,15 which is involved in the biosynthesis of the angucycline polyketide jadomycin, and both complete and partial kinamycin (kin) gene clusters (Table S4). The lom cluster contains homologs of all type II PKS components (KSα lom58, CLF lom59, acyl carrier protein (ACP) lom60) and tailoring enzymes (ketoreductase lom22, cyclases lom21 and lom25) needed to construct UWM6, a common precursor to the angucycline polyketides (Scheme 1A).16 Homologs of these genes are also encoded in the jad and kin clusters. A distinguishing feature of the lomaiviticin polyketide core is the presence of an ethyl substituent on the D-ring in place of the methyl group found in kinamycin and jadomycin. We propose that this group arises from use of a propionyl starter unit by the type II PKS, which will generate ‘ethyl’-UWM6. Interestingly, homologs of the propionyl starter unit-generating enzymes employed in other type II PKS pathways, such as doxorubicin biosynthesis,17 are not encoded in the lom cluster. Instead the cluster contains an acyltransferase/decarboxylase (AT/DC) (lom62) related to LnmK, an enzyme that converts methylmalonyl-CoA to propionyl-ACP in type I modular PKS pathways, and a second ACP (lom63). We hypothesized that Lom62 is responsible for propionyl starter unit formation, and we have recently confirmed the ability of this enzyme to generate propionyl-ACP in vitro with a strong preference for Lom63 as a partner ACP.8
Scheme 1.

Proposed biosynthetic pathway for the production of lomaiviticin A. (A) Biosynthetic hypothesis for synthesis of ‘ethyl’-kinobscurinone. (B) Biosynthetic hypothesis for tailoring of the core benzo[b]fluorene scaffold. The order of biosynthetic steps has not been confirmed. Abbreviations: PKS = polyketide synthase, CoA = coenzyme A, KS = ketosynthase, CLF = chain length factor, ACP = acyl carrier protein, AT = acyltransferase, KR = ketoreductase, TDP = thymidine diphosphate.
Following the production of ‘ethyl’-UWM6, our biosynthetic hypothesis differs in several aspects from the previous scheme provided by Moore and co-workers.7 Oxidative tailoring of this scaffold by a series of three flavin-dependent oxygenases (lom26–28), in analogy with jadomycin biosynthesis, should mediate ring opening and formation of a biaryl intermediate (Scheme 1A).18 Interestingly, both the lom and kin gene clusters contain an additional oxygenase not found in the jad cluster; we hypothesize that this additional flavin-dependent enzyme (lom24, SAMT0159/kinO2) may be involved in cyclization and oxidation of the biaryl intermediate to form the characteristic benzo[b]fluorene ring system shared by these natural products. In the case of lomaiviticin biosynthesis, this cyclization would produce an ethyl-substituted analog of kinobscurinone, a confirmed intermediate in kinamycin biosynthesis.9e
After formation of either ethyl-kinobscurinone or kinobscurinone, a complex series of tailoring events is required for elaboration to the final lomaiviticin or kinamycin structure (Scheme 1B). We identified putative tailoring enzymes by comparing the chemical modifications involved in constructing both classes of diazofluorenes and examining the contents of the lom cluster and both the full length and partial kin clusters. Using this approach, we uncovered candidate genes for diazo formation (lom29,30,32–35), dimerization (lom19), A- and D-ring oxidations (lom13,16,17), D-ring ketone reduction (lom18), and glycosylations (lom48,55). We also located all of the enzymatic machinery needed to construct both L-oleandrose and N,N-dimethyl-L-pyrrolosamine sugars (lom9,49–54,56,57,65), and proposed a biosynthetic hypothesis for their assembly from a common intermediate based upon the homologous L-oleandrose biosynthetic pathway employed in avermectin production (Scheme S1).19,20 Although the precise order of tailoring events in the late stages of lomaiviticin biosynthesis is currently unclear, the recent isolation of monomeric benzo[b]fluorene (−)-homoseongomycin from S. pacifica DPJ-0019, a metabolite which is likely derived from a prelomaiviticin intermediate, is consistent with our proposed timing of transformations.21 Overall, we have identified putative enzymes for each required tailoring event. The extent of this annotation exceeds that reported previously, and this work will greatly facilitate further examination of the unusual enzymatic chemistry employed in this pathway.
2.3 Identification of candidate diazo-forming enzymes
We were particularly interested in identifying candidate enzymes for two key tailoring events: diazo formation and dimerization of the core diazofluorene scaffold. The biosynthetic origins of naturally-occurring diazo groups22 has remained a mystery since the discovery of the first diazo-containing metabolite azaserine in 1954.23 Feeding studies with the labeled aminobenzo[b]fluorene stealthin C resulted in incorporation into kinamycin, indicating that the two nitrogen atoms of the diazo group are introduced into the natural product scaffold in a stepwise manner.9f A similar result was obtained from feeding experiments exploring the biosynthesis of azamerone, a natural product derived from a diazo-containing intermediate.24 We recognized that the availability of complete kin and lom gene clusters presented an opportunity to connect specific genes to diazo production. Reasoning that these pathways should employ a common mechanism for diazo installation, we evaluated genes shared by both clusters for their potential involvement in this tailoring event.
After eliminating the genes likely involved in constructing ‘ethyl’-kinobscurinone, the remaining candidates were two FAD-dependent monooxygenases (lom16,17), a short chain dehydrogenase (lom18), a carboxymuconolactone decarboxylase (lom11), a peptidase (lom12), and a small sub-cluster of genes (lom29,30,32–35) encoding homologs of several C–N bond-forming and bond-cleaving enzymes as well as enzymes that may perform redox chemistry. Because two oxygenation events and reduction of a ketone are required D-ring modifications for both lomaiviticins and kinamycins, we assigned those functions to Lom16, 17, and 18. From the remaining shared genes, we identified the lom29–35 sub-cluster as the best candidate genes for diazo group formation. Notably, homologs of these genes are absent from the partial kin cluster of S. murayamaensis, which is consistent with its inability to produce diazo-containing metabolites during heterologous expression in S. lividans.
The lom29–35 sub-cluster encodes homologs of an N-acetyltransferase (lom35), an adenylosuccinate lyase (lom34), an amidase (lom33), and a glutamine synthase (lom32), as well as two proteins we hypothesize perform redox chemistry, a ferredoxin (lom30) and a hypothetical protein that has no sequence homology to characterized enzymes (lom29) (Figure 3A). The predicted biochemical functions of these enzymes (C–N bond formation, C–N bond cleavage, redox) are consistent with general types of transformations required to construct a diazo group from two individual nitrogen-containing metabolites. Intriguingly, a recent study by van der Donk, Metcalf, and co-workers identified homologs of lom32–35 in a putative fosfazinomycin (fzm) biosynthetic gene cluster.25 Fosfazinomycin contains a hydrazido linkage, and the presence of these lom genes in the fzm cluster provides further evidence for their involvement in N–N bond formation (Figure 3B). We also noted that fznP displays significant sequence homology to hypothetical protein lom29 (30% amino acid identity, 43% similarity). The lack of a lom30 homolog in the fzm cluster could potentially indicate an impairment of redox function, which would be consistent with the difference in oxidation state between the hydrazido and diazo functional groups. This observation may also suggest that homologs of lom32–35 generate a common intermediate containing an N–N single bond, which is subsequently processed differently in these two biosynthetic pathways. Homologs of additional fzm genes postulated to be involved in N–N bond formation, flavin-dependent oxidoreductase FzmM and asparagine synthase FzmA, are not found in the S. pacifica DPJ-0019 genome, further suggesting differences between the biosynthetic logic of diazo and hydrazido assembly.
Figure 3.

Identification of candidate diazo-forming enzymes and potential involvement in fluostatin biosynthesis. (A) Genes encoding putative diazo-forming enzymes and their distribution in characterized natural product biosynthetic gene clusters. (B) Structure of hydrazide-containing natural product fosfazinomycin A. (C) Structures of the fluostatins isolated from heterologous expression of BAC AB649/1850. (D) Biosynthetic hypothesis for formation of the fluostatins from diazo-containing precursors.
We searched for homologs of the putative diazo forming enzymes in sequenced bacterial genomes and discovered similar sub-clusters in 57 other secondary metabolite biosynthetic gene clusters (Tables S6 and S7). This finding suggests that the potential to synthesize natural products containing N–N bonds may be more widespread than has been previously appreciated. The majority of the identified gene clusters possessed homologs of all six genes (lom29, 30, 32–35). In an analogous manner to fosfazinomycin biosynthesis, 13 of the clusters encoded only a portion of the sub-cluster (Table S7). Interestingly, we were not able to locate homologs of these genes within the genome of the azamerone producer Streptomyces sp. CNQ-766.24 This unexpected finding may indicate that microorganisms have evolved multiple, distinct pathways for diazo group biosynthesis.
Among the gene clusters containing homologs of all six genes were 34 homologous lom clusters from other Salinispora strains, the kin cluster from S. ambofaciens, nine clusters encoding unknown metabolites, and one reconstructed environmental gene cluster that produces fluostatins C, F, G, and H when heterologously expressed in Streptomyces albus.26 This cluster also encoded homologs of all of the kinamycin biosynthetic machinery (Table S8). Our discovery of the putative diazo-forming gene cassette in this gene cluster was surprising because the fluostatins do not contain diazo groups (Figure 3C). However, we recognized a striking resemblance between the fluostatin carbon skeleton and that of isoprekinamycin, a secondary metabolite isolated from the kinamycin producer S. murayamaensis.27 Dimitrienko and co-workers previously hypothesized that isoprekinamycin is derived from kinamycin via a rearrangement process that involves a 1,2-carbon shift.27c We hypothesize that the presence of diazo-synthesizing genes in the fluostatin cluster may indicate that these metabolites are derived biosynthetically from kinamycin via isoprekinamycin (Figure 3D). Further D-ring tailoring could provide ‘isokinamycins’, which upon reduction of the diazo group would give the observed fluostatin products. This reduction could have occurred during fermentation or isolation, as is hypothesized for the biosynthesis of lomaiviticins C–E.4
Overall, the presence of the putative diazo-forming gene cassette in environmental DNA and multiple cryptic gene clusters suggests that this modification may be found in more natural product scaffolds than have been identified to date. In addition, the presence of additional clusters encoding a portion of this enzymatic machinery, including the fosfazinomycin gene cluster, indicates its involvement in constructing additional scaffolds containing N–N bonds. As diazo-containing molecules are important intermediates in chemical synthesis, elucidation of the enzymatic chemistry involved in installing this functional group could pave the way for applications in biocatalysis and is an important objective for future studies.
2.4 Identification of a candidate dimerization enzyme
A central question in lomaiviticin biosynthesis is the nature and timing of the dimerization event which gives rise to the C2-symmetric core structure of the lomaiviticins, one of the most striking differences between these natural products and the kinamycins. This structural modification has powerful consequences for biological activity, with lomaiviticin A displaying substantially more cytotoxicity than the kinamycins.4 To identify possible dimerization enzymes, we searched for genes in the lom cluster that were absent from the complete kin cluster and were not hypothesized to be involved in the other tailoring events (A- and D-ring tailoring, sugar biosynthesis). This analysis revealed one particularly strong candidate gene for dimerization, lom19, which displayed striking homology to actVA-orf4, a gene from the actinorhodin biosynthetic gene cluster (51% identity, 63% similarity at the amino acid level). Both genes encode homologs of the NmrA family of nicotinamide cofactor-binding regulatory proteins and may therefore perform NAD+/NADP+-based redox chemistry.28,29
Using genetics, Ichinose and co-workers recently demonstrated that the actVA-orf4 gene product is involved in formation of the distinctive biaryl framework of actinorhodin and that this key coupling reaction proceeds via a hydroquinone intermediate (Scheme 2).30 The presence of a related gene in the lom cluster strongly suggests that the dimerization reaction in lomaiviticin assembly employs similar biosynthetic logic, utilizing a hydroquinone-containing substrate and generating a biaryl product (Scheme 1B). Moreover, the use of a hydroquinone intermediate would necessitate that D-ring tailoring and glycosidation steps occur after dimerization. This order of events is intriguing from the standpoint of reactivity, as it requires that these additional tailoring reactions take place on an extremely hindered biaryl framework. Indeed, Moore and coworkers also identified lom19 as the candidate dimerizing enzyme but proposed that the monomeric precursor substrate is a cyclohexanone derivative rather than a hydroquinone due to concerns with the plausibility of functionalizing a sterically encumbered biaryl scaffold. We do not favor this hypothesis, as it is unclear whether a nicotinamide-dependent enzyme could possess the reactivity needed to dimerize a less reactive cyclohexanone substrate.29 It is also important to emphasize that Lom19 and ActVA-orf4 do not resemble other microbial enzymes that catalyze biaryl coupling reactions.31 Efforts to chemically synthesize lomaiviticin A have been hampered both by an inability to functionalize sterically hindered biaryl intermediates and by difficulties in dimerizing functionalized monomeric precursors.1 If our proposed sequence of events proves correct, the characterization of Lom19 will underscore Nature’s ability to evolve biosynthetic machinery possessing reactivity that remains elusive to synthetic chemists.
Scheme 2.
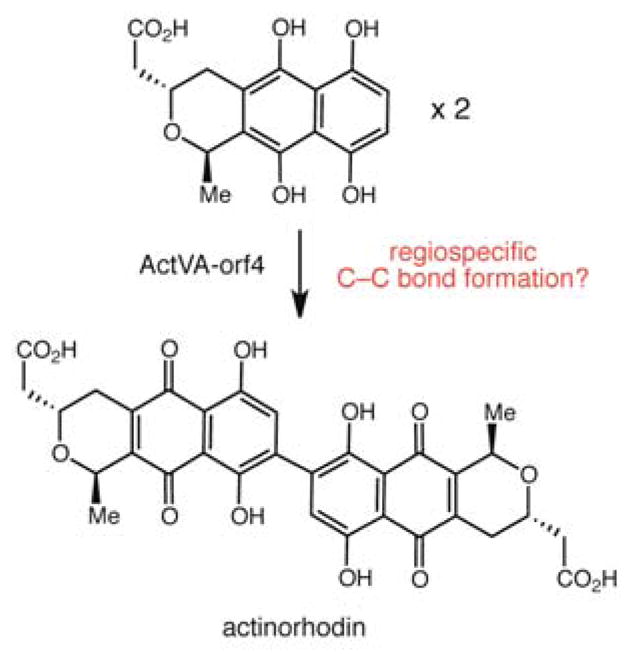
Predicted biochemical function of ActVA-orf4 a homolog of Lom19 from Streptomyces coelicolor that is involved in actinorhodin biosynthesis.
3. Conclusion
In summary, we have elucidated the genetic basis for lomaiviticin biosynthesis in two strains of S. pacifica using complementary approaches and annotated these biosynthetic clusters using a comparative genomic strategy, revealing a wealth of chemically unusual tailoring enzymes that includes genes hypothesized to be involved in diazo formation. A major challenge we still face in characterizing lomaiviticin biosynthesis is establishing the order of biosynthetic transformations, particularly the timing of diazo formation, dimerization, and glycosidation. Solving this problem will likely require a combination of genetic studies, feeding experiments, and in vitro biochemical characterization of tailoring enzymes. We will also investigate the roles of the putative diazo-forming and dimerization enzymes and attempt to elucidate the mechanisms of these unusual functionalization events. Overall, this work represents a critical step in understanding the unique biochemical transformations and logic employed in lomaiviticin assembly and lays the groundwork for future efforts to generate analogs of this natural product by combining the power of chemo- and biosynthesis.
4. Experimental
4.1 General materials and methods
Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA). Recombinant plasmid DNA was purified with a Qiaprep Kit from Qiagen. Purification of PCR reactions and gel extraction of DNA fragments for restriction endonuclease clean up were performed using an Illustra GFX PCR DNA and Gel Band Purification Kit from GE Healthcare. DNA sequencing was performed by Genewiz (Boston, MA) and Beckman Coulter Genomics (Danvers, MA). Optical densities of E. coli cultures were determined with a DU 730 Life Sciences UV/Vis spectrophotometer (Beckman Coulter) by measuring absorbance at 600 nm.
4.2 Cultivation of bacterial strains
S. pacifica DPJ-0016 and S. pacifica DPJ-0019 were routinely cultured on M1 artificial sea water-based agar (10 g/L Difco soluble starch, 4 g/L Bacto™ yeast extract, 2 g/L Bacto™ peptone, 18 g/L Bacto™ agar, 500 mL deionized water, 500 mL artificial seawater). DPJ-0019-lom58::acc(3)IV-MT1 (DPJ-0019-MT1) was cultured on the same medium containing 50 μg/mL apramycin. Escherichia coli strains EPI100™-T1R (Epicentre), TOP10 (Invitrogen), and ET12567/pUZ800232 used for mutagenesis experiments were cultured in Luria-Bertani (LB) media supplemented with the appropriate antibiotics.
4.3 Identification of lom gene clusters
Two complementary methods were used to discover lom gene clusters. For S. pacifica DPJ-0016, a cosmid library was generated from genomic DNA, while for S. pacifica DPJ-0019 the whole genome was sequenced. In both cases the sequencing data was then mined using genetic probes designed based on the likely biosynthetic machinery contained in the lom cluster.
S. pacifica DPJ-0016
A genomic library was prepared using the pWEB cosmid cloning kit (Epicentre, Madison, WI). A dNDP-glucose dehydratase gene fragment was PCR-amplified from the genome of DPJ-0016 using previously reported primers,33 radiolabeled with [α-32P]dCTP (Amersham) using Ready-To-Go Labeling Beads (GE, Pittsburgh, PA), and used to probe the genomic library with standard colony hybridization protocols. Two hybridizing cosmids (2C7 and 5E8b) were found to overlap by 4.4 kb. A second radiolabeled probe was developed based on an open reading frame (ORF) from 5E8b encoding a ketoreductase (lom22). A third cosmid, p730497, hybridized with the ketoreductase probe and was found to overlap with 5E8b by 1.25 kb. Cosmids 2C7 and 5E8b were sequenced by 454 Life Sciences and p730497 was sequenced at Pfizer on an ABI 3730 sequencer with the ABI Prism DNA sequencing kit and Big Dye terminators version 3.1 (Applied Biosystems). All sequences were assembled in Vector NTI. ORFs were identified using Frameplot 2.3.234 and putative functions were assigned by BLASTp35 comparison to deduced amino acid sequences of ORFs contained in the GenBank database. The nucleotide sequence of the lom cluster in DPJ-0016 has been deposited to GenBank under accession number KF731828.
S. pacifica DPJ-0019
DNA for whole genome sequencing was isolated from S. pacifica DPJ-0019 with the UltraClean Microbial DNA Isolation Kit (Mo Bio). The genomic DNA isolation was performed according to the manufacturer’s protocol with the following modifications. The cell pellets were washed twice by resuspending the pellets in 600 μL of the MicroBead solution, centrifuging at 10,000 x g for 60 sec, and removing the supernatant. Due to the large size of the cell clumps, a 5-mL serological pipet was used to transfer the cells to the 2-mL Collection Tubes. After the addition of Solution MD1, the samples were heated at 70 °C for 10 min and then vortexed at maximum speed for 10 min to lyse the cells. After lysis, 4 μL of RNaseA (100 mg/mL) was added, and the samples were incubated at room temperature for 2 min. The wash step with 300 μL of Solution MD4 was performed twice. Genomic DNA was eluted with 50 μL of Solution MD5 (10 mM Tris, pH 8) and the samples were stored at −80 °C until submission for sequencing.
Genome sequencing and assembly were performed at Cofactor Genomics (St. Louis, MO). Short insert and long insert mate pair DNA libraries were sequenced using the HiSeq2000 (Ilumina). The short insert genomic DNA library was constructed according to the following procedure. Briefly, 2.5 μg of genomic DNA was sheared to 300 bp using the Covaris S2 (Covaris, Woburn, MA). Another 2.5 μg was sheared to a size of 500 bp on the Covaris using manufacturer recommended protocols. Following shearing, the DNA was end-repaired and A-tailed to prepare for adaptor ligation. Indexed adaptors were ligated to sample DNA, and the adaptor-ligated DNA was then size-selected on a 2% SizeSelect™ E-Gel (Invitrogen, Carlsbad, CA) and amplified by PCR. Library quality was assessed by measuring the yield using a Qubit DNA broad range assay (Invitrogen, Carlsbad, CA) and by measuring fragment size in base pairs using the Experion (Bio-Rad). Adapter containing library molecules were quantified by qPCR prior to paired end 100 bp sequencing on the HiSeq 2000. The long insert mate pair genomic DNA library was constructed according to the following procedure. Briefly, genomic DNA was sheared to an average size of 2000 bp using the Covaris S2 (Covaris, Woburn, MA). Sheared DNA was size selected on a 1% agarose gel, excised from the gel, purified, and quantified using the Qubit broad range assay (Invitrogen, Carlsbad, CA). Size-selected DNA was end repaired and prepared for circularization. Circularized DNA was subjected to nick translation. DNA was A-tailed and indexed adapters ligated, followed by PCR. Library quality was assessed by measuring the yield using a Qubit DNA broad range assay (Invitrogen, Carlsbad, CA) and by measuring fragment size in base pairs using the Experion (Bio-Rad). Adapter containing library molecules were quantified by qPCR prior to paired end 100 bp sequencing on the HiSeq 2000.
Cluster generation and the subsequent sequencing were performed according to the cluster generation manual and sequencing manual from Illumina (Cluster Station User Guide and Genome Analyzer Operations Guide). Base calls were generated using Casava 1.8.2 (Illumina), and the resulting demultiplexed sequence reads were filtered for low quality. Cofactor Genomics’ assembly pipeline was then run, comprising the following procedure. Sequence data from each library were sampled to determine assembly parameters with the aim of balancing specificity and sensitivity. Specifically, low occurrence observations were filtered and assembly joins were optimized based on the available coverage and the k-mer characteristics of each library. Using this information, assembly parameters were chosen and then used to perform an assembly on the sequence data using SOAPdenovo 1.05 (BGI).
Assembly resulted in 5.45 MB of non-redundant sequence distributed over 8 scaffolds. The assembled data were converted into a local BLAST database using Geneious. The putative lomaiviticin gene cluster was identified on scaffold 2 by searching for homologs of the ketosynthase (KSα) and chain length factor (CLF) components of type II polyketide biosynthetic machinery using BLASTp. The total size of the putative lom gene cluster was 62 kb. Sequencing of short stretches of the lom cluster not covered in this assembly was accomplished by cloning these sequences from genomic DNA. Sequence data bordering these unsequenced stretches were used to design specific PCR primers that would amplify these regions. Blunt-ended PCR products (amplified with Q5 High-Fidelity 2X Master Mix, New England Biolabs) were ligated into the linearized pCRII-Blunt-TOPO vector using the Zero Blunt TOPO PCR Cloning Kit (Invitrogen) and used to transform E. coli Top10 cells according to the manufacturer’s protocol. The sequences of the inserts enabled a complete assembly of the lom cluster. Open reading frames (ORFs) were detected using FGENESB (Softberry), GeneMark.hmm for Prokaryotes (V 2.8), and Prodigal (Prokaryotic Dynamic Programming Genefinding Algorithm). ORF lengths were refined by comparison with the homologous ORFs from S. pacifica DPJ-0016 and the lom gene cluster from Salinispora tropica CNB-440. The nucleotide sequence of the lom cluster in DPJ-0019 has been deposited to GenBank under accession number KF515737.
4.4 Construction of the lom58 mutant in S. pacifica DPJ-0019
Inactivation of the putative lomaiviticin gene cluster was performed by insertional inactivation of the KSα (lom58) in DPJ-0019.36 A 3.5 kb fragment that encodes for the KSα (lom58), CLF (lom59), and ACP (lom60) was amplified by PCR (primers Lom-DH-f and Lom-ACP-r) and cloned into pUC19 to generate pUC19-lom58. pUC19-lom58 was digested with ApaI and then ligated with the aminoglycoside-(3)-acetyltransferase IV gene (aac3IV) containing ApaI restrictions sites on the 5′ and 3′ ends. The construct was moved to the bifunctional E. coli/Streptomyces conjugative plasmid pNWA20037 and introduced into DPJ-0019 via conjugation from E. coli ET12567/pUZ8002.32 Colonies that were resistant to apramycin but sensitive to kanamycin were screened by colony PCR with primers TP1_lomKS and TP2_lomKS (Figure S2).
4.5 Comparative parallel fermentations of DPJ-0016, DPJ-0019, and DPJ-0019-MT1
For first stage seed cultures, DPJ-0016 and DPJ-0019 were each inoculated into 7 mL of WSB seed medium [5 g/L Soy hydrosylate (SE50MAF-UF), 15 g/L wheat hydrosylate (WGE80M-UF), 3 g/L Bacto™ yeast extract, 10 g/L Difco soluble starch, 500 mL deionized water, 500 mL artificial seawater) in 25×150 mm culture tubes. DPJ-0019-MT1 was inoculated in identical conditions with 50 μg/mL apramycin. Seed cultures were incubated at 30 °C and 220 rpm. After four days, first stage seeds were transferred to 40 mL of the same seed medium in 250 mL Erlenmeyer flasks and incubated at 30 °C and 220 rpm. The second stage seeds were cultured for 48 hours. For fermentation, DPJ-0016, DPJ-0019, and DPJ-0019-MT1 were cultured in PCAA medium (10 g/L Bacto™ peptone, 5 g/L Bacto™ casamino acids, 0.1 g/L sodium citrate), YESS medium (30 g/L Difco soluble starch, 7.5 g/L Bacto™ yeast extract) and SPYESS medium (30 g/L Difco soluble starch, 7.5 g/L Kerry Hy-Soy® soy peptone, 2.5 g/L Bacto™ yeast extract). All fermentation media were prepared in half strength artificial seawater with 50 g/L Diaion HP-20 added pre-autoclave. All fermentations were performed in duplicate at 30 °C and 220 rpm in 500 mL Erlenmeyer flasks each containing 100 mL of medium and inoculated with 4% (volume/volume) second stage seed.
After 14 days of incubation, duplicate samples (one milliliter each) were taken from all fermentations. The bacterial cells and HP-20 were collected by centrifugation. The supernatants were removed and discarded. One set of resin and cell pellets was double extracted (1 mL × 2) with methanol (MeOH). The other set was double extracted (1 mL × 2) with ethyl acetate (EtOAc). All crude extracts were dried in vacuo. Methanolic extracts were resolubilized at 5X in 1:4 acetonitrile (MeCN) to MeOH, and EtOAc extracts were resolubilized at 5X in 1:4 DMSO to MeCN. All extracts were analyzed on an Agilent 1100 Series HPLC with a linear gradient of 15–60% aqueous MeCN (0.01% trifluoroacetic acid) at a flow rate of 1.0 mL/min over 20 min on a reversed-phase C18 column (YMC-ODS-A, 4.6 mm × 150 mm, 5 mm).
Confirmation of production of lomaiviticins C, D and E as well as neolymphostin A was performed on a Waters Acuity UPLC- LCT Premier™ TOF MS with alternating positive-ion and negative-ion full scan (100–2000 mass units) mode and a linear gradient of 5–95% MeCN in water (0.1% formic acid) at a flow rate of 0.4 mL/min over 12 minutes on a reversed-phase C18 column (XBridge, 4.6 mm × 150 mm, 5 mm, 3.5 μm).
4.6 Annotation of the S. pacifica lom gene clusters and comparative genomic analyses
The closest homolog of each ORF in both lom clusters was identified using pBLASTn searches of the nonredundant protein database in NCBI. At the time of our annotation there was only one sequenced genome with a homologous lom cluster, Salinispora tropica CNB-440, so Table S3 contains hits only from this cluster. Further BLAST searches were performed using the protein databases in both NCBI and IMG at the DOE Joint Genome Institute.
4.7 Annotation of putative dimerizing enzyme Lom19
The amino acid sequences of Lom19, ActVA-orf4 from Streptomyces coelicolor (CAA41640), NmrA (AAC39442, PDB = 1K6I), and structurally characterized NmrA-like proteins Hscarg (AAG09721, PDB = 2EXX), QOR2 (AAC77168, PDB = 2ZCU) and TMR (AAW88298, PDB = 2JL1) were aligned using ClustalOmega (http://www.ebi.ac.uk/Tools/msa/clustalo/).38 The resulting alignment shows that many of the amino acids that contact the nicotinamide cofactor are conserved among the NmrA-like proteins (Figure S4). We were unable to detect any motifs for binding flavin cofactors in Lom19 through searches of the Conserved Domains Database (NCBI, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml).39
Supplementary Material
Biosynthetic hypothesis for assembly of L-oleandrose and N,N-dimethyl-L-pyrrolosamine sugar scaffolds by Lom9,49–54,56,57,65.
Acknowledgments
We gratefully acknowledge Drs. Jan Kieleczawa and Tony Li at Pfizer’s BioCore facility (Cambridge, MA) for DNA sequencing of cosmid p730497, Dr. N. Fusetani (Hokkaido University) for providing D. proliferum tissue from which DPJ-0016 and DPJ-0019 were isolated, and Dr. Seth Herzon (Yale University) for providing a sample of lomaiviticin C and for helpful discussions. We received financial support from the National Institutes of Health (NIH) (DP2 GM105434 and GM095450), the Searle Scholars Program, and Harvard University. H. N. is the recipient of a Herchel Smith Fellowship.
Footnotes
Supporting Information: Figures, tables, and schemes illustrating phylogentic tree analysis, mutant construction, comparative parallel fermentations, gene cluster annotations, comparative genomic analyses, and biosynthetic hypotheses mentioned in the main text. Supplementary data associated with this article can be found in the online version, at http://...
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.For a review of the lomaiviticin and kinamycin families of natural products see: Woo CM, Herzon SB. Nat Prod Rep. 2012;29:87. doi: 10.1039/c1np00052g.
- 2.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J Am Chem Soc. 2001;123:5362. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 3.Miyanaga A, Janso JE, McDonald L, He M, Liu H, Barbieri L, Eustáquio AS, Fielding EN, Carter GT, Jensen PR, Feng X, Leighton M, Koehn FE, Moore BS. J Am Chem Soc. 2011;133:13311. doi: 10.1021/ja205655w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woo CM, Beizer NE, Janso JE, Herzon SB. J Am Chem Soc. 2012;134:15285. doi: 10.1021/ja3074984. [DOI] [PubMed] [Google Scholar]
- 5.(a) Herzon SB, Lu L, Woo CM, Gholap SL. J Am Chem Soc. 2011;113:7260. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Woo CM, Gholap SL, Lu L, Kaneko M, Li Z, Ravikumar PC, Herzon SB. J Am Chem Soc. 2012;134:17362. doi: 10.1021/ja307497h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Krygowski ES, Murphy-Benenato K, Shair MD. Angew Chem Int Ed. 2008;47:1680. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]; (b) Lee HG, Ahn JY, Lee AS, Shair MD. Chem Eur J. 2010;16:13058. doi: 10.1002/chem.201002157. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee AS, Shair MD. Org Lett. 2013;15:2390. doi: 10.1021/ol400832r. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Denton RM, Lenzen A, Edmonds DJ, Li A, Milburn RR, Harrison ST. Angew Chem Int Ed. 2006;45:2076. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]; (e) Nicolaou KC, Nold AL, Li H. Angew Chem Int Ed. 2009;48:5860. doi: 10.1002/anie.200902509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gholap SL, Woo CM, Ravikumar PC, Herzon SB. Org Lett. 2009;11:4322. doi: 10.1021/ol901710b. [DOI] [PubMed] [Google Scholar]; (g) Zhang W, Baranczak A, Sulikowski GA. Org Lett. 2008;10:1939. doi: 10.1021/ol800460a. [DOI] [PubMed] [Google Scholar]; (h) Feldman KS, Selfridge BR. Org Lett. 2013;78:4499. doi: 10.1021/jo4005074. [DOI] [PubMed] [Google Scholar]
- 7.While our initial manuscript describing this work was in preparation, Moore and coworkers disclosed the production of lomaiviticin by a formerly cryptic gene cluster from Salinispora tropica CNB-440: Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. Chem Bio Chem. 2013;14:955. doi: 10.1002/cbic.201300147.
- 8.Waldman AJ, Balskus EP. Org Lett. 2014;16:3640. doi: 10.1021/ol403714g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Ajisaka K, Takeshima H, Omura S. J Chem Soc Chem Commun. 1976:571. [Google Scholar]; (b) Sato Y, Gould SJ. Tetrahedron Lett. 1985;26:4023. [Google Scholar]; (c) Seaton PJ, Gould SJ. J Am Chem Soc. 1987;109:5282. [Google Scholar]; (d) Seaton PJ, Gould SJ. J Am Chem Soc. 1988;110:5912. [Google Scholar]; (e) Gould SJ, Melville CR. Bioorg Med Chem Lett. 1995;6:51. [Google Scholar]; (f) Gould SJ, Melville CR, Cone MC, Chen J, Carney JR. J Org Chem. 1997;62:320. doi: 10.1021/jo961486y. [DOI] [PubMed] [Google Scholar]
- 10.Gould SJ, Hong ST, Carney JR. J Antibiot. 1998;51:50. doi: 10.7164/antibiotics.51.50. [DOI] [PubMed] [Google Scholar]
- 11.(a) Bunet R, Song L, Mendes MV, Corre C, Hotel L, Rouhier N, Framboisier X, Leblond P, Challis GL, Aigle B. J Bacteriol. 2011;193:1142. doi: 10.1128/JB.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Aigle B, Lautru S, Spiteller D, Dickschat JS, Challis GL, Leblond P, Pernodet J-L. J Ind Microbiol Biotechnol. 2013 doi: 10.1007/s10295-013-1379-y. [DOI] [PubMed] [Google Scholar]
- 12.Decker H, Gaisser S, Pelzer S, Schneider P, Westrich L, Wohlleben W, Bechtold A. FEMS Microbiol Lett. 1996;141:195. doi: 10.1111/j.1574-6968.1996.tb08384.x. [DOI] [PubMed] [Google Scholar]
- 13.Lomaiviticin A was originally produced by fermentation of strain LL-37I366 in a medium containing 0.5% Bacto peptone and 1% HyCase SF (acid hydrolysis of casein).2
- 14.Kharel MK, Pahari P, Shepherd MD, Tibrewal N, Nybo SE, Shaaban KA, Rohr J. Nat Prod Rep. 2012;29:264. doi: 10.1039/c1np00068c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han L, Yang KQ, Ramalingam E, Mosher RH, Vining LC. Microbiology. 1994;140:3379. doi: 10.1099/13500872-140-12-3379. [DOI] [PubMed] [Google Scholar]
- 16.Kulowski K, Wendt-Pienkowski E, Han L, Yang K, Vining LC, Hutchinson CR. J Am Chem Soc. 1999;121:1786. [Google Scholar]
- 17.(a) Rajgarhia VB, Priestley ND, Strohl WR. Metab Eng. 2001;3:49. doi: 10.1006/mben.2000.0173. [DOI] [PubMed] [Google Scholar]; (b) Bao WL, Sheldon PJ, Hutchinson CR. Biochemistry. 1999;38:9752. doi: 10.1021/bi990751h. [DOI] [PubMed] [Google Scholar]; (c) Bao W, Sheldon PJ, Wendt-Pienkowski E, Hutchinson CR. J Bacteriol. 1999;181:4690. doi: 10.1128/jb.181.15.4690-4695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Fan K, Pan G, Peng X, Zheng J, Gao W, Wang J, Wang W, Li Y, Yang K. Chem Biol. 2012;19:1381. doi: 10.1016/j.chembiol.2012.09.009. [DOI] [PubMed] [Google Scholar]; (b) Tibrewal N, Pahari P, Wang G, Kharel MK, Morris C, Downey T, Hou Y, Bugni TS, Rohr J. J Am Chem Soc. 2012;134:18181. doi: 10.1021/ja3081154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rix U, Wang C, Chen Y, Lipata FM, Rix LLR, Greenwell LM, Vining LC, Yang K, Rohr J. Chem Bio Chem. 2005;6:838. doi: 10.1002/cbic.200400395. [DOI] [PubMed] [Google Scholar]
- 19.Wohlert S, Lomovskaya N, Kulowski K, Fonstein L, Occi JL, Gewain KM, MacNeil DJ, Hutchinson CR. Chem Biol. 2001;8:681. doi: 10.1016/s1074-5521(01)00043-6. [DOI] [PubMed] [Google Scholar]
- 20.The biosynthesis of N,N-dimethyl-L-pyrrolosamine by enzymes encoded in the lom gene cluster has been studied previously: Zhang H. PhD Thesis. Tufts University; 2011.
- 21.Woo CM, Gholap SL, Herzon SB. J Nat Prod. 2013;76:1238. doi: 10.1021/np400355h. [DOI] [PubMed] [Google Scholar]
- 22.Nawrat CC, Moody CJ. Nat Prod Rep. 2011;28:1426. doi: 10.1039/c1np00031d. [DOI] [PubMed] [Google Scholar]
- 23.Bartz QR, Haskell TH, Elder CC, Johannessen DW, Frohardt RP, Ryder A, Fusari SA. Nature. 1954;173:72. doi: 10.1038/173072b0. [DOI] [PubMed] [Google Scholar]
- 24.Winter JM, Jansma AL, Handel TM, Moore BS. Angew Chem Intl Ed. 2009;48:767. doi: 10.1002/anie.200805140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Ju K-S, Yu X, Velasquez JE, Mukherjee S, Lee J, Zhao C, Evans BS, Doroghazi JR, Metcalf WW, van der Donk WA. Angew Chem Intl Ed. 2014;53 doi: 10.1002/anie.201308363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Z, Kim JH, Brady SF. J Am Chem Soc. 2010;132:11902. doi: 10.1021/ja104550p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Cone MC, Seaton PJ, Halley KA, Gould SJ. J Antibiot. 1989;42:179. doi: 10.7164/antibiotics.42.179. [DOI] [PubMed] [Google Scholar]; (b) Seaton PJ, Gould SJ. J Antibiot. 1989;42:189. doi: 10.7164/antibiotics.42.189. [DOI] [PubMed] [Google Scholar]; (c) Proteau PJ, Li Y, Chen J, Williamson RT, Gould SJ, Laufer RS, Dmitrienko GI. J Am Chem Soc. 2000;122:8325. [Google Scholar]
- 28.Stammers DK, Ren J, Leslie K, Nichols CE, Lamb HK, Cocklin S, Dodds A, Hawkins AR. EMBO J. 2001;20:6619. doi: 10.1093/emboj/20.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.In reference 7, Moore and co-workers annotated Lom19 homolog Strop_2191 as an FAD-dependent monooxygenase. For bioinformatic analyses supporting our assignment, see Figure S4.
- 30.(a) Taguchi T, Yabe M, Odaki H, Shinozaki M, Metsa-Ketela M, Arai T, Okamoto S, Ichinose K. Chem Biol. 2013;20:510. doi: 10.1016/j.chembiol.2013.03.007. [DOI] [PubMed] [Google Scholar]; (b) Taguchi T, Ebihara T, Furukawa A, Hidaka Y, Ariga R, Okamoto S, Ichinose K. Bioorg Med Chem Lett. 2012;22:5041. doi: 10.1016/j.bmcl.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 31.For other microbial enzymes involved in biaryl coupling, see: Yamanaka K, Ryan KS, Gulder TA, Hughes CC, Moore BS. J Am Chem Soc. 2012;134:12434. doi: 10.1021/ja305670f.Gil Girol C, Fisch KM, Heinekamp T, Gunther S, Huttel W, Piel J, Brakhage AA, Muller M. Angew Chem Int Ed. 2012;51:9788. doi: 10.1002/anie.201203603.Baunach M, Ding L, Bruhn T, Bringmann G, Hertweck C. Angew Chem Int Ed. 2013;52:9040. doi: 10.1002/anie.201303733.
- 32.Paget MS, Chamberlin L, Atrih A, Foster SJ, Buttner MJ. J Bacteriol. 1999;181:204. doi: 10.1128/jb.181.1.204-211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decker H, Gaisser S, Pelzer S, Schneider P, Westrich L, Wohlleben W, Bechtold A. FEMS Microbiol Lett. 1996;141:195. doi: 10.1111/j.1574-6968.1996.tb08384.x. [DOI] [PubMed] [Google Scholar]
- 34.Ishikawa J, Hotta K. FEMS Microbiol Lett. 1999;174:251. doi: 10.1111/j.1574-6968.1999.tb13576.x. [DOI] [PubMed] [Google Scholar]
- 35.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, U.K: 2000. [Google Scholar]
- 37.Magarvey NA, Haltli B, He M, Greenstein M, Hucul JA. Antimicrob Agents Chemother. 2006;50:2167. doi: 10.1128/AAC.01545-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marchler-Bauer A, Bryant SH. Nucleic Acids Res. 2004;32:327. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Biosynthetic hypothesis for assembly of L-oleandrose and N,N-dimethyl-L-pyrrolosamine sugar scaffolds by Lom9,49–54,56,57,65.