

Abstract
Heart failure (HF) is frequently the consequence of sustained, abnormal neurohormonal, and mechanical stress and remains a leading cause of death worldwide. The key pathophysiological process leading to HF is cardiac remodeling, a term referring to maladaptation to cardiac stress at the molecular, cellular, tissue, and organ levels. HF and many of the conditions that predispose one to HF are associated with oxidative stress. Increased generation of reactive oxygen species (ROS) in the heart can directly lead to increased necrosis and apoptosis of cardiomyocytes which subsequently induce cardiac remodeling and dysfunction. Nuclear factor-erythroid-2- (NF-E2-) related factor 2 (Nrf2) is a transcription factor that controls the basal and inducible expression of a battery of antioxidant genes and other cytoprotective phase II detoxifying enzymes that are ubiquitously expressed in the cardiovascular system. Emerging evidence has revealed that Nrf2 and its target genes are critical regulators of cardiovascular homeostasis via the suppression of oxidative stress, which is the key player in the development and progression of HF. The purpose of this review is to summarize evidence that activation of Nrf2 enhances endogenous antioxidant defenses and counteracts oxidative stress-associated cardiac remodeling and HF.
1. Introduction
Despite recent advances in treatment, the morbidity, mortality, and economic burden of heart failure (HF) still remain very high. Hypertension, ischemia, diabetes, and some anticancer drugs used in the clinic are common causes of cardiac remodeling and HF. Cardiac remodeling, a term that refers to cardiac maladaptation at the molecular, cellular, tissue, and organ levels, is the key pathophysiological process leading to HF. It has been well established that oxidative stress is a major cause of HF [1–8].
Free radicals and other reactive small molecules have emerged as important regulators of many physiological and pathological processes [9]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) serve as signaling messengers to mediate various biological responses [10, 11], including numerous cardiovascular diseases, such as HF, coronary heart disease, and cardiac arrhythmias [12]. Whether the effects of ROS/RNS are beneficial or harmful depends on the site, type, and amount of ROS/RNS production and the activity of the organism's antioxidant defense system [13]. As a rule, heart and cardiovascular diseases are characterized by ROS overproduction, whereas the formation of major RNS, nitric oxide (a free radical) and peroxynitrite (a diamagnetic molecule), can decrease or increase depending on the nature of the heart injury [12]. ROS include superoxide anion (∙O2−), hydrogen peroxide (H2O2), hydroxyl radical (∙OH), and hypochlorite (OCl∙−), which impair cardiac function [14] and increase susceptibility to cardiac arrhythmia [15] through direct toxic effect, resulting in increased necrosis and apoptosis of cardiomyocytes [16]. Critical components of the cellular antioxidative defense mechanisms are the ROS scavengers, phase II detoxification enzymes, and other detoxification proteins which contain antioxidant response elements (AREs) in their promoter regions. A major regulator of the AREs is the highly conserved transcription factor, nuclear factor-erythroid-2- (NF-E2-) related factor 2 (Nrf2). Many of the Nrf2-regulated enzymes are essential in the pathogenesis of cardiovascular diseases [17] and are strongly associated with HF; in addition, they serve as sensitive and specific markers to reflect the ventricular function in HF patients [18]. Nrf2 also can prolong graft survival and modulate innate and adaptive immune responses after heart transplantation [19]. Emerging evidence has revealed that the Nrf2/ARE signaling pathway plays an important role in preventing oxidative cardiac cell injury in vitro [20, 21] as well as protecting the heart from maladaptive remodeling and cardiac dysfunction [7, 22–27].
The main purpose of this review is to discuss the current evidence for the cardioprotective role of Nrf2 and its target genes in the development of cardiac remodeling and HF caused by hypertension, ischemia, diabetes, and anticancer drugs.
2. ROS and Cardiac Remodeling
ROS refer to a group of small reactive molecules that include O2·−, H2O2, OH·−, and OCl·−. These molecules react avidly with other molecules such as cellular lipids, proteins, and nucleic acids [28]. The fine balance between ROS generated during normal physiological processes and endogenous antioxidants presented in the body is essential for redox homeostasis. Increased production of ROS or impaired endogenous antioxidant defense of the body that leads to oxidative stress may cause adverse effects due to irreversible modification of macromolecules, membranes, proteins, and DNA [29]. Cardiac remodeling is an adaptive, regulatory process of cardiomyocytes that occurs over time in order to maintain homeostasis against external stress [30]. Progression of cardiac hypertrophy, cardiomyocyte apoptosis, and interstitial collagen deposition into systolic dysfunction has been reported in numerous clinical and animal studies [31–35]. ROS can activate specific pathways leading to adaptive or maladaptive cardiac remodeling processes [28]. In experimental studies, oxidative stress has been identified as one of the molecular mechanisms and the key players involved in the development of cardiac hypertrophy [36]. In addition, antioxidants have been found to prevent the development of cardiac hypertrophy [37]. Various studies have suggested that oxidative stress is also an important regulator of profibrotic processes in the myocardium [34, 35]. NADPH oxidase-dependent ROS production contributes to the development of left ventricular (LV) interstitial fibrosis, reduction of ROS generation, and restoration of the redox balance; thus, it may be important in preventing or treating myocardial fibrosis in HF [38]. Programmed cell death (apoptosis) of cardiomyocytes has been identified as an essential process in the progression to HF [39]. ROS and the resulting oxidative stress also play a pivotal role in this pathological process. Antioxidants and thiol reductants, such as N-acetylcysteine, and overexpression of manganese superoxide dismutase (MnSOD) can block or delay apoptosis [40]. Taken together, ROS play an important role in the development of cardiac remodeling and HF. The findings of the majority of the studies suggest that antioxidants may be possible therapeutic candidates against cardiac remodeling.
3. The Nrf2-ARE Signaling Pathway
Nrf2 is the master regulator of oxidative stress signaling. It is a member of the cap-n-collar (CNC) family of transcription factors, which include Nrf1-3 and Bach1-2 [41–43]. These genes encode transcription factors that belong to the CNC-type of the basic region leucine zipper factor family (CNC-bZIP). CNC-bZIP factors are characterized by a highly conserved 43-amino acid homology region that lies in the immediate N-terminus of the basic DNA-binding domain and is referred to as the “CNC” domain after the Drosophila cap-n-collar protein. There is evidence indicating that CNC-bZIP factors function as obligate heterodimers by forming dimers with small Maf proteins (Maf G, Maf K, and Maf F) for DNA binding [44–46].
Under normal conditions, Nrf2 is kept in the cytoplasm by Kelch-like-ECH-associated protein 1 (Keap1) and Cullin 3 [47]. Cullin 3 ubiquitinates its substrate, Nrf2; and Keap1 serves as a substrate adaptor, which facilitates the ubiquitination of Nrf2 by Cullin 3. As a result, Nrf2 has a short half-life that lasts only 20 min under normal conditions [48]. As illustrated in Figure 1, oxidative stress destroys critical cysteine residues in Keap1, disrupting the Keap1-Cul3 ubiquitination system. If Nrf2 is not ubiquitinated, it builds up in the cytoplasm [49] and is translocated into the nucleus. In the nucleus, Nrf2 combines with a small protein called Maf to form a heterodimer, and, by binding to the ARE in the upstream promoter region, it initiates the transcription of a number of antioxidative genes, including heme oxygenase-1 (HO-1), NAD(P)H dehydrogenase (quinone 1) (NQO1), superoxide dismutases (SODs), catalase (CAT), glutathione-S-transferase (GST), γ-glutamylcysteine synthase (γ-GCS), and glutathione peroxidase (GPx) [50, 51] (Figure 1). These are the first line of defense mechanism against ROS-mediated cardiac injury. HO-1 is a rapidly inducible cytoprotective protein that degrades heme to biliverdin, ferrous iron, and carbon monoxide (CO) [52]. HO-1 mitigates cellular injury by exerting antioxidant, antiapoptotic, and anti-inflammatory effects [52, 53]. SOD catalyzes the dismutation of O2− into H2O2 and O2. H2O2 is a product of SOD activity and is handled by GPx and CAT. Three SOD isozymes have been identified, including copper/zinc-containing SOD (CuZn-SOD, also SOD1), which is primarily cytosolic in location, Mn-SOD (also SOD2), and extracellular SOD (EC-SOD, also SOD3) [54]. The GPx/glutathione (GSH) system is important in low-level oxidative stress. It scavenges hydroxyl radicals and singlet oxygen directly, detoxifying H2O2 and lipid peroxides by the catalytic action of GPx [55]. GPx not only scavenges H2O2 but also prevents the formation of other more toxic radicals, such as ·OH [56]. CAT is mainly located in cellular peroxisomes and, to some extent, in the cytosol and catalyzes the reaction of H2O2 to water and molecular oxygen [57]. Overexpression of mitochondrial CAT has shown protection against cytotoxic drugs [58].
Figure 1.
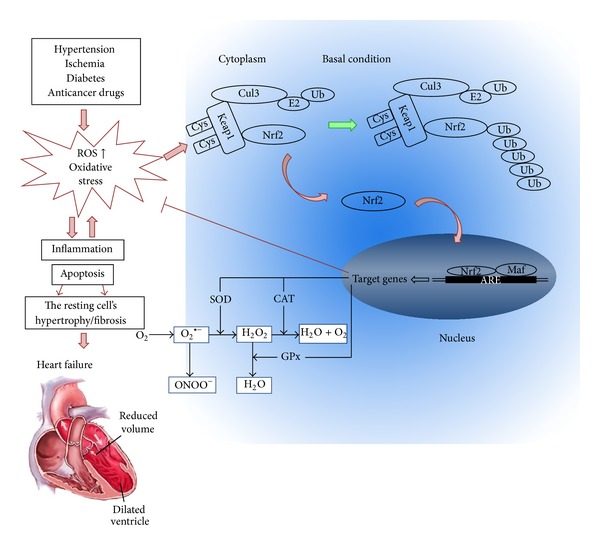
Roles of oxidative stress in cardiac remodeling and the potential protection by Nrf2 from oxidative damage. Hypertension, ischemia, diabetes, and anticancer drugs all induce additional generation of reactive oxygen and/or nitrogen species (ROS and/or RNS), leading to oxidative stress. Oxidative stress accelerates inflammation and apoptosis, which in turn causes cardiomyocyte hypertrophy and/or fibroblast proliferation, resulting in the cardiac remodeling (fibrosis). Meanwhile, ROS and/or RNS interact with cysteine residues in Keap1, disrupting the Keap1-Cul3 ubiquitination system. At the early stage of these pathological conditions, the released Nrf2 from Keap1 translocates to nucleus and combines with Maf and ARE to initiate the transcription of a number of antioxidative genes, such as SOD, CAT, and GPx, which are performing a wide range of cell defense processes against this pathological oxidative stress in the heart; however, at the late stage, Nrf2 may be exhausted or downregulated by its abnormal Nrf2 gene expression, leading to the failure to maintain the redox homeostasis by increasing ARE-mediated expression of phase II and antioxidant enzymes. Consequently, the persistently oxidative stress induces cardiac remodeling and finally heart failure.
Through inducing the expression of this battery of genes, Nrf2 is able to augment a wide range of cell defense processes, thereby enhancing the overall capacity of cells to detoxify potentially harmful substances. As such, the Nrf2-Keap1 pathway is generally considered to be a major cellular defense pathway. In addition, recent experimental and clinical studies have shown the important roles that Nrf2 and its downstream genes play in the pathogenesis of cardiac remodeling and HF induced by a number of factors.
4. Role of the Nrf2 Pathway in Hypertension-Induced Cardiac Remodeling
Many factors are involved in the pathophysiology of hypertension, such as upregulation of the renin-angiotensin-aldosterone system and activation of the sympathetic nervous system [59]. Common to these processes are the increased bioavailability of ROS due to excess ROS generation, decreased nitric oxide level, and reduced antioxidative capacity in the cardiovascular, renal, and nervous systems [60, 61]. In the cardiovascular system, ROS activate a broad variety of hypertrophy-promoting kinases and transcription factors, and oxidative stress has been identified as one of the key contributing factors in the development of cardiac hypertrophy [29]. In a pathologically hypertrophied myocardium, fibrosis and induction of fetal gene expression promote the development of cardiac dysfunction [62–65]. Activation of Nrf2 and its target genes provides a novel mechanism to protect the heart against pathological cardiac remodeling through suppressing oxidative stress [7, 25, 26, 66].
After transverse aortic constriction (TAC), Nrf2 expression was transiently increased and then declined to the basal level [7]. Compared to wild-type mice, Nrf2-knockout mice after TAC developed pathological cardiac hypertrophy, significant myocardial fibrosis and apoptosis, overt HF, and increased mortality, which were associated with elevated myocardial levels of 4-hydroxy-2-nonenal and 8-hydroxydeoxyguanosine and a complete blockade of the myocardial expression of several antioxidant genes. Overexpression of Nrf2 dramatically inhibited hypertrophic factor-induced ROS production and growth in both cardiomyocytes and cardiac fibroblasts, whereas knockdown of Nrf2 exerted the opposite effects in both cells [7]. Thus, Nrf2 is a critical regulator for maintaining structural and functional integrity of the heart that is abnormally stressed, as shown in Figure 2. Activating transcription factor 3 (ATF3) is a cAMP response element-binding protein/ATF family transcription factors member and has been implicated in the cardiovascular and inflammatory systems [67]. Infusion of tert-butylhydroquinone (tBHQ), a selective ATF3 inducer, increased the expression of ATF3 via the Nrf2-related transcriptional factor, inhibited transverse aortic banding-induced cardiac dilatation, and increased LV contractility, thereby rescuing from HF [67].
Figure 2.
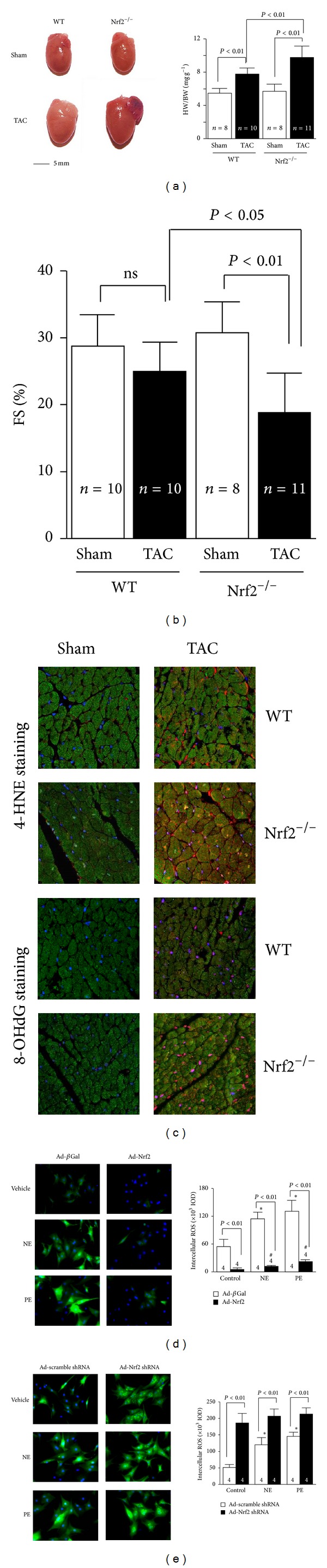
Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. (a) The left panel shows representative pictures of hearts of wild-type (WT) and Nrf2−/− mice after TAC. The right panel shows the heart weight/body weight (HW/BW) ratio. (b) Left ventricle fractional shortening (FS) (%) of WT and Nrf2−/− mice 2 weeks after TAC. (c) Representative confocal microscopic images of LV 4-HNE staining. 4-HNE-positive staining is shown in red. Nuclei are shown in blue (×630). Nrf2 gain- and loss-of-function on hypertrophic factor-induced ROS production in rat neonatal cardiomyocytes. Representative images and quantitative analysis of intracellular ROS production in norepinephrine (NE, 200 μmol/L) or phenylephrine (PE, 100 μmol/L) treated cardiac myocytes that were infected with adenovirus of Nrf2 (d) or rat Nrf2 shRNA (e). *P < 0.05 or # P < 0.05 versus control ad-βGal- or ad-Nrf2-infected cells that were treated with vehicles. The combined figures here were collected by the authors from different figures published in the study by Li et al. [7].
In addition, HO-1 protects against cardiac hypertrophy [24, 25, 68]. For example, Hu et al. reported that HO-1 attenuated angiotensin II- (Ang II-) induced cardiac hypertrophy both in vitro and in vivo [25]. Furthermore, Ndisang and Jadhav reported that upregulating the heme oxygenase system suppressed LV hypertrophy in spontaneously hypertensive adult rats and was accompanied with attenuated extracellular matrix remodeling, whereas HO-1 was blocked with chromium mesoporphyrin-exacerbated cardiac fibrosis/hypertrophy [68]. Deletion of Bach1 caused upregulation of cytoprotective HO-1 and also inhibited TAC-induced LV hypertrophy and remodeling [69]. Another group reported that HO-1 gene transfer to cardiomyocytes attenuated Ang II-mediated apoptosis but not hypertrophy [70]. However, Chen et al. reported that, after TAC, more calcineurin protein expression was induced in systemic HO-1 transgenic overexpressing mice than in wild-type mice, and it aggravated pressure overload-induced cardiac hypertrophy [71]. Therefore, the role of HO-1 in pressure overload-induced cardiac remodeling is controversial and further investigation is needed.
Myocardial SOD activity was suppressed in LV remodeling induced by TAC [26, 72]. In addition, Ang II could stimulate collagen production and in the meantime inhibit total SOD, SOD1, and SOD2 activity in rat cardiac fibroblasts [73]. Reduced expression of the manganese-dependent SOD2 resulted in increased cardiac oxidative stress and induced maladaptive cardiac hypertrophy [74]. Moreover, SOD3 deficiency exacerbated TAC-induced myocardial oxidative stress, hypertrophy, fibrosis, and HF [26]. These findings demonstrate that activation of Nrf2 and its downstream genes provides a novel mechanism to protect the heart against pressure overload-induced pathological cardiac hypertrophy, fibrosis, apoptosis, and HF via suppressing oxidative stress.
5. Role of the Nrf2 Pathway in Ischemia-Induced Cardiac Remodeling
Coronary artery disease and ischemic heart disease are prevalent worldwide. Cardiac hypertrophy, apoptosis, and fibrosis after myocardial infarction (MI) have been identified as key detrimental factors in the development of HF. The development of percutaneous coronary intervention and surgical revascularization has brought marked benefits to patients with acute MI. However, ischemia/reperfusion (I/R) injury during revascularization can cause further cardiac injury [75, 76]. Once blood flow is restored, ROS can be produced either by the myocardium itself or by infiltrating inflammatory cells [77]. ROS can subsequently lead to cellular damage through a number of pathways, including direct damage to membranes and proteins or indirect damage through activation of proapoptotic pathways [77]. This damage can further cause cardiac remodeling which leads to progressive heart chamber dilation, ventricular wall thinning, and eventually HF [78]. Nrf2 and its target genes have been shown to play a protective role in cardiac ischemia-associated injury [79, 80].
Some antioxidants protect the heart from ischemia-induced cardiac injury via the Nrf2 pathway. For example, α-lipoic acid and prostaglandin D2 significantly increased Nrf2 nuclear translocation and the expression of its downstream genes reduced lactate dehydrogenase (LDH) and creatine kinase (CK) release, attenuated myocardial infarct size, decreased cardiomyocyte apoptosis, and partially preserved heart function; and this effect was at least partially PI3K/Akt signaling pathway dependent [79, 81]. The phenomenon of ischemic preconditioning (IPC) has been recognized as one of the most potent mechanisms to protect against myocardial ischemic injury, reducing infarct size, attenuating the incidence and severity of reperfusion-induced arrhythmias, and preventing endothelial cell dysfunction [82]. In rat hearts, 30 minutes of left anterior descending coronary artery occlusion resulted in a reduction in the Nrf2 protein level, which was prevented by IPC of the myocardium [83]. Recently, Zhang et al. [23] reported IPC-induced activation of protein kinase C, which then promoted the translocation of Nrf2 and the inductions of antioxidant genes HO-1 and MnSOD and decreased tissue malondialdehyde content compared to control hearts.
Prior studies have reported that HO-1 is upregulated in human failing hearts [84] and in animal models of right ventricular failure [85]. The HO-1 expression in the noninfarct myocardium was increased four weeks after coronary ligation, and cardiomyocyte-specific HO-1 transgenic mice showed improved postinfarction survival and attenuated cardiac hypertrophy, interstitial fibrosis, oxidative stress, and apoptosis [86, 87]. Heterozygous HO-1+/− mice exhibited exaggerated cardiac injury and dysfunction after I/R, patially rescued by antioxidants [88]. In contrast, mice with cardiac-restricted HO-1 overexpression were resistant to I/R injury, with improved contractile recovery and reduced infarct size, inflammatory cell infiltration, oxidative damage, and apoptosis [89]. Similar results were obtained in rat hearts subjected to I/R 8 weeks after human HO-1 gene transfer [90]; the improvement in LV function was maintained for up to 1 year after injury [91]. Kuzuya et al. [92, 93] have shown that the infarct limitation observed in the canine myocardium 24 hours after IPC was accompanied by a significant increase in SOD2 activity. In addition, a recent study demonstrated that SOD3 expression was decreased in MI-induced HF [54]. SOD3-knockout mice had greater increases of nitrotyrosine in the peri-infarct myocardium, and this was associated with a greater reduction of LV ejection fraction, a greater decrease of sarcoplasmic or endoplasmic reticulum Calcium2+ ATPase, and a greater increase of atrial natriuretic peptide in the peri-infarct zone compared to wild-type mice at 8 weeks after MI, which means that mice with SOD3 gene deletion developed more severe LV hypertrophy, more fibrosis, and worse cardiac function [54]. Moreover, it has been demonstrated that GPx-knockout mice were more susceptible to myocardial I/R injury and transgenic mice were more resistant to myocardial I/R injury [94–96]. Overexpression of GPx also attenuated post-MI cardiac failure and cardiac remodeling by decreasing myocyte hypertrophy, apoptosis, and interstitial fibrosis in the noninfarct LV [56], which may be related to GPx preserving electron transport chain complex activities [96]. Overexpression of Nrf2 and its downstream genes inhibited ischemia-induced LV remodeling and HF; thus, therapies designed to interfere with oxidative stress might be beneficial to prevent ischemia-induced heart injury.
6. Role of the Nrf2 Pathway in Diabetes-Induced Cardiac Remodeling
Diabetic cardiomyopathy is a very common complication of diabetes and also one of the major causes of mortality in diabetic patients [97]. Evidence suggests that diabetes is associated with a reduced overall antioxidant defense system and increased oxidative stress [98]. Extra production of ROS and RNS causes the development of diabetic complications, including cardiomyopathy; on the other hand, antioxidants can prevent or treat diabetic complications [97, 99, 100]. Therefore, the activation of endogenous antioxidative components has been proposed as an appealing strategy to alleviate diabetic complications [101]. Nrf2 and its target genes may prevent or inhibit this pathological process through their antioxidative stress properties.
Emerging evidence has indicated that high glucose (HG) not only induces ROS and/or RNS production, but also enhances the expression and activation of Nrf2 and its downstream genes, both in vivo and in vitro [103, 115]. However, Tan et al. have reported a different finding [116]. In tissue sections of left ventricles obtained from autopsied heart specimens of humans with or without diabetes, Nrf2 expression in the nuclei was significantly downregulated compared to control hearts [116]. Tan et al. also demonstrated that Nrf2 protein expression was slightly increased in the hearts of mice with two-month hyperglycemia but significantly decreased in the hearts of mice with five-month hyperglycemia [116], which suggests that Nrf2 was adaptively overexpressed to combat diabetic damages at the early stage of diabetes; at the late stage, however, cardiac antioxidant function was so severely impaired that it led to a decrease in cardiac Nrf2 expression [117]. Glucose at high concentrations induced ROS production in both primary neonatal and adult cardiomyocytes from the Nrf2 wild-type mouse heart, whereas, in Nrf2-knockout cells, the amount of ROS was significantly greater under basal conditions and high glucose markedly further increased ROS production in concentration- and time-dependent manners. Concomitantly, high glucose induced significantly greater levels of apoptosis at lower concentrations and in a shorter time in Nrf2-knockout cells than in wild-type cells [115].
HO-1 and its reaction products have been shown to have both antioxidative and anti-inflammatory properties [89]. Association of a microsatellite polymorphism in the promoter region of the human HO-1 gene with the risk of coronary artery disease in type 2 diabetic patients has been reported [118]. Patients with type 2 diabetes with longer repeats of the HO-1 gene promoter (with lower HO-1 inducibility) were shown to have more oxidative stress and increased susceptibility to coronary artery disease [118]. In streptozotocin- (STZ-) induced diabetes rodent models, overexpression of HO-1 ameliorated LV dysfunction, myofibril structure disarray, oxidative stress, inflammation, apoptosis, and autophagy [119]. Furthermore, deficiency of HO-1 significantly increased the infarct size in normoglycemic mice and exacerbated myocardial infarction in diabetic mice [120]. In addition to a larger infarct size, mortality was two-fold greater in diabetic HO-1-knockout mice than in wild-type mice after I/R injury, and 55% of the diabetic HO-1-knockout mice survived LV thrombi induced by I/R [120]. Myocardial SOD activity and the GSH level were also significantly increased in the hearts of diabetic rats [114]. Overexpression of SOD2 protected the cardiac morphological changes induced by diabetes and completely normalized the impaired contractility in diabetic cardiomyocytes [121]. Moreover, the plasma type of GPx, GPx-3, was significantly upregulated in diabetic mice compared with control mice [122]. Furthermore, GPx overexpression inhibited the development of LV remodeling and diastolic dysfunction associated with diabetes [27]. These beneficial effects of GPx overexpression are thought to be associated with the attenuation of hypertrophy, apoptosis, and interstitial fibrosis of cardiomyocytes [27]. Thus, Nrf2 and its downstream genes play an essential protective role in the adaptation of diabetic cardiomyopathy. Taken together, these studies may provide a new target for developing therapeutic strategies to treat/prevent diabetic cardiomyopathy.
7. Role of the Nrf2 Pathway in Anticancer Drug-Induced Cardiac Remodeling
There are rising concerns among both cardiologists and oncologists about cardiotoxicity induced by cancer treatment, since it has a significant impact on the management and outcomes of cancer patients. The most typical manifestation of cardiotoxicity is hypokinetic cardiomyopathy leading to HF [123]. Nrf2 and its target genes have been suggested to be useful for protection against cardiotoxicity of anticancer drugs [124, 125].
Anthracycline anticancer drugs (e.g., doxorubicin or daunorubicin) can induce chronic cardiotoxicity and HF, both of which are believed to be caused by oxidative injury and mitochondrial damage [126]. GPx activity was significantly increased in both a daunorubicin-induced rabbit HF model and rat cardiomyocytes exposed to daunorubicin [127]. Incubation of H9c2 rat cardiomyoblasts with doxorubicin resulted in a two-fold increase in Nrf2 protein content and enhanced transcription of several of the Nrf2-regulated downstream genes, including GSTp1 and NQO1 [128]. Chronic arsenic exposure also has been linked to increased risks of vascular diseases. Arsenic exposure affects the activity of Nrf2 [125], and HO-1 has been identified as a response biomarker [129]. H9c2 rat cardiomyocytes exposed to arsenic showed a modest activation of Nrf2 and lower GSH availability [130]. However, the results have been contrastive. Recently, in a daunorubicin-induced rabbit HF model, Jirkovský et al. showed that Nrf2 and its target genes did not show upregulation and that global oxidative stress may not be a factor for the development of anthracycline-induced HF [131]. Therefore, the role of Nrf2 and its target genes in anticancer drug-induced cardiotoxicity is still controversial and more investigations are needed.
8. Potential Clinical Interventions of Cardiac Remodeling by Targeting Nrf2 and Its Target Genes
The exploration of mechanisms underlying the regulation of the Nrf2 pathway has led to the development of agents manipulating Nrf2 to treat HF. In fact, compounds that increase the activity of Nrf2 and its downstream genes are currently being tested for disease prevention [111, 132–134] (Table 1).
Table 1.
Activation of Nrf2 and its downstream genes to treat cardiac remodeling and HF.
| Name | Target disease model | Species | Function | Ways and volume | References | |
|---|---|---|---|---|---|---|
| Nrf2 activators | Sulforaphane | Ischemia (I/R, Langendorff) | SD rats | Prevent cardiac apoptosis | 0.5 mg/kg/d, 3 days, IP | [102] |
| Diabetes (type 1, STZ) | Mice | Prevent cardiac hypertrophy, fibrosis, and apoptosis | 0.5 mg/kg/d, 3 months, IP | [103] | ||
| MG132 | Hypertension (abdominal aortic banding) | SD rats | Prevent cardiac hypertrophy and fibrosis | 0.1 mg/kg/d, 8 or 16 weeks, IP | [104, 105] | |
| Diabetes (type 1, OVE) | Mice | Prevent cardiac hypertrophy and apoptosis | 10 μg/kg/d, 3 months, IP | [106] | ||
| Resveratrol | Ischemia (MI, LAD occlusion) | SD rats | Improve cardiac stem cells' survival, proliferation, and differentiation | 2.5 mg/kg/d, 2 weeks, gavage | [107] | |
| Allicin | Hypertension (Ang II) | SD rats | Prevent cardiac hypertrophy and fibrosis | 180 mg/kg/d, 8 weeks, receiving a diet in addition to standard chow | [108] | |
| α-Lipoic acid | Ischemia (I/R, coronary artery ligation followed by reperfusion) | SD rats | Prevent cardiac apoptosis | 15 mg/kg, one time, IV | [79] | |
| Hydrogen sulfate | Ischemia (I/R, coronary artery ligation followed by reperfusion) | Mice | Prevent cardiac apoptosis | 0.1 mg/kg, one time, IV | [109] | |
| 4-HNE | Ischemia (MI, Langendorff) | Mice | Improve cardiac function | 4 mg/kg, one time, IV | [110] | |
| HO-1 activators | CoPP | Hypertension (Ang II) | Wistar rats | Prevent cardiac hypertrophy | 1 mg/kg, every 2 days, 2 weeks, IP | [25] |
| Hypertension plus ischemia (spontaneous hypertensive rats + LAD occlusion) | Wistar rats | Prevent cardiac hypertrophy | 4.5 mg/kg, one/week, 6 weeks, IP | [111] | ||
| Hemin | Ischemia (MI, LAD occlusion) | SD rats | Prevent cardiac apoptosis | 4 mg/kg, every 2 days, 4 weeks, IP | [112] | |
| SOD activators | EUK-8 | Hypertension (transverse aortic banding) | Mice | Prevent cardiac hypertrophy, fibrosis, and apoptosis | 25 mg/kg/d, 4 weeks, IP | [113] |
| N-Acetylcysteine | Diabetes (STZ) | Wistar rats | Improve cardiac function | 1.4-1.5 g/kg/d, 8 weeks, in drinking water | [114] |
8.1. Activation of Nrf2 to Treat Cardiac Remodeling and HF
Many Nrf2 activators are natural products and plant-derived phytochemicals. Some examples of natural Nrf2 activators include sulforaphane, curcumin, resveratrol, and garlic organosulfur compounds [135]. Several synthetic Nrf2 activators have also been developed, such as carbobenzoxy-Leu-Leu-leucinal (MG132), 4-hydroxynonenal (4-HNE), α-lipoic acid, hydrogen sulfate, and 17β-estradiol (E2) [79, 109, 110]. These chemopreventive compounds exert their effects by inducing an Nrf2-mediated defense response as well as activation of phase II detoxification enzymes, antioxidants, and transporters that protect cells from oxidative stress.
8.1.1. Natural Nrf2 Activators
One of the most extensively studied natural products that target the Nrf2-Keap1 signaling pathway is sulforaphane, an isothiocyanate present in cruciferous vegetables such as broccoli [136]. Treatment with sulforaphane decreased infarct size, inhibited an increase in the LV end-diastolic pressure, and improved coronary flow in mice after MI [102]. This protective effect may be partly mediated through HO-1, SOD, and CAT expression [102].
Curcumin is another well-studied chemopreventive natural product that is capable of activating Nrf2 [132]. Curcumin has been used to attenuate acute doxorubicin-induced cardiomyopathy in rats [133]. In this study, curcumin pretreatment reversed the increase in lipid peroxidation and CAT content, as well as the decrease in GSH content and GPx activity in rat cardiac tissues induced by doxorubicin [133].
By treating rat cardiac stem cells with resveratrol in a rat left anterior descending occlusion model, Gorbunov et al. found that resveratrol significantly improved cardiac function through enhancing Nrf2 expression, as well as significantly increasing the survival and engraftment of implanted cardiac stem cells in the host [134]. Resveratrol can also achieve the same effect by gavage in the same animal model [107].
In an Ang II-induced cardiac remodeling and HF rat model, allicin treatment could prevent the development of cardiac remodeling and the progression of cardiac hypertrophy to cardiac dysfunction, by enhancing the Nrf2 pathway [108]. In addition, dietary phytochemical intake could upregulate the cardiac Nrf2 transcriptome and reduce oxidative damage and HF in hypertensive rats [137]. The protective effect of Nrf2 in myocardial remodeling and HF has been suggested to be mediated through Nox4 [138], which is known to be an important regulator of reduction-oxidation signaling in many cell types including cardiomyocytes [139].
8.1.2. Synthetic Nrf2 Activators
Accumulating evidence has demonstrated that MG132 can protect cells and tissues against oxidative damage, since it can activate the Nrf2-ARE signaling pathway, leading to upregulation of detoxifying and antioxidant genes [140, 141]. Both sulforaphane and MG132 could prevent diabetes-induced high blood pressure and cardiac dysfunction, as well as cardiac hypertrophy, fibrosis, oxidative damage, and inflammation [103, 106]. In pressure-overload-induced rodent HF models, treatment with MG132 significantly attenuated cardiac hypertrophy and cardiac fibrosis as well as improving cardiac function [104, 105]. Mechanistically, MG132 may enhance Nrf2-mediated antioxidative function and inhibit NF-κB-mediated inflammation [142].
4-HNE is an α, β-unsaturated hydroxyalkenal that is produced by lipid peroxidation in cells. α-Lipoic acid is an organosulfur compound derived from octanoic acid. E2 is a sex hormone. All three of these compounds could protect the heart from ischemia-induced cardiac remodeling and HF by activating Nrf2 and its target genes. Treatment with 4-HNE [110], α-lipoic acid [79], or hydrogen sulfate [109] could activate Nrf2 in the heart and increase the intramyocardial GSH content, consequently improving the functional recovery of the LV following I/R in Langendorff-perfused hearts [110] or reducing infarct size, decreasing cardiomyocyte apoptosis in vivo [79, 109]. The cardioprotective effect of 4-HNE was not observed for Nrf2-knockout mice [110], indicating that this protection is Nrf2 dependent. In an in vitro study, Yu et al. cultured primary cardiomyocytes and established a hypoxia/reoxygenation (H/R) model to simulate myocardial I/R injury [143]. They found that E2 could upregulate Nrf2 expression in nuclear extracts and also increased the expression of HO-1, SOD1, GST, and glutamate-cysteine ligase (GCL) dramatically during H/R injury [143]. These findings indicate that Nrf2 plays a pivotal role in preventing and alleviating cardiac I/R injury-induced oxidative stress.
8.2. Role of Nrf2 Target Genes Activation in Cardiac Remodeling
8.2.1. Role of HO-1 Activation in Cardiac Remodeling
It has been reported that elevation of HO-1 by treatment with cobalt protoporphyrin IX (CoPP IX) or a recombinant adenovirus carrying the human HO-1 gene attenuated cardiac hypertrophy and apoptosis, in both an Ang-II-induced HF model and a spontaneously hypertensive rat model [25, 70], while this pathological process was exacerbated in the presence of tin protoporphyrin, an inhibitor of HO activity [70]. In a spontaneous hypertensive rat model, HO-1 upregulation by CoPP IX administration decreased blood pressure, inhibited inflammation, and improved the ventricular remodeling process and postinfarct cardiac function [111]. In addition, cardiac stem cells (CSCs) were protected by pretreatment with CoPP against apoptosis through activation of the extracellular signal-regulated kinase (ERK)/Nrf2 signaling pathway and cytokine release [144].
Hemin could upregulate HO activity, reduce hypertension, suppress oxidative stress, and attenuate cardiac fibrosis, apoptosis, and oxidative stress. This effect was modulated through enhanced expression of the PI3K p85 regulatory subunit [145]. In an acute I/R with STZ-induced hyperglycemic rat model, intraperitoneal administration of hemin 18 h before I/R increased the levels of HO-1, providing marked protection against myocardial injury [146]. Moreover, zinc protoporphyrin IX (an inhibitor of HO activity) abolished the protective effect by hemin [146]. Furthermore, chronic HO-1 activation by prolonged administration of hemin improved postinfarction survival and exerted protective effects in a rat model of myocardial ischemia, through a potent antioxidant activity [112].
Taurine treatment significantly improved LV systolic and diastolic function in an STZ-induced diabetes rat model, and there were persistent increases in activities of Akt/PKB and SOD, as well as the level of HO-1 protein [147].
Mesenchymal stem cells (MSCs) have been reported to have the potential to release several kinds of cytokines, which could induce angiogenesis [148, 149]. However, almost all transplanted cells seemed to be lost by 6 weeks after transplantation, making it impossible for the limited number of MSCs to achieve a maximum proangiogenesis effect [150]. Due to its multiple catalytic byproducts, HO-1 has been proposed to be involved in a number of cytoprotective effects. HO-1 has been administered to improve the survival environment of MSCs and to achieve maximum functional benefits of MSCs [151]. HO-1 transduced by MSCs can induce angiogenic effects, reduce cardiac remodeling, and improve heart function after acute MI [152, 153]. Intramyocardial delivery of the HO-1 gene using adeno-associated virus-2 (AAV-2) before I/R also prevented cardiomyocyte apoptosis and reduced infarct size and cardiac dysfunction after myocardial I/R in mice [90]. To evaluate the long-term effects of HO-1 gene delivery, Liu et al. have showed that delivery of the HO-1 gene reduced mortality and preserved LV function and chamber dimensions 1 year after acute MI [91]. These results suggest that preemptive HO-1 gene delivery may be useful as a therapeutic strategy to reduce post-MI LV remodeling and HF.
8.2.2. Role of SOD and GSH Activation in Cardiac Remodeling
EUK-8, as a SOD mimetic, improved LV end-systolic dimensions and fractional shortening, prevented myocardial oxidant stress, attenuated necrotic and apoptotic cell death, and attenuated cardiac hypertrophy and fibrosis in mice subjected to pressure overload [113]. In vitro, the SOD mimetics tempol and EUK-8 could also reduce collagen production in Ang-II-treated fibroblasts [73]. Moreover, N-acetylcysteine, an antioxidant and GSH precursor, attenuated diabetic myocardial dysfunction via upregulating myocardial GSH and SOD2 activity [114]. Treatment with polyethylene glycol-conjugated SOD ameliorated doxorubicin-induced cardiac dysfunction, and this effect was mediated by inhibition of doxorubicin-induced upregulation of NF-κB signaling, lowering the levels of hexanoyl-lysine (a marker of free radical-induced lipid peroxidation) and suppressing the activation of Akt and Akt-regulated gene expression [124].
Recombinant SOD3 preserved cardiac function following I/R in isolated rat hearts [154–156] and reduced the infarct size when given just prior to coronary reperfusion in pigs [157]. Cardiac-selective expression of SOD3 from the cardiac troponin-T promoter after systemic administration of AAV-9 provided significant protection against both acute MI and LV remodeling [158]. Liu et al. also have demonstrated that encapsulation of CSCs in SOD-loaded alginate hydrogel enhanced CSC survival in the presence of doxorubicin, indicating its potential application as a novel therapy for the treatment of acute and early-onset doxorubicin-induced cardiotoxicity [159].
9. Advantages of Nrf2 Activation in Cardiac Remodeling and HF
Nrf2 dimerizes with members of the small Maf family to bind to antioxidant or electrophile response elements (AREs/EpREs) located in the regulatory regions of cellular defense enzyme genes [50]. The target genes of Nrf2 include many antioxidant genes, such as HO-1, NQO1, SODs, CAT, GST, γ-GCS, and GPx. By inducing the expression of these genes, Nrf2 activates a wide range of cell defense processes, thereby enhancing the overall capacity of cells to detoxify and eliminate harmful substances. As mentioned above, activated Nrf2 and its target genes, such as HO-1, SOD, and GPx, all play important roles in preventing cardiac remodeling. Although it cannot be excluded that some Nrf2 target genes may protect the heart through an Nrf2-independent pathway, Nrf2-induced cell defense processes remain to be the main driving force. Because Nrf2 could induce transcriptional activation of a number of ARE-bearing antioxidants, we speculate that, acting as an upstream gene, activation of Nrf2 may rescue from cardiac remodeling and HF induced by deficiency of an individual downstream gene. So, activation of Nrf2 may have an advantage over its downstream target genes in protecting one from oxidative stress-induced HF, but further investigations are needed before a conclusion can be drawn.
10. Conclusions
Increased generation of ROS in cardiomyocytes leads to increased necrosis and apoptosis, which induce cardiac remodeling and dysfunction. Nrf2 and its target genes are key components to maintain cellular redox homeostasis by attenuating oxidative stress-associated pathological processes. Patients with insufficient Nrf2 levels in their cardiovascular system are more vulnerable to HF development. It is conceivable that Nrf2 orchestrates a group of antioxidative and other cytoprotective genes to provide a protective mechanism against detrimental stress-induced cardiac dysfunction. However, further work is needed to understand the role of Nrf2 in the pathogenesis of cardiac remodeling and HF in more detail before it can be seriously considered as a therapeutic target for HF.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (no. 81370318, to Yang Zheng).
Abbreviations
- HF:
Heart failure
- ROS:
Reactive oxygen species
- RNS:
Reactive nitrogen species
- Nrf2:
Nuclear factor-erythroid-2- (NF-E2-) related factor 2
- LV:
Left ventricular
- AREs:
Antioxidant response elements
- CNC:
Cap-n-collar
- CNC-bZIP:
CNC type of basic region leucine zipper
- Keap1:
Kelch-like-ECH-associated protein 1
- HO-1:
Heme oxygenase-1
- NQO1:
NAD(P)H dehydrogenase (quinone 1)
- SODs:
Superoxide dismutases
- CAT:
Catalase
- GST:
Glutathione-S-transferases
- γ-GCS:
γ-Glutamylcysteine synthase
- GPx:
Glutathione peroxidases
- TAC:
Transverse aortic constriction
- STZ:
Streptozotocin
- ATF3:
Activating transcription factor 3
- LDH:
Lactate dehydrogenase
- GCL:
Glutamate-cysteine ligase
- GSH:
Glutathione
- MG132:
Carbobenzoxy-Leu-Leu-leucinal
- 4-HNE:
4-Hydroxynonenal
- E2:
17β-Estradiol
- H/R:
Hypoxia/reoxygenation
- CSCs:
Cardiac stem cells
- ERK:
Extracellular signal-regulated kinase
- MSCs:
Mesenchymal stem cells
- AAV-2:
Adeno-associated virus-2
- EpREs:
Electrophile response elements
- ∙O2−:
Superoxide anion
- ∙OH:
Hydroxyl radical
- H2O2:
Hydrogen peroxide
- OCl∙−:
Hypochlorite
- Ang II:
Angiotensin II
- MI:
Myocardial infarction
- I/R:
Ischemia/reperfusion
- HG:
High glucose
- tBHQ:
Tert-Butylhydroquinone
- CK:
Creatine kinase.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circulation Research. 2006;99(9):924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 2.Guzik TJ, Harrison DG. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discovery Today. 2006;11(11-12):524–533. doi: 10.1016/j.drudis.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Jialal I, Devaraj S. Antioxidants and atherosclerosis: don't throw out the baby with the bath water. Circulation. 2003;107(7):926–928. doi: 10.1161/01.cir.0000048966.26216.4c. [DOI] [PubMed] [Google Scholar]
- 4.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(1):29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 5.Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovascular Research. 2006;71(2):208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 6.Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49(2):241–248. doi: 10.1161/01.HYP.0000254415.31362.a7. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Ichikawa T, Villacorta L, et al. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(11):1843–1850. doi: 10.1161/ATVBAHA.109.189480. [DOI] [PubMed] [Google Scholar]
- 8.Santos CXC, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radical Biology and Medicine. 2011;50(7):777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas DD, Ridnour LA, Isenberg JS, et al. The chemical biology of nitric oxide: implications in cellular signaling. Free Radical Biology & Medicine. 2008;45(1):18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowling DK, Simmons LW. Reactive oxygen species as universal constraints in life-history evolution. Proceedings of the Royal Society B: Biological Sciences. 2009;276(1663):1737–1745. doi: 10.1098/rspb.2008.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends in Biochemical Sciences. 2011;36(1):30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Afanas'ev I. ROS and RNS signaling in heart disorders: could antioxidant treatment be successful? Oxidative Medicine and Cellular Longevity. 2011;2011:13 pages. doi: 10.1155/2011/293769.293769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology and Medicine. 2010;48(6):749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolli R, Zhu WX, Hartley CJ, et al. Attenuation of dysfunction in the postischemic “stunned” myocardium by dimethylthiourea. Circulation. 1987;76(2):458–468. doi: 10.1161/01.cir.76.2.458. [DOI] [PubMed] [Google Scholar]
- 15.Beresewicz A, Horackova M. Alterations in electrical and contractile behavior of isolated cardiomyocytes by hydrogen peroxide: Possible ionic mechanisms. Journal of Molecular and Cellular Cardiology. 1991;23(8):899–918. doi: 10.1016/0022-2828(91)90133-7. [DOI] [PubMed] [Google Scholar]
- 16.Chesley A, Lundberg MS, Asai T, et al. The β 2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3'-kinase. Circulation Research. 2000;87(12):1172–1179. doi: 10.1161/01.res.87.12.1172. [DOI] [PubMed] [Google Scholar]
- 17.Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Molecular Aspects of Medicine. 2011;32(4–6):234–246. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Andrukhova O, Salama M, Rosenhek R, et al. Serum glutathione S-transferase P1 1 in prediction of cardiac function. Journal of Cardiac Failure. 2012;18(3):253–261. doi: 10.1016/j.cardfail.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu W, Qiu Q, Wang H, et al. Nrf2 is crucial to graft survival in a rodent model of heart transplantation. Oxidative Medicine and Cellular Longevity. 2013;2013: 9 pages. doi: 10.1155/2013/919313.919313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Letters. 2005;579(14):3029–3036. doi: 10.1016/j.febslet.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H, Jia Z, Misra BR, et al. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovascular Toxicology. 2008;8(2):71–85. doi: 10.1007/s12012-008-9016-0. [DOI] [PubMed] [Google Scholar]
- 22.Peake BF, Nicholson CK, Lambert JP, et al. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. American Journal of Physiology: Heart and Circulatory Physiology. 2013;304(9):H1215–H1224. doi: 10.1152/ajpheart.00796.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Xiao Z, Yao J, Zhao G, Fa X, Niu J. Participation of protein kinase C in the activation of Nrf2 signaling by ischemic preconditioning in the isolated rabbit heart. Molecular and Cellular Biochemistry. 2013;372(1-2):169–179. doi: 10.1007/s11010-012-1458-9. [DOI] [PubMed] [Google Scholar]
- 24.Wiesel P, Patel AP, Carvajal IM, et al. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circulation Research. 2001;88(10):1088–1094. doi: 10.1161/hh1001.091521. [DOI] [PubMed] [Google Scholar]
- 25.Hu CM, Chen YH, Chiang MT, Chau LY. Heme oxygenase-1 inhibits angiotensin II-induced cardiac hypertrophy in vitro and in vivo. Circulation. 2004;110(3):309–316. doi: 10.1161/01.CIR.0000135475.35758.23. [DOI] [PubMed] [Google Scholar]
- 26.Lu Z, Xu X, Hu X, et al. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension. 2008;51(1):19–25. doi: 10.1161/HYPERTENSIONAHA.107.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushima S, Kinugawa S, Ide T, et al. Overexpression of glutathione peroxidase attenuates myocardial remodeling and preserves diastolic function in diabetic heart. American Journal of Physiology—Heart and Circulatory Physiology. 2006;291(5):H2237–H2245. doi: 10.1152/ajpheart.00427.2006. [DOI] [PubMed] [Google Scholar]
- 28.Hafstad AD, Nabeebaccus AA, Shah AM. Novel aspects of ROS signalling in heart failure. Basic Research in Cardiology. 2013;108(4, article 359) doi: 10.1007/s00395-013-0359-8. [DOI] [PubMed] [Google Scholar]
- 29.Maulik SK, Kumar S. Oxidative stress and cardiac hypertrophy: a review. Toxicology Mechanisms and Methods. 2012;22(5):359–366. doi: 10.3109/15376516.2012.666650. [DOI] [PubMed] [Google Scholar]
- 30.Nattel S. Electrophysiologic remodeling: are ion channels static players or dynamic movers? Journal of Cardiovascular Electrophysiology. 1999;10(11):1553–1556. doi: 10.1111/j.1540-8167.1999.tb00214.x. [DOI] [PubMed] [Google Scholar]
- 31.Yu C, Lin H, Yang H, Kong S, Zhang Q, Lee SW. Progression of systolic abnormalities in patients with “isolated” diastolic heart failure and diastolic dysfunction. Circulation. 2002;105(10):1195–1201. doi: 10.1161/hc1002.105185. [DOI] [PubMed] [Google Scholar]
- 32.Cai L, Li W, Wang G, Guo L, Jiang Y, James Kang Y. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome c-mediated caspase-3 activation pathway. Diabetes. 2002;51(6):1938–1948. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- 33.Chang GL, Zhang DY, Yu H, et al. Cardioprotective effects of exenatide against oxidative stress-induced injury. International Journal of Molecular Medicine. 2013;32(5):1011–1020. doi: 10.3892/ijmm.2013.1475. [DOI] [PubMed] [Google Scholar]
- 34.Zhao WY, Zhao TQ, Chen YJ, Ahokas RA, Sun Y. Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Molecular and Cellular Biochemistry. 2008;317(1-2):43–50. doi: 10.1007/s11010-008-9803-8. [DOI] [PubMed] [Google Scholar]
- 35.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. The FASEB Journal. 2006;20(9):1546–1548. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 36.Gupta S, Das B, Sen S. Cardiac hypertrophy: mechanisms and therapeutic opportunities. Antioxidants and Redox Signaling. 2007;9(6):623–652. doi: 10.1089/ars.2007.1474. [DOI] [PubMed] [Google Scholar]
- 37.Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxidants and Redox Signaling. 2003;5(6):731–740. doi: 10.1089/152308603770380034. [DOI] [PubMed] [Google Scholar]
- 38.Huynh K, Bernardo BC, McMullen. JR, et al. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacology & Therapeutics. 2014;142(3):375–415. doi: 10.1016/j.pharmthera.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 39.van Empel VPM, Bertrand ATA, Hofstra L, Crijns HJ, Doevendans PA, de Windt LJ. Myocyte apoptosis in heart failure. Cardiovascular Research. 2005;67(1):21–29. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 40.Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7(3):153–163. doi: 10.1016/s0928-4680(00)00053-5. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Advances in Enzyme Regulation. 2006;46(1):113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: Implications for the impact of particulate pollutants on asthma. Antioxidants and Redox Signaling. 2006;8(1-2):88–98. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 43.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual Review of Pharmacology and Toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi A, Ito E, Toki T, et al. Molecular cloning and functional characterization of a new Cap’n’ collar family transcription factor Nrf3. The Journal of Biological Chemistry. 1999;274(10):6443–6452. doi: 10.1074/jbc.274.10.6443. [DOI] [PubMed] [Google Scholar]
- 45.Johnsen Ø, Murphy P, Prydz H, Kolstø A. Interaction of the CNC-bZIP factor TCF11/LCR-F1/Nrf1 with MafG: binding-site selection and regulation of transcription. Nucleic Acids Research. 1998;26(2):512–520. doi: 10.1093/nar/26.2.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marini MG, Chan K, Casula L, Kan YW, Cao A, Moi P. hMAF, a small human transcription factor that heterodimerizes specifically with Nrf1 and Nrf2. The Journal of Biological Chemistry. 1997;272(26):16490–16497. doi: 10.1074/jbc.272.26.16490. [DOI] [PubMed] [Google Scholar]
- 47.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes and Development. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi A, Kang M, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular and Cellular Biology. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto T, Suzuki T, Kobayashi A, et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Molecular and Cellular Biology. 2008;28(8):2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochemical and Biophysical Research Communications. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 51.Howden R. Nrf2 and cardiovascular defense. Oxidative Medicine and Cellular Longevity. 2013;2013:10 pages. doi: 10.1155/2013/104308.104308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otterbein LE, Choi AMK. Heme oxygenase: colors of defense against cellular stress. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2000;279(6):L1029–L1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 53.Abraham NG, Kappas A. Heme oxygenase and the cardiovascular-renal system. Free Radical Biology and Medicine. 2005;39(1):1–25. doi: 10.1016/j.freeradbiomed.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 54.van Deel ED, Lu Z, Xu X, et al. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radical Biology and Medicine. 2008;44(7):1305–1313. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Molecular Aspects of Medicine. 2009;30(1-2):1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shiomi T, Tsutsui H, Matsusaka H, et al. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2004;109(4):544–549. doi: 10.1161/01.CIR.0000109701.77059.E9. [DOI] [PubMed] [Google Scholar]
- 57.Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovascular Research. 2005;68(1):26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 58.Kohler JJ, Cucoranu I, Fields E, et al. Transgenic mitochondrial superoxide dismutase and mitochondrially targeted catalase prevent antiretroviral-induced oxidative stress and cardiomyopathy. Laboratory Investigation. 2009;89(7):782–790. doi: 10.1038/labinvest.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Savoia C, Burger D, Nishigaki N, Montezano A, Touyz RM. Angiotensin II and the vascular phenotype in hypertension. Expert Reviews in Molecular Medicine. 2011;13, article e11 doi: 10.1017/S1462399411001815. [DOI] [PubMed] [Google Scholar]
- 60.Sugamura K, Keaney JF., Jr. Reactive oxygen species in cardiovascular disease. Free Radical Biology and Medicine. 2011;51(5):978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puddu P, Puddu GM, Cravero E, Rosati M, Muscari A. The molecular sources of reactive oxygen species in hypertension. Blood Pressure. 2008;17(2):70–77. doi: 10.1080/08037050802029954. [DOI] [PubMed] [Google Scholar]
- 62.Tardiff JC, Hewett TE, Factor SM, Vikstrom KL, Robbins J, Leinwand LA. Expression of the β (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects. The American Journal of Physiology—Heart and Circulatory Physiology. 2000;278(2):H412–H419. doi: 10.1152/ajpheart.2000.278.2.H412. [DOI] [PubMed] [Google Scholar]
- 63.Wakui H, Dejima T, Tamura K, et al. Activation of angiotensin II type 1 receptor-associated protein exerts an inhibitory effect on vascular hypertrophy and oxidative stress in angiotensin II-mediated hypertension. Cardiovascular Research. 2013;100(3):511–519. doi: 10.1093/cvr/cvt225. [DOI] [PubMed] [Google Scholar]
- 64.Wang HX, Yang H, Han QY, et al. Nadph oxidases mediate a cellular “memory” of angiotensin ii stress in hypertensive cardiac hypertrophy. Free Radical Biology & Medicine. 2013;65:897–907. doi: 10.1016/j.freeradbiomed.2013.08.179. [DOI] [PubMed] [Google Scholar]
- 65.Tsukamoto Y, Mano T, Sakata Y, et al. A novel heart failure mice model of hypertensive heart disease by angiotensin ii infusion, nephrectomy, and salt loading. The American Journal of Physiology—Heart and Circulatory Physiology. 2013;305:H1658–H1667. doi: 10.1152/ajpheart.00349.2013. [DOI] [PubMed] [Google Scholar]
- 66.Xing Y, Niu T, Wang W, et al. Triterpenoid dihydro-cddo-trifluoroethyl amide protects against maladaptive cardiac remodeling and dysfunction in mice: a critical role of Nrf2. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0044899.e44899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin H, Li HF, Chen HH, et al. Activating transcription factor 3 protects against pressure-overload heart failure via the autophagy molecule beclin-1 pathway. Molecular Pharmacology. 2014;85:682–691. doi: 10.1124/mol.113.090092. [DOI] [PubMed] [Google Scholar]
- 68.Ndisang JF, Jadhav A. Upregulating the heme oxygenase system suppresses left ventricular hypertrophy in adult spontaneously hypertensive rats for 3 months. Journal of Cardiac Failure. 2009;15(7):616–628. doi: 10.1016/j.cardfail.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 69.Mito S, Ozono R, Oshima T, et al. Myocardial protection against pressure overload in mice lacking bachl, a transcriptional repressor of heme oxygenase-1. Hypertension. 2008;51(6):1570–1577. doi: 10.1161/HYPERTENSIONAHA.107.102566. [DOI] [PubMed] [Google Scholar]
- 70.Foo RSY, Siow RCM, Brown MJ, Bennett MR. Heme oxygenase-1 gene transfer inhibits angiotensin II-mediated rat cardiac myocyte apoptosis but not hypertrophy. Journal of Cellular Physiology. 2006;209(1):1–7. doi: 10.1002/jcp.20723. [DOI] [PubMed] [Google Scholar]
- 71.Chen C, Huo R, Tong Y, et al. Systemic heme oxygenase-1 transgenic overexpression aggravates pressure overload-induced cardiac hypertrophy in mice. Cellular Physiology and Biochemistry. 2011;28(1):25–32. doi: 10.1159/000331710. [DOI] [PubMed] [Google Scholar]
- 72.Hikoso S, Yamaguchi O, Nakano Y, et al. The IκB Kinase β/Nuclear factor κb signaling pathway protects the heart from hemodynamic stress mediated by the regulation of manganese superoxide dismutase expression. Circulation Research. 2009;105(1):70–79. doi: 10.1161/CIRCRESAHA.108.193318. [DOI] [PubMed] [Google Scholar]
- 73.Lijnen P, Petrov V, van Pelt J, Fagard R. Inhibition of superoxide dismutase induces collagen production in cardiac fibroblasts. American Journal of Hypertension. 2008;21(10):1129–1136. doi: 10.1038/ajh.2008.242. [DOI] [PubMed] [Google Scholar]
- 74.Richters L, Lange N, Renner R, et al. Exercise-induced adaptations of cardiac redox homeostasis and remodeling in heterozygous SOD2-knockout mice. Journal of Applied Physiology. 2011;111(5):1431–1440. doi: 10.1152/japplphysiol.01392.2010. [DOI] [PubMed] [Google Scholar]
- 75.Takemura G, Nakagawa M, Kanamori H, Minatoguchi S, Fujiwara H. Benefits of reperfusion beyond infarct size limitation. Cardiovascular Research. 2009;83(2):269–276. doi: 10.1093/cvr/cvp032. [DOI] [PubMed] [Google Scholar]
- 76.Eefting F, Rensing B, Wigman J, et al. Role of apoptosis in reperfusion injury. Cardiovascular Research. 2004;61(3):414–426. doi: 10.1016/j.cardiores.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 77.Braunersreuther V, Jaquet V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Current Pharmaceutical Biotechnology. 2012;13(1):97–114. doi: 10.2174/138920112798868782. [DOI] [PubMed] [Google Scholar]
- 78.Sun Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovascular Research. 2009;81(3):482–490. doi: 10.1093/cvr/cvn333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deng C, Sun Z, Tong G, et al. Alpha-lipoic acid reduces infarct size and preserves cardiac function in rat myocardial ischemia/reperfusion injury through activation of pi3k/akt/nrf2 pathway. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0058371.e58371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ashrafian H, Czibik G, Bellahcene M, et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metabolism. 2012;15(3):361–371. doi: 10.1016/j.cmet.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Katsumata Y, Shinmura K, Sugiura Y, et al. Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia-reperfusion injury by activating Nrf2. Hypertension. 2014;63(1):80–87. doi: 10.1161/HYPERTENSIONAHA.113.01639. [DOI] [PubMed] [Google Scholar]
- 82.Iliodromitis EK, Lazou A, Kremastinos DT. Ischemic preconditioning: protection against myocardial necrosis and apoptosis. Vascular Health and Risk Management. 2007;3(5):629–637. [PMC free article] [PubMed] [Google Scholar]
- 83.Gurusamy N, Malik G, Gorbunov NV, Das DK. Redox activation of Ref-1 potentiates cell survival following myocardial ischemia reperfusion injury. Free Radical Biology & Medicine. 2007;43(3):397–407. doi: 10.1016/j.freeradbiomed.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 84.Grabellus F, Schmid C, Levkau B, et al. Reduction of hypoxia-inducible heme oxygenase-1 in the myocardium after left ventricular mechanical support. Journal of Pathology. 2002;197(2):230–237. doi: 10.1002/path.1106. [DOI] [PubMed] [Google Scholar]
- 85.Raju VS, Imai N, Liang CS. Chamber-specific regulation of heme oxygenase-1 (heat shock protein 32) in right-sided congestive heart failure. Journal of Molecular and Cellular Cardiology. 1999;31(8):1581–1589. doi: 10.1006/jmcc.1999.0995. [DOI] [PubMed] [Google Scholar]
- 86.Wang G, Hamid T, Keith RJ, et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation. 2010;121(17):1912–1925. doi: 10.1161/CIRCULATIONAHA.109.905471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vulapalli SR, Chen Z, Chua BHL, Wang T, Liang C. Cardioselective overexpression of HO-1 prevents I/R-induced cardiac dysfunction and apoptosis. American Journal of Physiology: Heart and Circulatory Physiology. 2002;283(2):H688–H694. doi: 10.1152/ajpheart.00133.2002. [DOI] [PubMed] [Google Scholar]
- 88.Yoshida T, Maulik N, Ho YS, Alam J, Das DK. H mox-1 constitutes an adaptive response to effect antioxidant cardioprotection: a study with transgenic mice heterozygous for targeted disruption of the heme oxygenase-1 gene. Circulation. 2001;103(12):1695–1701. doi: 10.1161/01.cir.103.12.1695. [DOI] [PubMed] [Google Scholar]
- 89.Yet S, Tian R, Layne MD, et al. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circulation Research. 2001;89(2):168–173. doi: 10.1161/hh1401.093314. [DOI] [PubMed] [Google Scholar]
- 90.Melo LG, Agrawal R, Zhang L, et al. Gene therapy strategy for long-term myocardial protection using adeno-associated virus-mediated delivery of heme oxygenase gene. Circulation. 2002;105(5):602–607. doi: 10.1161/hc0502.103363. [DOI] [PubMed] [Google Scholar]
- 91.Liu X, Simpson JA, Brunt KR, et al. Preemptive heme oxygenase-1 gene delivery reveals reduced mortality and preservation of left ventricular function 1 yr after acute myocardial infarction. American Journal of Physiology: Heart and Circulatory Physiology. 2007;293(1):H48–H59. doi: 10.1152/ajpheart.00741.2006. [DOI] [PubMed] [Google Scholar]
- 92.Kuzuya T, Hoshida S, Yamashita N, et al. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circulation Research. 1993;72(6):1293–1299. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 93.Dana A, Baxter GF, Yellon DM. Both protein kinase c and protein tyrosine kinase mediate adenosine induced delayed cardioprotection in rabbits. Circulation. 1997;96(6):1745–1749. [Google Scholar]
- 94.Yoshida T, Watanabe M, Eagelman DT, et al. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. Journal of Molecular and Cellular Cardiology. 1996;28(8):1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 95.Yoshida T, Maulik N, Engelman RM, et al. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation. 1997;96(9):II216–II220. [PubMed] [Google Scholar]
- 96.Dabkowski ER, Williamson CL, Hollander JM. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radical Biology and Medicine. 2008;45(6):855–865. doi: 10.1016/j.freeradbiomed.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 97.Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovascular Toxicology. 2001;1(3):181–193. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- 98.Ramprasath T, Selvam GS. Potential impact of genetic variants in nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Current Medicinal Chemistry. 2013;20:4680–4693. doi: 10.2174/09298673113209990154. [DOI] [PubMed] [Google Scholar]
- 99.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 100.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 101.Cai L. Diabetic cardiomyopathy and its prevention by metallothionein: experimental evidence, possible mechanisms and clinical implications. Current Medicinal Chemistry. 2007;14(20):2193–2203. doi: 10.2174/092986707781389646. [DOI] [PubMed] [Google Scholar]
- 102.Piao CS, Gao S, Lee G, et al. Sulforaphane protects ischemic injury of hearts through antioxidant pathway and mitochondrial KATP channels. Pharmacological Research. 2010;61(4):342–348. doi: 10.1016/j.phrs.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 103.Bai Y, Cui W, Xin Y, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. Journal of Molecular and Cellular Cardiology. 2013;57(1):82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 104.Chen B, Ma Y, Meng R, et al. MG132, a proteasome inhibitor, attenuates pressure-overload-induced cardiac hypertrophy in rats by modulation of mitogen-activated protein kinase signals. Acta Biochimica et Biophysica Sinica (Shanghai) 2010;42(4):253–258. doi: 10.1093/abbs/gmq012. [DOI] [PubMed] [Google Scholar]
- 105.Ma Y, Chen Y, Yang Y, et al. Proteasome inhibition attenuates heart failure during the late stages of pressure overload through alterations in collagen expression. Biochemical Pharmacology. 2013;85(2):223–233. doi: 10.1016/j.bcp.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 106.Wang Y, Sun W, Du B, et al. Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-κB. American Journal of Physiology: Heart and Circulatory Physiology. 2013;304(4):H567–H578. doi: 10.1152/ajpheart.00650.2012. [DOI] [PubMed] [Google Scholar]
- 107.Gurusamy N, Ray D, Lekli I, Das DK. Red wine antioxidant resveratrol-modified cardiac stem cells regenerate infarcted myocardium. Journal of Cellular and Molecular Medicine. 2010;14(9):2235–2239. doi: 10.1111/j.1582-4934.2010.01140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li X, Li C, Xiang Z, et al. Allicin ameliorates cardiac hypertrophy and fibrosis through enhancing of Nrf2 antioxidant signaling pathways. Cardiovascular Drugs and Therapy. 2012;26(6):457–465. doi: 10.1007/s10557-012-6415-z. [DOI] [PubMed] [Google Scholar]
- 109.Calvert JW, Jha S, Gundewar S, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circulation Research. 2009;105(4):365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Y, Sano M, Shinmura K, et al. 4-Hydroxy-2-nonenal protects against cardiac ischemia-reperfusion injury via the Nrf2-dependent pathway. Journal of Molecular and Cellular Cardiology. 2010;49(4):576–586. doi: 10.1016/j.yjmcc.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 111.Chen TM, Li J, Liu L, et al. Effects of heme oxygenase-1 upregulation on blood pressure and cardiac function in an animal model of hypertensive myocardial infarction. International Journal of Molecular Sciences. 2013;14(2):2684–2706. doi: 10.3390/ijms14022684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Collino M, Pini A, Mugelli N, et al. Beneficial effect of prolonged heme oxygenase 1 activation in a rat model of chronic heart failure. Disease Models and Mechanisms. 2013;6(4):1012–1020. doi: 10.1242/dmm.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Empel VPM, Bertrand AT, van Oort RJ, et al. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. Journal of the American College of Cardiology. 2006;48(4):824–832. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- 114.Xia Z, Guo Z, Nagareddy PR, Yuen V, Yeung E, McNeill JH. Antioxidant N-acetylcysteine restores myocardial Mn-SOD activity and attenuates myocardial dysfunction in diabetic rats. European Journal of Pharmacology. 2006;544(1–3):118–125. doi: 10.1016/j.ejphar.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 115.He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2009;46(1):47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 116.Tan Y, Ichikawa T, Li J, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. 2011;60(2):625–633. doi: 10.2337/db10-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li B, Liu S, Miao L, Cai L. Prevention of diabetic complications by activation of Nrf2: diabetic cardiomyopathy and nephropathy. Experimental Diabetes Research. 2012;2012:7 pages. doi: 10.1155/2012/216512.216512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen Y, Lin S, Lin M, et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Human Genetics. 2002;111(1):1–8. doi: 10.1007/s00439-002-0769-4. [DOI] [PubMed] [Google Scholar]
- 119.Zhao Y, Zhang L, Qiao Y, et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE. 2013;8(9) doi: 10.1371/journal.pone.0075927.e75927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu X, Wei J, Peng DH, Layne MD, Yet S. Absence of heme oxygenase-1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes. 2005;54(3):778–784. doi: 10.2337/diabetes.54.3.778. [DOI] [PubMed] [Google Scholar]
- 121.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55(3):798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 122.Iwata K, Nishinaka T, Matsuno K, Yabe-Nishimura C. Increased gene expression of glutathione peroxidase-3 in diabetic mouse heart. Biological and Pharmaceutical Bulletin. 2006;29(5):1042–1045. doi: 10.1248/bpb.29.1042. [DOI] [PubMed] [Google Scholar]
- 123.Cardinale D, Bacchiani G, Beggiato M, Colombo A, Cipolla CM. Strategies to prevent and treat cardiovascular risk in cancer patients. Seminars in Oncology. 2013;40(2):186–198. doi: 10.1053/j.seminoncol.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 124.Ichihara S, Yamada Y, Kawai Y, et al. Roles of oxidative stress and Akt signaling in doxorubicin cardiotoxicity. Biochemical and Biophysical Research Communications. 2007;359(1):27–33. doi: 10.1016/j.bbrc.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 125.Sumi D, Sasaki T, Miyataka H, Himeno S. Rat H9c2 cardiac myocytes are sensitive to arsenite due to a modest activation of transcription factor Nrf2. Archives of Toxicology. 2011;85(12):1509–1516. doi: 10.1007/s00204-011-0700-7. [DOI] [PubMed] [Google Scholar]
- 126.Shenkenberg TD, VonHoff DD. Mitoxantrone: a new anticancer drug with significant clinical activity. Annals of Internal Medicine. 1986;105(1):67–81. doi: 10.7326/0003-4819-105-1-67. [DOI] [PubMed] [Google Scholar]
- 127.Vávrová A, Popelová O, Štěrba M, et al. In vivo and in vitro assessment of the role of glutathione antioxidant system in anthracycline-induced cardiotoxicity. Archives of Toxicology. 2011;85(5):525–535. doi: 10.1007/s00204-010-0615-8. [DOI] [PubMed] [Google Scholar]
- 128.Nordgren KK, Wallace KB. Keap1 redox-dependent regulation of doxorubicin-induced oxidative stress response in cardiac myoblasts. Toxicology and Applied Pharmacology. 2013;274:107–116. doi: 10.1016/j.taap.2013.10.023. [DOI] [PubMed] [Google Scholar]
- 129.Menzel DB, Rasmussen RE, Lee E, et al. Human lymphocyte home oxygenase 1 as a response biomarker to inorganic arsenic. Biochemical and Biophysical Research Communications. 1998;250(3):653–656. doi: 10.1006/bbrc.1998.9363. [DOI] [PubMed] [Google Scholar]
- 130.Kumagai Y, Sumi D. Arsenic: signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annual Review of Pharmacology and Toxicology. 2007;47:243–262. doi: 10.1146/annurev.pharmtox.47.120505.105144. [DOI] [PubMed] [Google Scholar]
- 131.Jirkovský E, Popelová O, Křiváková-Staňková P, et al. Chronic anthracycline cardiotoxicity: molecular and functional analysis with focus on nuclear factor erythroid 2-related factor 2 and mitochondrial biogenesis pathways. Journal of Pharmacology and Experimental Therapeutics. 2012;343(2):468–478. doi: 10.1124/jpet.112.198358. [DOI] [PubMed] [Google Scholar]
- 132.Balogun E, Hoque M, Gong P, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochemical Journal. 2003;371(3):887–895. doi: 10.1042/BJ20021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Venkatesan N. Curcumin attenuation of acute adriamycin myocardial toxicity in rats. British Journal of Pharmacology. 1998;124(3):425–427. doi: 10.1038/sj.bjp.0701877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gorbunov N, Petrovski G, Gurusamy N, Ray D, Kim DH, Das DK. Regeneration of infarcted myocardium with resveratrol-modified cardiac stem cells. Journal of Cellular and Molecular Medicine. 2012;16(1):174–184. doi: 10.1111/j.1582-4934.2011.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jeong WS, Jun M, Kong ANT. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxidants and Redox Signaling. 2006;8(1-2):99–106. doi: 10.1089/ars.2006.8.99. [DOI] [PubMed] [Google Scholar]
- 136.Kensler TW, Curphey TJ, Maxiutenko Y, Roebuck BD. Chemoprotection by organosulfur inducers of phase 2 enzymes: dithiolethiones and dithiins. Drug Metabolism and Drug Interactions. 2000;17(1–4):3–22. doi: 10.1515/dmdi.2000.17.1-4.3. [DOI] [PubMed] [Google Scholar]
- 137.Seymour EM, Bennink MR, Bolling SF. Diet-relevant phytochemical intake affects the cardiac AhR and Nrf2 transcriptome and reduces heart failure in hypertensive rats. Journal of Nutritional Biochemistry. 2013;24(9):1580–1586. doi: 10.1016/j.jnutbio.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brewer AC, Murray TVA, Arno M, et al. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo . Free Radical Biology and Medicine. 2011;51(1):205–215. doi: 10.1016/j.freeradbiomed.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(35):15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dreger H, Westphal K, Weller A, et al. Nrf2-dependent upregulation of antioxidative enzymes: a novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiovascular Research. 2009;83(2):354–361. doi: 10.1093/cvr/cvp107. [DOI] [PubMed] [Google Scholar]
- 141.Sahni SK, Rydkina E, Sahni A. The proteasome inhibitor MG132 induces nuclear translocation of erythroid transcription factor Nrf2 and cyclooxygenase-2 expression in human vascular endothelial cells. Thrombosis Research. 2008;122(6):820–825. doi: 10.1016/j.thromres.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 142.Cui W, Bai Y, Luo P, Miao L, Cai L. Preventive and therapeutic effects of mg132 by activating nrf2-are signaling pathway on oxidative stress-induced cardiovascular and renal injury. Oxidative Medicine and Cellular Longevity. 2013;2013:10 pages. doi: 10.1155/2013/306073.306073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yu J, Zhao Y, Li B, Sun L, Huo H. 17β-Estradiol regulates the expression of antioxidant enzymes in myocardial cells by increasing Nrf2 translocation. Journal of Biochemical and Molecular Toxicology. 2012;26(7):264–269. doi: 10.1002/jbt.21417. [DOI] [PubMed] [Google Scholar]
- 144.Cai C, Teng L, Vu D, et al. The heme oxygenase 1 inducer (CoPP) protects human cardiac stem cells against apoptosis through activation of the extracellular signal-regulated kinase (ERK)/NRF2 signaling pathway and cytokine release. Journal of Biological Chemistry. 2012;287(40):33720–33732. doi: 10.1074/jbc.M112.385542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Worou ME, Belmokhtar K, Bonnet P, et al. Hemin decreases cardiac oxidative stress and fibrosis in a rat model of systemic hypertension via PI3K/Akt signalling. Cardiovascular Research. 2011;91(2):320–329. doi: 10.1093/cvr/cvr072. [DOI] [PubMed] [Google Scholar]
- 146.Di Filippo C, Marfella R, Cuzzocrea S, et al. Hyperglycemia in streptozotocin-induced diabetic rat increases infarct size associated with low levels of myocardial H0-1 during ischemia/reperfusion. Diabetes. 2005;54(3):803–810. doi: 10.2337/diabetes.54.3.803. [DOI] [PubMed] [Google Scholar]
- 147.Wang GG, Li W, Lu XH, Zhao X, Xu L. Taurine attenuates oxidative stress and alleviates cardiac failure in type 1 diabetic rats. Croatian Medical Journal. 2013;54(2):171–179. doi: 10.3325/cmj.2013.54.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gnecchi M, He H, Noiseux N, et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. The FASEB Journal. 2006;20(6):661–669. doi: 10.1096/fj.05-5211com. [DOI] [PubMed] [Google Scholar]
- 149.Zeng B, Ren X, Lin G, et al. Paracrine action of HO-1-modified mesenchymal stem cells mediates cardiac protection and functional improvement. Cell Biology International. 2008;32(10):1256–1264. doi: 10.1016/j.cellbi.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 150.Müller-Ehmsen J, Krausgrill B, Burst V, et al. Effective engraftment but poor mid-term persistence of mononuclear and mesenchymal bone marrow cells in acute and chronic rat myocardial infarction. Journal of Molecular and Cellular Cardiology. 2006;41(5):876–884. doi: 10.1016/j.yjmcc.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 151.Tang YL, Zhang YC, Qian K, Shen L, Phillips MI. Improved graft mesenchymal stem cell survival in ischemic heart with a hypoxia-regulated heme oxygenase-1 vector. Journal of the American College of Cardiology. 2005;46(7):1339–1350. doi: 10.1016/j.jacc.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 152.Shu T, Zeng B, Ren X, Li Y. HO-1 modified mesenchymal stem cells modulate MMPs/TIMPs system and adverse remodeling in infarcted myocardium. Tissue and Cell. 2010;42(4):217–222. doi: 10.1016/j.tice.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 153.Zeng B, Lin G, Ren X, Zhang Y, Chen H. Over-expression of HO-1 on mesenchymal stem cells promotes angiogenesis and improves myocardial function in infarcted myocardium. Journal of Biomedical Science. 2010;17(1, article 80) doi: 10.1186/1423-0127-17-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hatori N, Sjoquist P-, Marklund SL, Pehrsson SK, Ryden L. Effects of recombinant human extracellular-superoxide dismutase type C on myocardial reperfusion injury in isolated cold-arrested rat hearts. Free Radical Biology and Medicine. 1992;13(2):137–142. doi: 10.1016/0891-5849(92)90075-r. [DOI] [PubMed] [Google Scholar]
- 155.Sjoquist PO, Carlsson L, Jonason G, Marklund SL, Abrahamsson T. Cardioprotective effects of recombinant human extracellular-superoxide dismutase type C in rat isolated heart subjected to ischemia and reperfusion. Journal of Cardiovascular Pharmacology. 1991;17(4):678–683. doi: 10.1097/00005344-199104000-00023. [DOI] [PubMed] [Google Scholar]
- 156.Sjoquist P-O, Marklund SL. Endothelium bound extracellular superoxide dismutase type C reduces damage in reperfused ischaemic rat hearts. Cardiovascular Research. 1992;26(4):347–350. doi: 10.1093/cvr/26.4.347. [DOI] [PubMed] [Google Scholar]
- 157.Hatori N, Sjöquist P-O, Marklund SL, Ryden L. Effects of recombinant human extracellular-superoxide dismutase type C on myocardial infarct size in pigs. Free Radical Biology and Medicine. 1992;13(3):221–230. doi: 10.1016/0891-5849(92)90018-c. [DOI] [PubMed] [Google Scholar]
- 158.Konkalmatt PR, Beyers RJ, O'Connor DM, Xu Y, Seaman ME, French BA. Cardiac-selective expression of extracellular superoxide dismutase after systemic injection of adeno-associated virus 9 protects the heart against post-myocardial infarction left ventricular remodeling. Circulation: Cardiovascular imaging. 2013;6(3):478–486. doi: 10.1161/CIRCIMAGING.112.000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu TCK, Ismail S, Brennan O, Hastings C, Duffy GP. Encapsulation of cardiac stem cells in superoxide dismutase-loaded alginate prevents doxorubicin-mediated toxicity. Journal of Tissue Engineering and Regenerative Medicine. 2013;7(4):302–311. doi: 10.1002/term.523. [DOI] [PubMed] [Google Scholar]