

Abstract
Atopic dermatitis (AD) is associated with the effects of Th2 and Th22 cytokines. Recent studies, however, have also implicated Th17 in acute AD. Functional studies of Th2 and Th22 cytokines revealed their roles in generating AD’s molecular changes; IL-17A’s role has yet to be defined. In this issue, Nakajima et al. (2014) begin to define that role by demonstrating IL-17A’s ability to induce Th2 inflammation in acute disease.
Atopic dermatitis (AD) is a chronic inflammatory skin disease characterized by acute and chronic stages. The pathogenesis of AD is still unclear; both immune- and barrier-driven mechanisms have been demonstrated in murine models and in human disease. AD has been recognized predominantly as a Th2/Th22-centered disease (Guttman-Yassky et al., 2011). Lesional AD skin is characterized primarily by increased expression of Th2 cytokines and chemokines (IL-4/IL-13, CCL17, OX40), Th22-related markers (IL-22), as well as Th1-(CXCL10), Th17- (Elafin/PI3, CXCL1), and Th17/Th22-mediated S100 epidermal responses. While the pathogenic role of Th17 in psoriasis is well-established and supported by disease antagonism with anti-IL-17 biologics (i.e. brodalumab, ixekizumab, and secukinumab), its role in the pathogenesis of AD remains unclear. Although IL-17 is increased in skin and peripheral blood in patients with acute disease (Gittler et al., 2012), IL-17 and its associated antimicrobial peptides are much lower than in psoriasis, possibly explaining the increased risk of infections in AD (reviewed in Guttman-Yassky et al., 2011). A recent study by our group compared non-lesional tissue with that of acute and chronic lesions from the same patient, which revealed increased expression of IL-17 and several associated markers (CCL20, PI3) in acute AD compared with non-lesional skin. Interestingly, levels remained insignificantly changed as disease progressed to the chronic stage (Gittler et al. JACI 2012; Figure 1). Obtaining a functional understanding for IL-17A in AD has remained elusive until now. In this issue, Nakajima et al. (2014) utilized two murine models of AD, including hapten-mediated induction and flaky-tailed mice with spontaneous lesions, in the setting of IL-17A deficiency, to evaluate its role in disease pathogenesis.
Figure 1. A schematic illustration of initiation of acute AD and progression to chronic skin lesions.
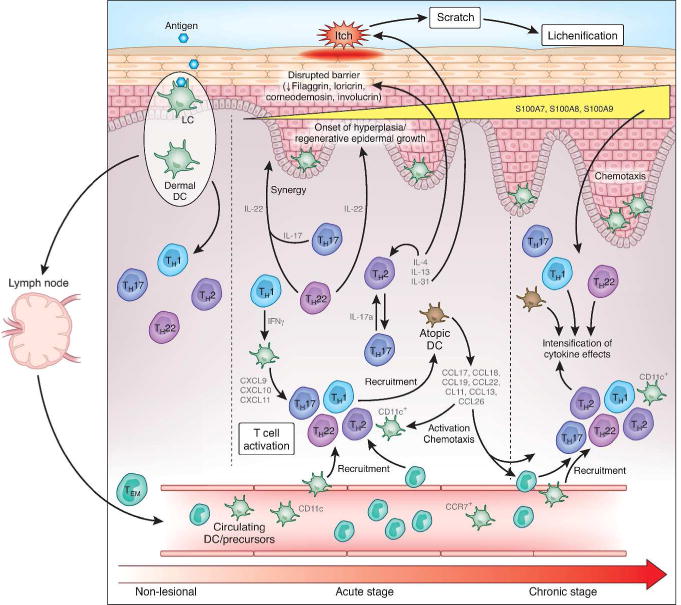
Non-lesional AD skin lesions show some immune infiltrates that produce inflammatory mediators, which might contribute to a defective epidermal barrier. Barrier defects lead to penetration by epicutaneous antigens that encounter Langerhans cells in the epidermis and dermal dendritic cells (DCs) in the dermis, inducing marked immune activation and recruitment of inflammatory cells in acute AD lesions. Nakajima et al. (2014)’s finding regarding the role of IL-17A might potentially bridge the gap between non-lesional skin following antigen penetration and onset of acute disease, where Th17 T-cells have been identified in limited quantities. This relatively small Th17 activity could potentially lead to the marked activation of Th2 axis in acute disease onset; Th22 activity also increases. A progressive activation of Th2, Th22, and Th1 pathways is characteristic of the chronic stage of AD; Th17 activity remains constant from acute disease. The relative induction of each T-cell subset according to disease stage is represented pictorially by their size relative to the other T-cell subsets. Cytokines (i.e., IL-4 and IL-13) and chemokines (i.e., CCL17, CCL18, CCL19, CXCL9, CXCL10, and CXCL11) produced by various T-cells and DCs induce further activation and recruitment of additional immune cells. With the onset of acute disease, Th22 cells release IL-22, which induces epidermal hyperplasia and, synergistically with the Th17 cytokine IL-17, drives an abrupt increase in a subset of terminal differentiation genes, specifically S100A7, S100A8, and S100A9 proteins. The increases in these barrier proteins contrast with the uniformly disrupted epidermal differentiation gene products (e.g., filaggrin, loricrin, and corneodesmosin) throughout nonlesional, acute, and chronic AD skin. The Th2 and Th22 cytokines contribute to inhibition of the terminal differentiation proteins. IL-31 is abruptly upregulated in acute disease, potentially reflecting its role as an itch mediator in patients with AD. Figure and legend adapted and modified from Gittler et al., 2012 with permission from Elsevier/RightsLink.
Th2 induction by IL-17A in a murine model of AD-like inflammation
Using both models, Nakajima et al. demonstrated that IL-17A deficiency attenuated Th2 induction during the acute phase of inflammation, implicating IL-17A as an inducer or modifier that drives IL-17A T-cells towards Th2 in the early inflammation of susceptible skin. The hapten-induced mouse model of AD relies on the concept that chronic hapten exposure across an impaired barrier establishes a pro-inflammatory milieu (the “hapten-atopy hypothesis”; McFadden et al., 2011) dominated by Th2 T-cells, mimicking the inflammation in AD skin. Nakajima et al.’s hapten model may explain how an immune response to an external allergic agent, via IL-17A-mediated Th2 induction, ultimately polarizes towards the acute AD cytokine milieu that has been described previously. In addition, Nakajima et al.’s demonstration of similar immune findings in barrier-deficient mice with spontaneous AD-like lesions helps to bridge our understanding of why some patients develop lesions only in the setting of a sensitizer while others develop them spontaneously. Their finding, that contact allergen-triggered disease and barrier deficiency both result in similar IL-17A-mediated induction of Th2 inflammation, may explain how this clinical phenomenon occurs.
Interestingly, in a recent report, Newell et al. (2013) described use of the potent hapten dinitrochlorobenzene (DNCB) to study contact sensitization in humans, revealing an interesting dichotomy between patients with AD and volunteers without AD. Those with a history of AD exhibited preferential Th2 induction whereas those without AD exhibited a Th1 response primarily. Thus, it can be postulated that in human AD, an allergen penetrating across a defective barrier might trigger low-grade IL-17A release; with ongoing stimulation by chronic happen exposure, it may induce Th2-polarizing molecules (TSLP, CCL17), promoting naïve T-cells towards Th2 and establishing the predominant immune polarization of AD. Interestingly, a similar concept may be applied to the role of Staphyloccoccus aureus, another implicated extrinsic agent, in triggering AD exacerbations. S. aureus superantigen, like the haptens in Nakajima et al.’s study, was also demonstrated to induce IL-17A expression (Boguniewicz and Leung, 2010), potentially suggesting a common molecular link between the two major external inducers of disease flares.
Limitations of the murine contact hypersensitivity (CHS) model and our current understanding of IL-17 signaling introduce new questions
The AD mouse models used by Nakajima et al. have sseveral inherent limitations with respect to their translatability to humans. Specifically, at present, no single mouse model represents acute and chronic AD with true accuracy; rather, these models are able to represent select characteristics of the disease process and generate AD-like clinical and molecular inflammation. The murine CHS model, for instance, can only be induced via potent sensitizing agents such as DNCB, which have an inherent irritating capacity and thus induce non-specific immune activation (i.e. of the innate immune response) in addition to antigen-specific activation. Conversely, clinically-relevant sensitizers that cause allergic contact dermatitis, and may trigger human AD, do not exhibit an initial irritant response, thus making it difficult to translate the generated immunological data fully to human models. Another important limitation relates to γδ T-cells, which Nakajima et al. found to be chief producers of the IL-17A in their study; the role of these T-cells in human IL-17 production is unknown at present, and thus it is unclear whether a similar mechanism may exist in humans.
Only a few studies prior to those of Nakajima et al. evaluated the human effector interplay between IL-17A and the Th2 pathway, particularly in skin disease, and a select subset from other tissue types reported results contradictory to those in this study. One such work, by Xu et al. (2009), on cultured nasal epithelial cells revealed an inhibitory effect of IL-17A on TSLP expression, while Th2-sourced IL-25 (also known as IL-17E) had the opposite effect. Studies in skin, too, have produced dissimilar results from Nakajima et al.’s findings. Bogiatzi et al. (2012) demonstrated, using human skin explants and cultured dendritic cells, that IL-17A induced marked decreases in TSLP and Th2-related cytokine production. Eyerich et al. (2009) complicated this picture further, postulating that IL-4 was capable of inhibiting the IL-17 pathway, as well.
A final point of consideration is revealed in recent studies that identified a subset of Th2 T-cells capable of producing IL-17A itself. Wang et al. (2010) demonstrated the presence of these IL-4 and IL-17-producing Th2 T-cells in peripheral blood of allergic asthmatics. It can be postulated that IL-17A produced by these dual-function cells may signal back on naive T-cells, augmenting Th2 activity. Interestingly, they reported that these cells could be stimulated to produce IL-17 by IL-1β, IL-6, and IL-21 but not IL-23, suggesting a distinct mechanism from classical Th17-mediated release. The same study found that in murine models of allergic asthma, mice with allergen-specific IL-17-producing Th2 cells exhibited increased airway inflammation following allergen exposure, suggesting some correlation between clinical disease activity and stimulation of these IL-17-producing Th2 T-cells. Similarly in skin, Eyerich et al. (2009) used eczema samples and identified that approximately one-third of IL-17-producing cells exhibited a concurrent Th0 or Th2 profile (both IL-4-producing without or with interferon γ/IFNγ, respectively). Thus, additional studies, based off Nakajima et al.’s work, will be required to evaluate the possibility that IL-17A is itself being sourced from a subset of Th2 T-cells rather than from the classically presumed Th17 cells.
Upcoming targeted therapeutics for AD may reveal further insights into the role of cytokine interplay in disease pathogenesis
AD research has recently entered a new era in which our collective understanding of the immune mechanisms driving disease has led to the development and testing of agents targeting key molecules in pathogenic pathways. A clinical trial that is currently being conducted by our group testing ustekinumab (anti-IL-23p40) (clinicaltrials.gov, NCT01806662) in AD patients may help elucidate the effects of IL-17A inhibition on Th2 cytokine induction. By inhibiting Th17 T-cell activation (and thus significant IL-17 production) with this biologic, we will be able to evaluate the post-treatment effects of such a blockade on all the major inflammatory axes, including Th2 T-cells and their associated markers. In doing so, our study may bridge Nakajima et al.’s murine work to humans, potentially providing insight into whether IL-17 inhibition truly reduces Th2 activity. Conversely, dupilumab (clinicaltrials.gov, NCT01859988), a monoclonal antibody that inhibits IL-4Rα, dually blocking IL-4 and IL-13 signaling, may shed light on the effects of Th2 inhibition on IL-17 pathway genes in addition to the other major T-cell axes. Moving forward, a bedside-to-bench approach with pathway-targeted therapeutics will enable us to not only test these drugs’ efficacies but also potentially to unveil additional novel targets in the major inflammatory pathways. Nakajima et al.’s work in this issue of the Journal, however, presents a fascinating model for the initiation of Th2 inflammation in AD, providing a potential therapeutic target and an exciting clue into the early stages of the slowly unraveling AD pathogenesis model.
Clinically Relevant Points.
Nakajima et al. (2014)’s demonstration of IL-17A-mediated induction of Th2 inflammation in the setting of hapten exposure or barrier disruption in murine models introduces a new facet into AD pathogenesis models.
Their finding provides a better understanding of the primary immunological processes that initiate acute and chronic disease.
Ongoing studies in humans using IL-4 and IL-17 targeted biologics will provide additional insight into the translatability of this finding.
Abbreviations
- AD
Atopic Dermatitis
- CHS
Contact hypersensitivity
- DC
Dendritic cell
- DNCB
Dinitrochlorobenzene
- IL
Interleukin
- CCL
Chemokine (C-C motif) ligand
- CXCL
Chemokine (CXC motif) ligand
- Th1/2/17/22
Helper T-cell subtypes 1, 2, 17, and 22
- TSLP
Thymic stromal lymphopoietin
References
- Bogiatzi SI, Guillot-Delost M, Cappuccio A, et al. Multiple-checkpoint inhibition of thymic stromal lymphopoietin-induced TH2 response by TH17-related cytokines. J Allergy Clin Immunol. 2012;130:233–40 e5. doi: 10.1016/j.jaci.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Boguniewicz M, Leung DY. Recent insights into atopic dermatitis and implications for management of infectious complications. J Allergy Clin Immunol. 2010;125:4–13. doi: 10.1016/j.jaci.2009.11.027. quiz 4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyerich K, Pennino D, Scarponi C, et al. IL-17 in atopic eczema: linking allergen-specific adaptive and microbial-triggered innate immune response. J Allergy Clin Immunol. 2009;123:59–66 e4. doi: 10.1016/j.jaci.2008.10.031. [DOI] [PubMed] [Google Scholar]
- Gittler JK, Shemer A, Suarez-Farinas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344–54. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis–part II: immune cell subsets and therapeutic concepts. J Allergy Clin Immunol. 2011;127:1420–32. doi: 10.1016/j.jaci.2011.01.054. [DOI] [PubMed] [Google Scholar]
- McFadden JP, Dearman RJ, White JM, et al. The Hapten-Atopy hypothesis II: the ‘cutaneous hapten paradox’. Clin Exp Allergy. 2011;41:327–37. doi: 10.1111/j.1365-2222.2010.03684.x. [DOI] [PubMed] [Google Scholar]
- Nakajima S, Kitoh A, Egawa G, et al. IL-17A as an Inducer for Th2 Immune Responses in Murine Atopic Dermatitis Models. J Invest Dermatol. 2014 doi: 10.1038/jid.2014.51. [DOI] [PubMed] [Google Scholar]
- Newell L, Polak ME, Perera J, et al. Sensitization via healthy skin programs Th2 responses in individuals with atopic dermatitis. J Invest Dermatol. 2013;133:2372–80. doi: 10.1038/jid.2013.148. [DOI] [PubMed] [Google Scholar]
- Wang YH, Voo KS, Liu B, et al. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. 2010;207:2479–91. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Zhang L, Wang DY, et al. Opposing roles of IL-17A and IL-25 in the regulation of TSLP production in human nasal epithelial cells. Allergy. 2010;65:581–9. doi: 10.1111/j.1398-9995.2009.02252.x. [DOI] [PubMed] [Google Scholar]