

Abstract
Background
Most previous studies of morphological and molecular data have consistently supported the monophyly of the true water bugs (Hemiptera: Nepomorpha). An exception is a recent study by Hua et al. (BMC Evol Biol 9: 134, 2009) based on nine nepomorphan mitochondrial genomes. In the analysis of Hua et al. (BMC Evol Biol 9: 134, 2009), the water bugs in the group Pleoidea formed the sister group to a clade that consisted of Nepomorpha (the remaining true water bugs) + Leptopodomorpha (shore bugs) + Cimicomorpha (assassin bugs and relatives) + Pentatomomorpha (stink bugs and relatives), thereby suggesting that fully aquatic hemipterans evolved independently at least twice. Based on these results, Hua et al. (BMC Evol Biol 9: 134, 2009) elevated the Pleoidea to a new infraorder, the Plemorpha.
Results
Our reanalysis suggests that the lack of support for the monophyly of the true water bugs (including Pleoidea) by Hua et al. (BMC Evol Biol 9: 134, 2009) likely resulted from inadequate taxon sampling. In particular, long-branch attraction (LBA) between the distant outgroup taxa and Pleoidea, as well as LBA among taxa in the ingroup, made Nepomorpha appear to be polyphyletic. We used three complementary strategies to test and alleviate the effects of LBA: (1) the removal of distant outgroups from the analysis; (2) the addition of closely related outgroups; and (3) the addition of a mitochondrial genome from a second family of Pleoidea. We also performed likelihood-ratio tests to examine the support for monophyly of Nepomorpha with different combinations of taxa included in the analysis. Furthermore, we found that specimens of Helotrephes sp. were misidentified as Paraplea frontalis (Fieber, 1844) by Hua et al. (BMC Evol Biol 9: 134, 2009).
Conclusions
All analyses that included the addition of more taxa significantly and consistently supported the placement of Pleoidea within the Nepomorpha (i.e., supported the monophyly of the traditional true water bugs). Our analyses further support a close relationship between Notonectoidea and Pleoidea within Nepomorpha, and the superfamilies Nepoidea, Ochteroidea, Naucoroidea, and Pleoidea are resolved as monophyletic in all trees with strong support. Our results also confirmed that monophyly of Nepomorpha clearly is not refuted by the mitochondrial genome data.
Keywords: Long-branch attraction, Nepomorpha, Mitochondrial genome, Taxon sampling, Likelihood-ratio test
Background
Long-branch attraction (LBA) is a bias that results in spurious support for relationships between two (or more) long branches in an estimated phylogenetic tree when the assumed model of evolution is too simplistic [1,2]. Biases associated with LBA have been identified in many phylogenetic studies, including analyses of mammals [3,4], birds [5], arthropods [6-8], and seed plants [9,10]. The most common problem occurs when distantly related ingroup taxa are poorly sampled and one or a few distant outgroup taxa are included to root the tree. Under these conditions, a simplistic model of evolution is unlikely to sufficiently account for homoplasy, and long branches will be connected (or attracted to one another) in the inferred tree based on homoplastic similarities [11]. One method for detecting this problem involves conducting phylogenetic analyses with and without outgroups [12]. If the inclusion of a distant outgroup changes the inferred relationships of the ingroup, it may be better to infer ingroup relationships separately and consider other methods for rooting the resulting tree, or to use more closely related outgroups [13]. In addition, several strategies have been suggested to reduce the effects of LBA, including: (1) excluding long-branch taxa from the analysis, (2) replacing the long-branch taxa with slow-evolving close relatives, (3) removing fast-evolving proteins or sites, (4) improving the models of character evolution assumed in the analysis, and (5) sampling more taxa to break up long branches in the tree [14-16]. Among these methods, adding taxa to break up long branches is one of the most widely suggested strategies to reduce the effects of LBA bias [17,18]. Appropriate and thorough taxon sampling is thus one of the most important considerations for accurate phylogenetic estimation [16-19]. Phylogenetic analyses based on relatively few distantly related taxa (but with each taxon represented by many characters, such as from a mitochondrial genome) are particularly prone to problems with LBA; such analyses are likely to produce high support values for incorrect phylogenetic relationships [16,20].
The relationships of the true water bugs (Hemiptera: Nepomorpha) within heteropteran insects [21] have been the subject of many studies of molecular and morphological data. The monophyly of Nepomorpha has been consistently and strongly supported by studies based on morphological characters [22-25], molecular data (partial sequences of 16S rDNA and 28S rDNA [26], and four Hox genes [27]), and by combined data analyses [26]. In contrast, the monophyly of Nepomorpha has only been disputed in the study of Hua et al. [28], who based their analysis on nine nepomorphan mitochondrial genomes (mt-genomes). In the study by Hua et al. [28], Pleoidea was not supported as part of Nepomorpha, but instead was resolved as the sister-group of a clade that included the remaining species of Nepomorpha plus Leptopodomorpha, Cimicomorpha, and Pentatomomorpha (Figure 1). As a result of these analyses, Hua et al. [28] suggested that Pleoidea should be raised from a superfamily within Nepomorpha to the infraorder Plemorpha, outside of Nepomorpha. Their conclusions were supported by high Bayesian posterior probabilities (BPP) and maximum likelihood (ML) bootstrap proportions in five of eight phylogenetic analyses.
Figure 1.
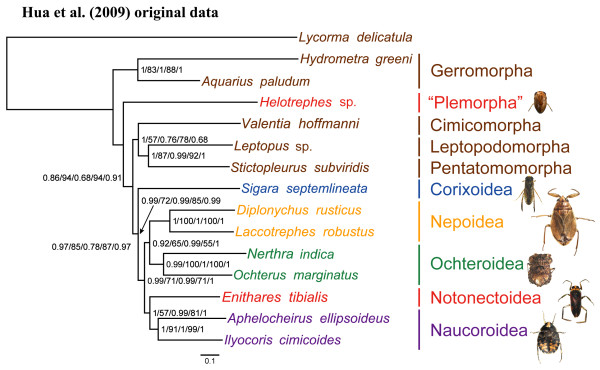
The consensus phylogeny based on the data sets analyzed by Hua et al. [[28]]. Five of the eight phylogenetic analyses they conducted supported this tree. Numbers at the nodes indicate the BPP and ML support values for each data matrix analyzed by Hua et al. [28] in the following order: PP and BP for PCG123RT, PP and BP for PCG12RT, and PP for PCG12. Branch lengths are similar across analyses; these branch lengths represent the analysis of the PCG123RT data set. The scale bar represents the number of expected substitutions per site.
The study by Hua et al. [28] has both strengths and weaknesses when compared with previous studies of the phylogenetic relationships of Nepomorpha. Each taxon sampled by Hua et al. [28] was sampled for complete mitochondrial genomes, so the number of characters available for phylogenetic inference was large. In contrast, previous studies [22-27] examined fewer characters per taxon, but included more taxa in the analyses. Thorough taxon sampling can often lead to more accurate phylogenetic inference, even if the total number of characters in the analysis is decreased [29-32]. In particular, the position of Pleoidea in the study of Hua et al. [28] may have been affected by the inclusion of just one of two families in Pleoidea (Helotrephidae, without any representation of Pleidae; see Results and discussion). This made it more likely for the tree to be rooted by connection of the distantly related outgroup taxa to the long branch leading to Helotrephes sp. (Figure 1).
A second consideration is the selection of outgroups used by Hua et al. [28]. Fulgoromorpha is very distantly related to the ingroup Nepomorpha, making problems associated with LBA more likely [30,33]. Furthermore, in groups more closely related to Nepomorpha, Hua et al. [28] sampled only one representative for three different infraorders (Cimicomorpha, Leptopodomorpha and Pentatomomorpha). Thus, we examined the possibility that the findings of Hua et al. [28] resulted from biases associated with inadequate taxon sampling. Because the model-based methods used by Hua et al. [28] are less sensitive to the problems of LBA [34-36], these authors did not consider LBA to be a likely explanation of their results. However, models of evolution are never perfect, and poor taxon sampling exacerbates the problems of model insufficiency, so the use of model-based inference methods is not, by itself, a panacea for dealing with biases associated with LBA [11,16].
We undertook the current study to explore the conclusion of Hua et al. [28] that the Pleoidea evolved their fully aquatic lifestyle independently of the remaining true water bugs in Nepomorpha. Our hypothesis was that this conclusion was a result of LBA between the single sampled representative of Pleoidea and the distantly related outgroup, Fulgoromorpha. We tested this hypothesis by: (1) removing the outgroups and re-estimating the phylogeny of Nepomorpha only, to detect whether the ingroup topology is affected by the long-branch outgroup taxa [12,13]; (2) increasing taxon sampling of groups related to Nepomorpha, including Leptopodomorpha, Cimicomorpha, and Pentatomomorpha [37]; and (3) adding new mt-genome data for a representative of the second family within Pleoidea, namely Pleidae (the presumed sister-group of Helotrephidae).
Results and discussion
Misidentification of previously sampled taxa
To test our hypothesis that the conclusion of Hua et al. [28] (Pleidae outside of the remaining Nepomorpha) was an artifact of limited taxon sampling, we sampled a member of the family Helotrephidae. Helotrephidae is generally accepted as the sister-group of Pleidae [22,23,25,26], so we reasoned that including the sister-group of Pleidae was the best way to break up the long terminal branch leading to this taxon. We sequenced the mt-genome of Helotrephes semiglobosus semiglobosus Stål, 1860 (Nepomorpha: Helotrephidae). However, after we obtained a partial mt-genome sequence of Helotrephes semiglobosus semiglobosus (GenBank accession number: KJ027513) with the length of 8,876 bp, including 29 genes (two rRNAs, ten protein coding genes [PCGs] and 17 tRNAs) as well as the control region, we found extreme similarity (97.4%) between this species and the specimen previously identified by Hua et al. [28] as Paraplea frontalis (Fieber, 1844). As this level of sequence similarity was unexpected between species in these two families, we checked the specimens identified previously as Paraplea frontalis by Hua et al. [28]. We found that those specimens are properly identified as Helotrephes sp., and so represent a species in Helotrephidae rather than Pleidae. As the mt-genome of a species in Helotrephidae was already represented in the data set, we then sequenced a new mt-genome of Paraplea frontalis, as a true representative of Pleidae. Henceforth, we label the sample sequenced by Hua et al. [28] correctly as Helotrephes sp..
Removal of outgroups from the analysis
The most common problem of LBA is that distantly related outgroups have a biased attraction to long branches within the ingroup [3,4,38]. For this reason, a common suggestion is to conduct phylogenetic analyses both with and without the outgroups to compare whether the distantly related outgroup alters the ingroup topology [16]. To test if outgroup selection affected the topology of our ingroup, we ran analyses using only the ingroup taxa of Hua et al. [28]. Using Bayesian and ML analyses, all data matrices of Hua et al. [28] generated phylogenetic trees with the same topology (Figure 2). When the outgroups are removed, the ingroup topology is distinct from that obtained by Hua et al. [28] (Figure 1). In all of these analyses, Helotrephes sp. was connected to Enithares tibialis Liu et Zheng, 1991 (Nepomorpha: Notonectoidea).
Figure 2.
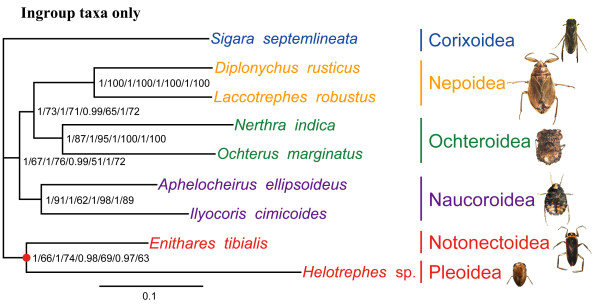
Phylogenetic results based on analyses of ingroup taxa only. Numbers at the nodes are BPP and ML support values in the following order: PP and BP for PCG12, PP and BP for PCG123, PP and BP for PCG12RT, and PP and BP for PCG123RT. The red dot on the tree indicates the clade of Notonectoidea + Pleoidea. The scale bar represents the number of expected substitutions per site based on analysis of the PCG12 data set.
Addition of outgroups
Outgroup selection is an important factor for reconstructing phylogenetic trees, because the choice of outgroup taxa can affect the ingroup topology [39]. However, outgroup selection is often not adequately considered [40,41]. Moreover, several authors have pointed out that adding more outgroup taxa in the sister-group to a phylogenetic analysis can improve the accuracy of phylogenetic estimation, and also should help break up the LBA between any long-branch members of the ingroup and the outgroup [38,42,43]. Therefore, we added three more taxa (selected from the sister-group of Nepomorpha) to the dataset of Hua et al. [28].
Both Bayesian inference and ML analyses resulted in the same topology (Figure 3A); the position of the long branch of Helotrephes sp. (Nepomorpha: Pleoidea) was supported within Nepomorpha rather than outside of Nepomorpha, in contrast to the findings of Hua et al. [28]. The monophyly of Nepomorpha (including both Helotrephidae and Pleidae) received strong support in Bayesian analyses (based on posterior probabilities: PP) but with relatively weak support in ML analyses (based on bootstrap proportions: BP). The monophyletic Nepoidea, Ochteroidea, and Naucoroidea were strongly supported by both PP and BP, similar to the results of Hua et al. [28]. Additionally, the topology of the infraordinal relationships of Heteroptera is similar to previous work [44] also based on mt-genomes, namely (Gerromorpha + (Pentatomomorpha + (Leptopodomorpha + (Cimicomorpha + Nepomorpha)))).
Figure 3.
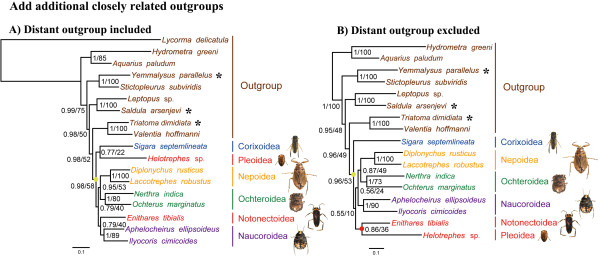
Phylogenetic trees based on the inclusion of additional closely related outgroups. (A) Analysis including the distant outgroup Lycorma delicatula (Hemiptera: Auchenorrhyncha: Fulgoromorpha). (B) Analysis excluding the distant outgroup Lycorma delicatula. Numbers at the nodes are BPP (left) and ML support values (right). Yellow dots on each phylogram indicate the clades of Nepomorpha, and red dot indicate the clades of Notonectoidea + Pleoidea. Asterisks indicate these additional closely related outgroups. The scale bar represents the number of expected substitutions per site.
We also estimated phylogenetic trees without the long-branched outgroup of Lycorma delicatula (White, 1845) (Hemiptera: Auchenorrhyncha: Fulgoromorpha). The major changes that resulted from deletion of this taxon were the position of Helotrephes sp. and Naucoroidea (Figure 3B). In both Bayesian and ML analyses, Helotrephes sp. (Nepomorpha: Pleoidea) was supported as the sister group of Enithares tibialis (Nepomorpha: Notonectoidea). The close relationship between the Notonectoidea and Pleoidea also has been supported in most previous studies [22-26]. Although the relationships among families of Nepomorpha varied among trees, all the analyses that excluded Fulgoromorpha supported the monophyly of Nepomorpha (including Helotrephidae as well as Pleidae, when the latter was added to the analyses). These analyses demonstrate that the conclusions of Hua et al. [28] were at least partly a result of their use of a very distant outgroup.
Addition of a new mitochondrial genome of Pleidae
We sequenced and assembled a new mt-genome for Paraplea frontalis (Fieber, 1844), except for small portions of 12S rRNA gene and the control region (polynucleotide sequences in these two regions proved difficult to resolve with certainty). This mt-genome was 14,143 bp in length and has been deposited in the GenBank (Accession number: KJ027516). The mt-genome of Paraplea frontalis contained the typical 37 genes (two rRNAs, 13 PCGs and 22 tRNAs), with the same gene order as observed in most other true bugs [44,45] (Table 1). Gene overlaps were found at 11 gene junctions and involved a total of 32 bp, which may make the genome relatively compact. Twelve of the 13 PCGs initiated with ATN as start codon, whereas the COI gene started with TTG. Eight PCGs ended with the termination codon TAA and one with TAG, whereas the remaining four were terminated with T. All of the 22 typical animal tRNA genes were observed in the Paraplea frontalis mt-genome, ranging from 63 to 74 bp. Most of the tRNAs could be folded into typical cloverleaf secondary structures, except that the stem of the dihydrouridine (DHU) arm simply formed a loop in tRNA-Ser (GCT) (see Additional file 1). There are 22 unmatched base pairs in the Paraplea frontalis mitochondrial tRNA secondary structures.
Table 1.
Organization of the Paraplea frontalis mitochondrial genome
| Gene | Strand | Position | Anticodon | Size (bp) | Start codon | Stop codon | Intergenic nucleotides a |
|---|---|---|---|---|---|---|---|
| tRNA-Ile |
J |
1-64 |
GAT |
64 |
|
|
|
| tRNA-Gln |
N |
62-130 |
TTG |
69 |
|
|
-3 |
| tRNA-Met |
J |
131-199 |
CAT |
69 |
|
|
0 |
| ND2 |
J |
200-1201 |
|
1002 |
ATT |
TAA |
0 |
| tRNA-Trp |
J |
1203-1268 |
TCA |
66 |
|
|
1 |
| tRNA-Cys |
N |
1261-1324 |
GCA |
64 |
|
|
-8 |
| tRNA-Tyr |
N |
1325-1391 |
GTA |
67 |
|
|
0 |
| COI |
J |
1393-2931 |
|
1539 |
TTG |
TAA |
1 |
| tRNA-Leu |
J |
2927-2991 |
TAA |
65 |
|
|
-5 |
| COII |
J |
2992-3670 |
|
679 |
ATA |
T- |
0 |
| tRNA-Lys |
J |
3671-3744 |
CTT |
74 |
|
|
0 |
| tRNA-Asp |
J |
3744-3806 |
GTC |
63 |
|
|
-1 |
| ATPase8 |
J |
3807-3962 |
|
156 |
ATA |
TAA |
0 |
| ATPase6 |
J |
3956-4666 |
|
667 |
ATG |
TAG |
-7 |
| COIII |
J |
4623-5409 |
|
787 |
ATG |
T- |
-44 |
| tRNA-Gly |
J |
5410-5472 |
TCC |
63 |
|
|
0 |
| ND3 |
J |
5473-5826 |
|
354 |
ATA |
TAA |
0 |
| tRNA-Ala |
J |
5850-5913 |
TGC |
64 |
|
|
23 |
| tRNA-Arg |
J |
5914-5979 |
TCG |
66 |
|
|
0 |
| tRNA-Asn |
J |
5979-6044 |
GTT |
66 |
|
|
-1 |
| tRNA-Ser |
J |
6044-6113 |
GCT |
70 |
|
|
-1 |
| tRNA-Glu |
J |
6114-6178 |
TTC |
65 |
|
|
0 |
| tRNA-Phe |
N |
6177-6242 |
GAA |
66 |
|
|
-2 |
| ND5 |
N |
6243-7944 |
|
1702 |
ATG |
T- |
0 |
| tRNA-His |
N |
7946-8009 |
GTG |
64 |
|
|
1 |
| ND4 |
N |
8009-9346 |
|
1338 |
ATG |
TAA |
-1 |
| ND4L |
N |
9349-9651 |
|
303 |
TTG |
TAA |
2 |
| tRNA-Thr |
J |
9654-9717 |
TGT |
64 |
|
|
2 |
| tRNA-Pro |
N |
9718-9782 |
TGG |
65 |
|
|
0 |
| ND6 |
J |
9785-10285 |
|
501 |
ATT |
TAA |
2 |
| CytB |
J |
10285-11421 |
|
1137 |
ATG |
TAG |
-1 |
| tRNA-Ser |
J |
11420-11487 |
TGA |
68 |
|
|
-2 |
| ND1 |
N |
11504-12421 |
|
918 |
ATT |
TAA |
16 |
| tRNA-Leu |
N |
12422-12485 |
TAG |
64 |
|
|
0 |
| 16S rRNA |
N |
12486-13757 |
|
1272 |
|
|
0 |
| tRNA-Val |
N |
13758-13827 |
TAC |
70 |
|
|
0 |
| 12S rRNA | N | 13828-14143 | 316 | 0 |
aNumbers correspond to nucleotides separating a gene from an upstream one; negative numbers indicate that adjacent genes overlap.
Increased taxon sampling, especially when it breaks up long branches in a tree, is the most effective strategy for reducing the effects of LBA [16,31,32]. We added the representative of Pleidae, which is thought to be the sister-group of Helotrophidae, to help reduce the length of the branch that led to the single sampled species of Helotrephidae sampled by Hua et al. [28]. We therefore added our mt-genome of Paraplea frontalis to the four data matrices of Hua et al. [28] and conducted new phylogenetic analyses (Figure 4).
Figure 4.

Phylogenetic trees based on the addition of a new mitochondrial genome of Paraplea frontalis (Nepomorpha: Pleoidea). With adding the new mt-genome of Paraplea frontalis (Fieber, 1844) to the data matrices of Hua et al. [28], we gathered four new data matrices of 16(PCG12), 16(PCG123), 16(PCG12RT), and 16(PCG123RT). (A) Numbers at the nodes are BPP for the data matrix of 16(PCG12) (left) and 16(PCG123) (right). (B) Numbers at the nodes are ML support values for the data matrix of 16(PCG12) (left), 16(PCG123) (middle), and 16(PCG123RT) (right). (C) Numbers at the nodes are BPP for 16(PCG12RT) (left), ML support values for 16(PCG12RT) (middle), and BPP for 16(PCG123RT) (right). Yellow dots on each phylogram indicate the clades of Nepomorpha, and Red dots indicate the clades of Notonectoidea + Pleoidea. The scale bar represents the number of expected substitutions per site.
As with our analyses that replaced the distant outgroup with more appropriate outgroups, the analyses that included a member of Pleidae supported monophyly of Nepomorpha (with strong PP support but weak BP support). Moreover, these analyses strongly supported Paraplea frontalis (Pleidae) as the sister group of Helotrephes sp. (Helotrephidae). Together, Pleidae and Helotrephidae were supported as the sister-group of Notonectidae. The monophyletic groups of Nepoidea, Ochteroidea, Naucoroidea, Pleoidea, and Notonectoidea + Pleoidea were strongly supported by both PP and BP in all analyses that included Pleidae.
Likelihood-ratio tests
We compared the likelihood ratios of the best solutions for each of our two alternative hypotheses (Pleoidea inside versus outside of Nepomorpha; see Additional file 2) for eight different combinations of taxa (Table 2). The monophyly of Nepomorpha (including Pleoidea) was strongly supported if we added Paraplea frontalis and/or three more outgroup taxa to the original data matrix of Hua et al. [28], as well as when we analyzed the data set without the distant outgroup consisting of Lycorma delicatula. The original conclusion of Hua et al. [28] (the polyphyly of true water bugs) was only supported with the specific combination of taxa analyzed in the original study. Even then, the likelihood-ratio support for this result over the alternative is weak (Table 2).
Table 2.
Likelihood-ratio tests for monophyly of Nepomorpha with eight different combinations of taxa
| Taxa added to analysis of Hua et al. [28] | Taxa deleted from analysis of Hua et al. [28] | ln L (Hypothesis 1) | ln L (Hypothesis 2) | 2ΔL | Hypothesis 1 a (Helotrephidae within Nepomorpha) | Hypothesis 2 a (Helotrephidae outside Nepomorpha) |
|---|---|---|---|---|---|---|
| None |
None |
-68913.45 |
-68909.21 |
8.48 |
|
Weak |
| None |
Lycorma delicatula |
-63056.47 |
-63052.68 |
7.58 |
|
Weak |
|
Paraplea frontalis |
None |
-72517.55 |
-72552.52 |
-69.94 |
Very strong |
|
|
Paraplea frontalis |
Lycorma delicatula |
-66486.5 |
-66522.49 |
-71.98 |
Very strong |
|
|
Triatoma dimidiata |
None |
-80054.05 |
-80074.67 |
-41.24 |
Very strong |
|
|
Yemmalysus parallelus | ||||||
|
Saldula arsenjevi | ||||||
|
Triatoma dimidiata |
Lycorma delicatula |
-74196.98 |
-74230.9 |
-67.84 |
Very strong |
|
|
Yemmalysus parallelus | ||||||
|
Saldula arsenjevi | ||||||
|
Paraplea frontalis |
None |
-83521.55 |
-83612.5 |
-181.9 |
Very strong |
|
|
Triatoma dimidiata | ||||||
|
Yemmalysus parallelus | ||||||
|
Saldula arsenjevi | ||||||
|
Paraplea frontalis |
Lycorma delicatula | -77682.65 | -77774.22 | -183.14 | Very strong | |
|
Triatoma dimidiata | ||||||
|
Yemmalysus parallelus | ||||||
| Saldula arsenjevi |
a: 2ΔL scores with an absolute value of 0 to 10 indicate weak support, >10 to 30 indicate strong support, and >30 indicate very strong support for the favored hypothesis [48]. Scores are calculated so that positive values indicate support for Hypothesis 2, and negative values indicate support for Hypothesis 1.
Phylogeny of nepomorpha
Given that the monophyly of Nepomorpha is consistently supported in all of our new analyses, we find no support for the new infraorder Plemorpha. Therefore, we recommend retaining Pleoidea as part of Nepomorpha. The superfamilies of Nepoidea (Belostomatidae + Nepidae), Ochteroidea (Gelastocoridae + Ochteridae), Naucoroidea (Aphelocheiridae + Naucoridae), and Pleoidea (Pleidae + Helotrephidae) are monophyletic groups in all our analyses with high support from both PP and BP. We also found strong support for the close relationship between Notonectoidea and Pleoidea. Several synapomorphies of biological and ecological traits also support some of these monophyletic groups [24-26,46]:
Nepomorpha: the short antennae are concealed below the eyes; all have an aquatic lifestyle, although Ochteroidea (including Ochteridae and Gelastocoridae) live along freshwater shores rather than underwater;
Nepoidea (including Nepidae and Belostomatidae): air-breathing through a siphon;
Naucoroidea: all Aphelocheiridae and some Naucoridae use plastron respiration;
Pleoidea (including Pleidae and Helotrephidae): also have plastron respiration, which allows them to stay permanently submerged;
Notonectoidea and Pleoidea (including Notonectidae, Pleidae, and Helotrephidae): swim on their backs in an inverted position.
Our principal goal in this study was to discuss the monophyly of Nepomorpha and the effects of adequate taxon sampling on this phylogenetic problem. As we did not sample all the families of Nepomorpha, a more thorough sampling of taxa is needed to adequately resolve the family relationships within Nepomorpha. In particular, more sampling of Potamocoridae, Micronectidae and Diaprepocoridae (Hemiptera: Nepomorpha) mt-genome sequences will be needed for a thorough analysis of the major groups within Nepomorpha.
Conclusions
This study provides a clear example of the importance of adequate sampling. We support the conclusion that investigators should be cautious about making major taxonomic rearrangements on the basis of limited taxon sampling, even (or especially) when the number of characters sampled per taxon is large [16,17,31,32]. Phylogenetic analyses that are based on even complete genomes of relatively few taxa are likely to result in strongly supported, but incorrect, evolutionary reconstructions [16,17,47]. In the study by Hua et al. [28], limited sampling of mt-genomes, coupled with the use of a distant outgroup, resulted in a conclusion that was at odds with a traditionally supported group (true water bugs, or Neopmorpha). But even minimal additional sampling to break up long branches in the tree, or the use of more closely related outgroups, results in trees in which the traditional group Nepomorpha is supported.
In the phylogenomic era [48], many papers are reporting surprising phylogenetic results that conflict with traditional hypotheses of relationships. Many (or even most) of these surprising results are based on analyses of many characters (even whole genomes) from very few taxa [16,47,49]. Strong “statistical support” for a given conclusion may come from strong underlying phylogenetic signal, but also from systematic bias that stems from assuming inadequate or inappropriate models of evolution [50]. Using large numbers of characters in a phylogenetic analysis means that even small systematic biases associated with overly simplistic methodological assumptions are likely to be mistaken as strong phylogenetic signal. Thorough taxon sampling allows the use of more simplistic models of evolution, because multiple changes at each nucleotide site can be appropriately reconstructed through the increased sampling of the tree [18]. If the sampling in a phylogenomic study is sparse, investigators should use appropriate caution before overturning analyses that are based on more thorough sampling of taxa.
Methods
Ethics statement
No specific permits were required for the insect collected for this study in Yunnan and Hubei Province, China. The insect specimens were collected with a sturdy aquatic net at the pond. The field studies did not involve endangered or protected species. The species in the genus of Paraplea and Helotrephes are common small insects and are not included in the “List of Protected Animals in China”.
Specimen collection
Adult specimens of Paraplea frontalis were collected from Tongbiguan Village (24°36.411 N, 97°39.349E), Yingjiang County, Dehong City, Yunnan Province, China, on May 18th, 2009. Adult specimens of Helotrephes semiglobosus semiglobosus were collected from Jin Ji Valley (29°22.339 N, 114°34.301E), Jiu Gong Shan, Tong Shan County, Hubei Province, China, on July 30th, 2010. Voucher specimens are deposited in the Insect Molecular Systematics Lab, Institute of Entomology, College of Life Sciences, Nankai University, Tianjin, China. All specimens were initially preserved in 95% ethanol in the field. After being transferred to the laboratory, they were stored at -20°C until used for DNA extraction.
PCR amplification and sequencing
Whole genomic DNA was extracted from thoracic muscle tissue by CTAB-based method [51]. The mt-genome of Paraplea frontalis was amplified in four overlapping PCR fragments by PCR amplification (see Additional file 3). The partial mt-genome of Helotrephes semiglobosus semiglobosus was sequenced with two fragments (see Additional file 4). Primer pairs were modified from previous work [28], and designed from sequenced fragments.
PCR reactions were performed with TaKaRa LA Taq under the following conditions: 1 min initial denaturation at 94°C, followed by 30 cycles of 20 s at 94°C, 1 min at 50°C, and 2–8 min at 68°C, and a final elongation for 10 min at 72°C. PCR products were electrophoresed in 1% agarose gel, purified, and then sequenced using an ABI 3730XL capillary sequencer with the BigDye Terminator Sequencing Kit (Applied Bio Systems). All fragments were sequenced with primer walking on both strands.
Sequence analysis and annotation
Sequence files were assembled into contigs using BioEdit version 7.0.5.2 [52]. Protein coding regions were determined via ORF Finder implemented at the NCBI website (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) with invertebrate mitochondrial genetic codes. Transfer RNA analysis was performed by tRNAscan-SE version 1.21 [53] with the invertebrate mitochondrial codon predictors and a cove score cut-off of 5. Few tRNA genes that could not be identified by tRNAscan-SE were determined by comparing to other heteropterans. Analyses of sequences were performed with MEGA version 5.0 [54].
Taxon sampling
In total, 19 taxa were sampled. These taxa included representatives of 10 out of 11 extant families of Nepomorpha [46,55] and 9 outgroups (Table 3). Among them, the mt-genome data of Paraplea frontalis is reported here for the first time. To make the results more directly comparable to the study of Hua et al. [28], we retrieved all mt-genomes of 15 taxa (including nine ingroups and six outgroups) from their work. According to the analysis of the heteropteran infraorders of Wheeler et al. [37], the phylogenetic relationships of Heteroptera are as follows: (Enicocephalomorpha + (Dipsocoromorpha + (Gerromorpha + (Nepomorpha + (Leptopodomorpha + (Cimicomorpha + Pentatomomorpha)))))). Therefore, we sampled another three taxa within the sister group to Nepomorpha as outgroups, with one representative from each of Leptopodomorpha, Cimicomorpha and Pentatomomorpha.
Table 3.
Taxonomy and GenBank accession numbers of mitochondrial genomes for species sampled in this study
| Suborder (bold) Infraorder (not bold) | Superfamily | Family | Species | Accession number |
|---|---|---|---|---|
|
Auchenorrhyncha |
|
|
|
|
| Fulgoromorpha |
Fulgoroidea |
Fulgoridae |
Lycorma delicatula |
NC_012835 |
|
Heteroptera |
|
|
|
|
| Gerromorpha |
Hydrometroidea |
Hydrometridae |
Hydrometra greeni |
NC_012842 |
| |
Gerroidea |
Gerridae |
Aquarius paludum |
NC_012841 |
| Leptopodomorpha |
Saldoidea |
Saldidae |
Saldula arsenjevi |
NC_012463 |
| |
Leptopodoidea |
Leptopodidae |
Leptopus sp. |
FJ456946 |
| Cimicomorpha |
Reduvioidea |
Reduviidae |
Triatoma dimidiata |
NC_002609 |
| |
Reduvioidea |
Reduviidae |
Valentia hoffmanni |
NC_012823 |
| Pentatomomorpha |
Lygaeoidea |
Berytidae |
Yemmalysus parallelus |
NC_012464 |
| |
Coreoidea |
Rhopalidae |
Stictopleurus subviridis |
NC_012888 |
| Nepomorpha |
Corixoidea |
Corixidae |
Sigara septemlineata |
FJ456941 |
| |
Nepoidea |
Belostomatidae |
Diplonychus rusticus |
FJ456940 |
| |
|
Nepidae |
Laccotrephes robustus |
FJ456948 |
| |
Ochteroidea |
Gelastocoridae |
Nerthra indica |
FJ456943 |
| |
|
Ochteridae |
Ochterus marginatus |
FJ456950 |
| |
Naucoroidea |
Naucoridae |
Ilyocoris cimicoides |
NC_012845 |
| |
|
Aphelocheiridae |
Aphelocheirus ellipsoideus |
FJ456939 |
| |
Notonectoidea |
Notonectidae |
Enithares tibialis |
NC_012819 |
| |
Pleoidea |
Helotrephidae |
Helotrephes sp. |
FJ456951 |
| Pleidae | Paraplea frontalis | KJ027516 |
Phylogenetic analyses
All PCGs were aligned based on their amino acid sequences using MUSCLE as implemented in the MEGA version 5.0 [54]. The rRNAs and tRNAs were aligned with CLUSTAL_X version 1.83 [56] under the default settings. The alignments of tRNA genes were corrected according to the secondary structures, especially the stem regions. The aligned nucleotide sequences, excluding stop codons, were then concatenated and used to reconstruct the phylogeny. All phylogenetic trees were built using only first and second codon positions of 13 PCGs, except in our analyses in which we removed or added taxa to the data matrices of Hua et al. [28], so that we could make a direct comparison using methods used in the original paper. Our analyses with added and deleted taxa used the same data sampling methods of Hua et al. [28]; these analyses contained four kinds of data matrices: (1) The PCG123RT matrix, including all three codon positions of PCGs, rRNA genes, and tRNA genes; (2) the PCG12RT matrix, including the first and the second codon positions of PCGs, rRNA genes, and tRNA genes; (3) the PCG123 matrix, including all the three codon positions of PCGs; and (4) the PCG12 matrix, including the first and the second codon positions of PCGs.
We used GPU MrBayes [57] for Bayesian inference and raxmlGUI 1.2 [58] for ML analyses to reconstruct phylogenetic trees. We used the GTR + I + Γ model, based on results from Modeltest Version 3.7 [59]. In Bayesian inference, two simultaneous runs of 10,000,000 generations were conducted for each matrix. Each set was sampled every 100 generations. Trees that were sampled prior to stationarity (at 25% of the run) were discarded as burnin, and the remaining trees were used to construct a 50% majority-rule consensus tree. For the ML analysis, we conducted 1000 bootstrap replicates with thorough ML search.
Tests of monophyly
Traditionally recognized taxonomic groups are usually challenged when there is strong statistical support for an alternative phylogeny [16,60]. Likelihood-ratio tests [61] can provide a powerful means of examining alternatives. We applied likelihood-ratio tests to compare the support of various data sets for two different hypotheses (see Additional file 2):
Hypothesis 1: Helotrephidae is nested within Nepomorpha (i.e., the true water bugs are monophyletic, and Helotrephidae is nested within the group).
Hypothesis 2: Helotrephidae is outside of the remaining species of Nepomorpha (i.e., true water bugs are only monophyletic if Helotrephidae is excluded from the group).
We conducted likelihood-ratio tests [61] of these two hypotheses for the original data set of Hua et al. [28], as well as with various additions and deletions of taxa, including both ingroups and outgroups. The likelihood-ratio tests were conducted using PAUP* 4 [62]. Heuristic searches were performed using the GTR + I + Γ model with 100 random addition replicates.
Availability of supporting data
The data sets supporting the results of this article are available in the Dryad repository, http://dx.doi.org/10.5061/dryad.tf25c[63].
Abbreviations
LBA: Long-branch attraction; Mt-genomes: Mitochondrial genomes; BPP: Bayesian posterior probabilities; ML: Maximum likelihood; PCGs: Protein coding genes; PP: Posterior probabilities; BP: Bootstrap proportions.
Competing interests
The authors have declared that no competing interests.
Authors’ contributions
TL designed the experiments, carried out the phylogenetic analyses, made all figures and drafted the manuscript. AMW and YC participated in the data analyses. JH and QX helped draft the manuscript. WB and DMH directed this study, designed and reviewed analyses, and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Putative secondary structure of the 22 tRNAs identified in the mitochondrial genome of Paraplea frontalis . The tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashes indicate Watson-Crick base pairing and asterisks indicate G-U base pairing.
Constraints for the two hypotheses used in the likelihood-ratio test regarding the monophyly of Nepomorpha.
Primers designed for Paraplea frontalis in this study.
Primers designed for Helotrephes semiglobosus semiglobosus in this study.
Contributor Information
Teng Li, Email: teng.chn@gmail.com.
Jimeng Hua, Email: nkhuajimeng@163.com.
April M Wright, Email: wright.aprilm@gmail.com.
Ying Cui, Email: cql8179270@126.com.
Qiang Xie, Email: qiangxie@nankai.edu.cn.
Wenjun Bu, Email: wenjunbu@nankai.edu.cn.
David M Hillis, Email: dhillis@austin.utexas.edu.
Acknowledgements
We are grateful to Dr. Ping-ping Chen and Nico Nieser (Netherlands Biodiversity Center Naturalis), and Mr. Zhen Ye and Tongyin Xie (Nankai University) for identifying our samples of Helotrephes sp., Paraplea frontalis, and Helotrephes semiglobosus semiglobosus. We thank Mr. Hongju Xia, and Profs. Xiaoguang Liu and Gang Wang (College of Information Technical Science, Nankai University) for help with the parallel implementations of the GPU MrBayes program. This project was supported by National Natural Sciences Foundation of China (No. 31372240, J1210005).
References
- Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool. 1978;27(4):401–410. doi: 10.2307/2412923. [DOI] [Google Scholar]
- Hendy MD, Penny D. A framework for the quantitative study of evolutionary trees. Syst Zool. 1989;38(4):297–309. doi: 10.2307/2992396. [DOI] [Google Scholar]
- Sullivan J, Swofford DL. Are guinea pigs rodents? The importance of adequate models in molecular phylogenetics. J Mamm Evol. 1997;4(2):77–86. doi: 10.1023/A:1027314112438. [DOI] [Google Scholar]
- Lin YH, McLenachan PA, Gore AR, Phillips MJ, Ota R, Hendy MD, Penny D. Four new mitochondrial genomes and the increased stability of evolutionary trees of mammals from improved taxon sampling. Mol Biol Evol. 2002;19(12):2060–2070. doi: 10.1093/oxfordjournals.molbev.a004031. [DOI] [PubMed] [Google Scholar]
- Garcia-Moreno J, Sorenson MD, Mindell DP. Congruent avian phylogenies inferred from mitochondrial and nuclear DNA sequences. J Mol Evol. 2003;57(1):27–37. doi: 10.1007/s00239-002-2443-9. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Phillips MJ, Penny D. Comment on “Hexapod origins: monophyletic or paraphyletic?”. Science. 2003;301(5639):1482. doi: 10.1126/science.1086558. [DOI] [PubMed] [Google Scholar]
- Chen W-J, Bu Y, Carapelli A, Dallai R, Li S, Yin W-Y, Luan Y-X. The mitochondrial genome of Sinentomon erythranum (Arthropoda: Hexapoda: Protura): an example of highly divergent evolution. BMC Evol Biol. 2011;11(1):246. doi: 10.1186/1471-2148-11-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz MP, Tierney SM, Cooper SJB, Bull NJ. Molecular phylogenetics of the allodapine bee genus Braunsapis: A–T bias and heterogeneous substitution parameters. Mol Phylogenet Evol. 2004;32(1):110–122. doi: 10.1016/j.ympev.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, Wojciechowski MF, Hu JM, Khan TS, Brady SG. Error, bias, and long-branch attraction in data for two chloroplast photosystem genes in seed plants. Mol Biol Evol. 2000;17(5):782–797. doi: 10.1093/oxfordjournals.molbev.a026357. [DOI] [PubMed] [Google Scholar]
- Zhong B, Yonezawa T, Zhong Y, Hasegawa M. The position of Gnetales among seed plants: overcoming pitfalls of chloroplast phylogenomics. Mol Biol Evol. 2010;27(12):2855–2863. doi: 10.1093/molbev/msq170. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Hillis DM. Success of phylogenetic methods in the 4-taxon case. Syst Biol. 1993;42(3):247–264. doi: 10.1093/sysbio/42.3.247. [DOI] [Google Scholar]
- Bergsten J. A review of long-branch attraction. Cladistics. 2005;21(2):163–193. doi: 10.1111/j.1096-0031.2005.00059.x. [DOI] [PubMed] [Google Scholar]
- Holland BR, Penny D, Hendy MD. Outgroup misplacement and phylogenetic inaccuracy under a molecular clock–a simulation study. Syst Biol. 2003;52(2):229–238. doi: 10.1080/10635150390192771. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Brinkmann H, Philippe H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol Biol. 2007;7(Suppl 1):S4. doi: 10.1186/1471-2148-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YW, Yu L, Zhang YP. “Long-branch Attraction” artifact in phylogenetic reconstruction. Hereditas(Beijing) 2007;29(6):659–667. doi: 10.1360/yc-007-0659. [DOI] [PubMed] [Google Scholar]
- Heath TA, Hedtke SM, Hillis DM. Taxon sampling and the accuracy of phylogenetic analyses. J Syst Evol. 2008;46(3):239–257. [Google Scholar]
- Hedtke SM, Townsend TM, Hillis DM. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst Biol. 2006;55(3):522–529. doi: 10.1080/10635150600697358. [DOI] [PubMed] [Google Scholar]
- Hillis DM. Inferring complex phylogenies. Nature. 1996;383(6596):130–131. doi: 10.1038/383130a0. [DOI] [PubMed] [Google Scholar]
- Nabhan AR, Sarkar IN. The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy. Brief Bioinform. 2012;13(1):122–134. doi: 10.1093/bib/bbr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG, Salipante SJ. Measures of clade confidence do not correlate with accuracy of phylogenetic trees. Plos Comput Biol. 2007;3(3):e51. doi: 10.1371/journal.pcbi.0030051. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Štys P, Kerzhner I. The rank and nomenclature of higher taxa in recent Heteroptera. Acta Entomol Bohemoslov. 1975;72(2):65–79. [Google Scholar]
- Popov YA. Trudy Paleontological Institute Academy of Science Volume 129. Nauk: USSR; 1971. Historical development of the hemipterous infraorder Nepomorpha; pp. 1–228. [Google Scholar]
- Rieger C. Skelett und muskulatur des kopfes und prothorax von Ochterus marginatus Latreille. Zoomorphology. 1976;83(2):109–191. doi: 10.1007/BF00993483. [DOI] [Google Scholar]
- China WE. Symposium on organic evolution. India: Bulletin of the National Institute of Science; 1955. The evolution of the water bugs; pp. 91–103. [Google Scholar]
- Mahner M. Systema cryptoceratum phylogeneticum (Insecta, Heteroptera) Zoologica. 1993;143 [Google Scholar]
- Hebsgaard MB, Andersen NM, Damgaard J. Phylogeny of the true water bugs (Nepomorpha: Hemiptera-Heteroptera) based on 16S and 28S rDNA and morphology. Syst Entomol. 2004;29(4):488–508. doi: 10.1111/j.0307-6970.2004.00254.x. [DOI] [Google Scholar]
- Li M, Wang J, Tian XX, Xie Q, Liu HX, Bu WJ. Phylogeny of the true water bugs (Hemiptera-Heteroptera: Nepomorpha) based on four Hox genes. Entomotaxonomia. 2012;34(1):35–44. [Google Scholar]
- Hua JM, Li M, Dong PZ, Cui Y, Xie Q, Bu WJ. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): evidence from mitochondrial genomes. BMC Evol Biol. 2009;9:134. doi: 10.1186/1471-2148-9-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Rice DW, Palmer JD. Long branch attraction, taxon sampling, and the earliest angiosperms: Amborella or monocots? BMC Evol Biol. 2004;4:35. doi: 10.1186/1471-2148-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis DM. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst Biol. 1998;47(1):3–8. doi: 10.1080/106351598260987. [DOI] [PubMed] [Google Scholar]
- Zwickl DJ, Hillis DM. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002;51(4):588–598. doi: 10.1080/10635150290102339. [DOI] [PubMed] [Google Scholar]
- Pollock DD, Zwickl DJ, McGuire JA, Hillis DM. Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol. 2002;51(4):664. doi: 10.1080/10635150290102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala B, Huelsenbeck JP, Yang Z, Nielsen R. Taxon sampling and the accuracy of large phylogenies. Syst Biol. 1998;47(4):702–710. doi: 10.1080/106351598260680. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Scally M, Madsen O, Stanhope MJ, de Jong WW, Catzeflis FM, Springer MS, Douzery EJ. Molecular phylogeny of living xenarthrans and the impact of character and taxon sampling on the placental tree rooting. Mol Biol Evol. 2002;19(10):1656–1671. doi: 10.1093/oxfordjournals.molbev.a003989. [DOI] [PubMed] [Google Scholar]
- Holder M, Lewis PO. Phylogeny estimation: traditional and Bayesian approaches. Nat Rev Genet. 2003;4(4):275–284. doi: 10.1038/nrg1044. [DOI] [PubMed] [Google Scholar]
- Saitoh K, Sado T, Mayden RL, Hanzawa N, Nakamura K, Nishida M, Miya M. Mitogenomic evolution and interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): the first evidence toward resolution of higher-level relationships of the world’s largest freshwater fish clade based on 59 whole mitogenome sequences. J Mol Evol. 2006;63(6):826–841. doi: 10.1007/s00239-005-0293-y. [DOI] [PubMed] [Google Scholar]
- Wheeler WC, Schuh RT, Bang R. Cladistic relationships among higher groups of Heteroptera: congruence between morphological and molecular data sets. Entomol Scand. 1993;24(2):121–137. doi: 10.1163/187631293X00235. [DOI] [Google Scholar]
- Graham SW, Olmstead RG, Barrett SC. Rooting phylogenetic trees with distant outgroups: a case study from the commelinoid monocots. Mol Biol Evol. 2002;19(10):1769–1781. doi: 10.1093/oxfordjournals.molbev.a003999. [DOI] [PubMed] [Google Scholar]
- Ware JL, Litman J, Klass K-D, Spearman LA. Relationships among the major lineages of Dictyoptera: the effect of outgroup selection on dictyopteran tree topology. Syst Entomol. 2008;33(3):429–450. doi: 10.1111/j.1365-3113.2008.00424.x. [DOI] [Google Scholar]
- Lyons-Weiler J, Hoelzer GA, Tausch RJ. Optimal outgroup analysis. Biol J Linn Soc. 1998;64(4):493–511. doi: 10.1111/j.1095-8312.1998.tb00346.x. [DOI] [Google Scholar]
- Luo AR, Zhang YZ, Qiao HJ, Shi WF, Murphy RW, Zhu CD. Outgroup selection in tree reconstruction: a case study of the family Halictidae (Hymenoptera: Apoidea) Acta Entomologica Sinica. 2010;53(2):192–201. [Google Scholar]
- Qiu YL, Lee J, Whitlock BA, Bernasconi-Quadroni F, Dombrovska O. Was the ANITA rooting of the angiosperm phylogeny affected by long-branch attraction? Mol Biol Evol. 2001;18(9):1745–1753. doi: 10.1093/oxfordjournals.molbev.a003962. [DOI] [PubMed] [Google Scholar]
- Smith AB. Rooting molecular trees - problems and strategies. Biol J Linn Soc. 1994;51(3):279–292. doi: 10.1111/j.1095-8312.1994.tb00962.x. [DOI] [Google Scholar]
- Li T, Gao CQ, Cui Y, Xie Q, Bu W. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera) Plos One. 2013;8(2):e55381. doi: 10.1371/journal.pone.0055381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua JM, Li M, Dong PZ, Cui Y, Xie Q, Bu WJ. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera) BMC Genomics. 2008;9:610. doi: 10.1186/1471-2164-9-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RT, Slater JA. True bugs of the world (Hemiptera: Heteroptera): classification and natural history: Cornell University Press. 1995.
- Soltis DE, Albert VA, Savolainen V, Hilu K, Qiu YL, Chase MW, Farris JS, Stefanović S, Rice DW, Palmer JD, Soltis PS. Genome-scale data, angiosperm relationships, and ‘ending incongruence’: a cautionary tale in phylogenetics. Trends Plant Sci. 2004;9(10):477–483. doi: 10.1016/j.tplants.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Philippe H. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 2005;6(5):361–375. doi: 10.1038/nrg1603. [DOI] [PubMed] [Google Scholar]
- Philippe H, Brinkmann H, Lavrov DV, Littlewood DT, Manuel M, Worheide G, Baurain D. Resolving difficult phylogenetic questions: why more sequences are not enough. Plos Biol. 2011;9(3):e1000602. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL, Olsen GJ, Waddell PJ, Hillis DM. In: Molecular systematics. 2. Hillis DM, Moritz C, Mable BK, editor. Sunderland, Massachusetts: Sinauer Associates; 1996. Phylogenetic inference; pp. 407–514. [Google Scholar]
- Reineke A, Karlovsky P, Zebitz CP. Preparation and purification of DNA from insects for AFLP analysis. Insect Mol Biol. 1998;7(1):95–99. doi: 10.1046/j.1365-2583.1998.71048.x. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štys P, Jansson A. Check-list of recent family-group and genus-group names of Nepomorpha (Heteroptera) of the world. Acta Entomol Fenn. 1988;50:1–44. [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liu X, Stones DS, Xie Q, Wang G. MrBayes on a graphics processing unit. Bioinformatics. 2011;27(9):1255–1261. doi: 10.1093/bioinformatics/btr140. [DOI] [PubMed] [Google Scholar]
- Silvestro D, Michalak I. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12(4):335–337. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- McVay JD, Carstens B. Testing monophyly without well-supported gene trees: evidence from multi-locus nuclear data conflicts with existing taxonomy in the snake tribe Thamnophiini. Mol Phylogenet Evol. 2013;68(3):425–431. doi: 10.1016/j.ympev.2013.04.028. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Hillis DM, Nielsen R. A likelihood-ratio test of monophyly. Syst Biol. 1996;45(4):546–558. doi: 10.1093/sysbio/45.4.546. [DOI] [Google Scholar]
- Swofford DL. PAUP*: Phylogenetic analysis using parsimony (* and other methods). Version 4. Sunderland, Massachusetts: Sinauer Associates; 2003. [Google Scholar]
- Li T, Hua J, Wright AM, Cui Y, Xie Q, Bu W, Hillis DM. Long-branch attraction and the phylogeny of true water bugs (Hemiptera: Nepomorpha) as estimated from mitochondrial genomes. Dryad Digital Repository. 2014. http://dx.doi.org/10.5061/dryad.tf25c. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Putative secondary structure of the 22 tRNAs identified in the mitochondrial genome of Paraplea frontalis . The tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashes indicate Watson-Crick base pairing and asterisks indicate G-U base pairing.
Constraints for the two hypotheses used in the likelihood-ratio test regarding the monophyly of Nepomorpha.
Primers designed for Paraplea frontalis in this study.
Primers designed for Helotrephes semiglobosus semiglobosus in this study.