

Abstract
Mitochondrial function is challenged by toxic byproducts of metabolism as well as by pathogen attack1,2. Caenorhabditis elegans normally responds to mitochondrial dysfunction with activation of mitochondrial repair, drug detoxification, and pathogen-response pathways1–7. From a genome-wide RNAi screen, we identified 45 C. elegans genes that are required to upregulate detoxification, pathogen-response, and mitochondrial repair pathways after inhibition of mitochondrial function by drugs or genetic disruption. Animals defective in ceramide biosynthesis are deficient in mitochondrial surveillance, and addition of particular ceramides can rescue the surveillance defects. Ceramide can also rescue the mitochondrial surveillance defects of other gene inactivations, mapping these gene activities upstream of ceramide. Inhibition of the mevalonate pathway, either by RNAi or statin drugs also disrupts mitochondrial surveillance. Growth of C. elegans with a significant fraction of bacterial species from their natural habitat causes mitochondrial dysfunction. Other bacterial species inhibit C. elegans defense responses to a mitochondrial toxin, revealing bacterial countermeasures to animal defense.
Mitochondria are almost entirely composed of proteins encoded by the nuclear genome8. A surveillance pathway detects mitochondrial defects to induce mitochondrial chaperone genes hsp-6 and hsp-60 and xenobiotic detoxification and pathogen-response pathways1–7. Disrupting C. elegans mitochondrial function induces drug detoxification genes such as cyp-14A3 and ugt-61 (Figure 1a-c), as well as a Pseudomonas pathogen-response gene irg-19 (Figure 1d). This suggests that animals interpret a disruption of mitochondrial activity as a xenobiotic or pathogen attack. Consistent with this, C. elegans exhibits bacterial avoidance behavior when mitochondria are inhibited by RNAi or antimycin, an inhibitor of mitochondrial electron transport produced by Streptomyces1 (Figure 1e and 1f). Animals also show aversive behaviors in the absence of any drug or mitochondrial gene inactivation if they carry a mutation of the nuclearly-encoded mitochondrial gene isp-1(qm150) (Figure 1g). This mutation also activates hsp-6 (Figure 1h).
Figure 1.

Mitochondrial dysfunction activates homeostatic, detoxification and pathogen responses. a, mitochondrial chaperone and detoxification transcripts in control vs. spg-7(RNAi) (n=3). Error bars represent s.d. b-d, hsp-6p::gfp (b), ugt-61p::gfp (c), and irg-1p::gfp (d) animals raised on control E. coli, or E. coli with antimycin or spg-7 (RNAi). e-g, Drug-, RNAi- or mutant allele-based mitochondrial inhibition causes food avoidance. h, wild type hsp-6p::gfp and isp-1(qm150); hsp-6p::gfp animals.
C. elegans live in microbe-rich environment10. We isolated microbes from habitats with wild C. elegans populations10, classified them based on their 16S ribosomal sequences (Samuel BS, Félix MA and Ruvkun G, unpublished), and found that 18% of the 560 strains tested caused mitochondrial stress as revealed by induction of hsp-6p::gfp (Figure 2a-c). This suggests that mitochondria are targeted by many bacterial species, and explains the evolution of coupling detection of C. elegans mitochondrial dysfunction to antibacterial gene expression and behavioral responses. Iron is a valuable resource to microbes which often produce siderophores to capture the iron; the eukaryotic mitochondrion, rich in heme and iron sulfur proteins, is an attractive iron depot for bacteria. Bacterial toxins, siderophores or virulence factors from these many bacterial strains may target the mitochondrion.
Figure 2.

Some bacteria from the C. elegans natural habitat antagonize the mitochondria. a, Genera that induce hsp-6p::gfp [100 of 560 tested microbes (18%)]. b, Proportion of bacterial genera tested that cause mitochondrial dysfunction and hsp-6p::gfp induction. c, hsp-6p::gfp animals raised on E. coli or natural microbial species. d, Six bacterial strains that render C. elegans defective in hsp-6 response to antimycin. e, hsp-6p::gfp animals exposed to antimycin and raised on control E. coli or the six microbial strains.
To identify the molecular elements of how animal mitochondrial dysfunction is detected and coupled to a detoxification response, we performed a genome-wide RNAi screen for gene inactivations that render C. elegans unresponsive to mitochondrial dysfunction (Extended Data Figure 1 and 2). Gene inactivations that caused a failure to induce hsp-6p::gfp in antimycin were re-screened for failure to upregulate other genes normally induced by mitochondrial dysfunction: the detoxification gene ugt-61::gfp in antimycin, hsp-6p::gfp induction in the isp-1(qm150), or hsp-6p::gfp induction by ATP synthase atp-2 RNAi. Mitochondrial surveillance and response was disrupted by 45 gene inactivations (Extended Data Table 1), including the known components atfs-1, clpp-1 and dve-1 4,6.
One of the gene inactivations that potently disrupts response to mitochondrial dysfunction, sptl-1, encodes serine palmitoyl transferase, in the sphingolipid biosynthesis pathway (Extended Data Figure 3). Inactivation of sptl-1 by RNAi inhibited the induction of hsp-6p::gfp upon antimycin treatment or spg-7(RNAi) (Figure 3a and 3b), whereas sptl-1(RNAi) alone without antimycin treatment did not induce the expression of hsp-6p::gfp (Extended Data Figure 4b). sptl-1(RNAi) did not affect the activation of ER stress reporter hsp-4p::gfp by the drug tunicamycin (Figure 3c), indicating a specific role in mitochondrial surveillance. A probable null mutation in sptl-1 (Extended Data Figure 5a) impaired the induction of hsp-6, cyp-14A3, and ugt-61 after mitochondrial disruption by spg-7(RNAi) (Figure 3d). A double mutant in the two ceramide synthase genes of the sphingolipid biosynthetic pathway also attenuated the induction of hsp-6 upon mitochondrial damage (Extended Data Figure 5b). Treatment with myriocin, a fungal inhibitor of mammalian serine palmitoyl transferase, disrupted antimycin-induced hsp-6p::gfp induction (Figure 3e). sptl-1(RNAi) (with normal movement, Supplementary Video S1 and S2) attenuated food avoidance induced by antimycin or spg-7(RNAi), suggesting that this behavioral response is also coupled to sptl-1 (Figure 3g, Extended Data Figure 4d and 4e). sptl-1 expression is up-regulated 2.5-fold by mitochondrial damage (Extended Data Figure 4a), suggesting that increased sphingolipids during mitochondrial disruption may act as a signal rather than as a membrane component required for another signal.
Figure 3.

The serine palmitoyl transferase sptl-1 is required for mitochondrial surveillance. a, hsp-6p::gfp animals raised on control or sptl-1(RNAi) in the presence or absence of antimycin. b, GFP immunoblot expressed by hsp-6p::gfp animals in the presence or absence of antimycin or spg-7(RNAi). c, hsp-4p::gfp animals raised on control or sptl-1(RNAi) in the presence or absence of the ER drug tunicamycin. d, C. elegans hsp-6 and xenobiotic detoxification gene transcripts in control or sptl-1(ok1693) mutant animals after exposure to spg-7(RNAi) (n=2). e, hsp-6p::gfp animals raised in the presence or absence of myriocin. f, Body wall muscle of sptl-1(RNAi) compared to wild type animals expressing a mitochondrially localized GFP reporter. g, Food avoidance phenotypes for control or sptl-1(RNAi) animals treated with antimycin. h, Wild type or isp-1(qm150) raised on control or sptl-1(RNAi).
The morphology of the normally extensive mitochondrial network of C. elegans and other animals is responsive to mutation or inactivation of mitochondrial components11. Mitochondria in sptl-1(RNAi) animals hyperfused into larger, longer mitochondria (Figure 3f), suggesting that sptl-1 is critical for mitochondrial homeostasis. The isp-1 mitochondrial mutant had a severe synthetic growth defect on sptl-1(RNAi), whereas wild type development was only slightly delayed on sptl-1(RNAi) (Figure 3h). Sphingolipid signaling may act in the homeostatic response to the mitochondrial defect caused by the isp-1(qm150) mutation; in the absence of that response, isp-1(qm150) may cause a more severe mitochondrial defect and growth arrest. sptl-1(RNAi) animals were also more sensitive to the mitochondrial inhibitor antimycin (Extended Data Figure 4f). Additionally, sptl-1(RNAi) animals were unable to sense mitochondrial inhibition and activate the mitochondrial surveillance pathway when challenged by a set of the wild microbes that disrupt mitochondrial function (Extended Data Figure 4g).
Ceramide supplementation rescued the sptl-1 defect in mitochondrial surveillance (Figure 4a and Extended Data Figure 5d), whereas ceramide in the absence of mitochondrial damage did not induce hsp-6p::gfp (Extended Data Figure 5e), suggesting that ceramide is not a sufficient signal to induce the suite of responses to mitochondrial dysfunction in the absence of true mitochondrial dysfunction. Ceramide supplementation also partially rescued the mitochondrial morphology defect (Figure 4b) and the impaired food avoidance behavior caused by sptl-1(RNAi) (Extended Data Figure 6a). Dihydroceramide, a ceramide precursor with no signaling function in mammals, could not rescue the sptl-1(RNAi) mitochondrial surveillance defect (Figure 4a). Two C. elegans ceramide synthases HYL-1 and HYL-2 synthesize different ceramide species, C24 to C26, and C20 to C22 ceramides, respectively12. Testing C16, C20, C22 and C24 ceramides, only C24 ceramide rescued the deficiency of mitochondrial surveillance caused by sptl-1(RNAi) (Figure 4c).
Figure 4.

Ceramide biogenesis is required for mitochondrial surveillance. a, hsp-6p::gfp animals exposed to antimycin in the presence or absence of dihydroceramide or ceramide. b, Body wall muscle of animals expressing a mitochondrially localized GFP reporter in sptl-1(RNAi) with and without added ceramide. c, hsp-6p::gfp sptl-1(RNAi) plus antimycin with different ceramide species. d, Dissected animals after antimycin or spg-7(RNAi) treatment were stained with anti-COX-IV (red) and anti-ceramide antibodies (green). Quantification represents proportion of mitochondria with contact of mitochondrial and ceramide staining (mean ± s.d., n=3). e, atfs-1(tm4525); hsp-60p::gfp animals alone or with antimycin. f, atfs-1(tm4525); hsp-16p::ATFS-1Δ1-32.myc; hsp-60p::gfp animals raised on control or sptl-1(RNAi).
During apoptosis and mitophagy, ceramide accumulates on the outer membrane of mitochondria13–15. Using antibodies to stain ceramide and the mitochondrial protein oxidative phosphorylation Complex IV (COX-IV), ceramide and mitochondrial protein colocalization dramatically increased after C. elegans mitochondrial inhibition (Figure 4d). The increased colocalization preceded induction of hsp-6p::gfp (Extended Data Figure 6b and 6c). Thus, ceramide may participate in an early step of mitochondrial surveillance by marking domains of dysfunction.
To map ceramide relative to other gene inactivations that render animals unable to respond to mitochondrial damage, we tested if ceramide could rescue the mitochondrial surveillance defects of other hits from the RNAi screen. Five other gene inactivations were rescued by ceramide: ran-4, a nuclear transport component; Y47G6A.29, a phosphatidyl inositol signaling component; F40F12.7, a zinc finger protein; Y54E10BR.5, a signal peptidase component; and ceh-20, a homeobox transcription factor (Extended Data Figure 6e). These gene inactivations may disrupt mitochondrial surveillance upstream of the production of ceramide.
ATFS-1 is a transcription factor that activates hsp-6 and hsp-60 during mitochondrial stress (Figure 4e)5. ATFS-1 contains an N-terminal mitochondrial targeting sequence as well as a nuclear localization signal; when mitochondria are damaged, nuclear accumulation of ATFS-1 is favored6. Deletion of the N-terminal 1–32 amino acid causes constitutive nuclear accumulation of ATFS-1 (Extended Data Figure 6d) and activates hsp-60p::gfp6. Activation of hsp-60p::gfp by ATFS-1Δ1–32.myc did not require sptl-1 gene activity (Figure 4f), suggesting that ceramide works upstream of ATFS-1 and plays a role in the early detection of mitochondrial dysfunction.
hmgs-1 which encodes HMG-CoA (3-hydroxy-3-methyl-glutaryl-CoA) synthase of the mevalonate synthetic pathway is also a strong hit from the genome-wide screen. hmgs-1 gene inactivation inhibited antimycin-induced hsp-6p::gfp induction and food avoidance (Extended Data Figure 7a-d). hmgs-1(RNAi) also induced abnormal mitochondrial morphology (Extended Data Figure 7e). Supplementing mevalonate to hmgs-1(RNAi) animals rescued the deficiency of antimycin-induced hsp-6p::gfp (Extended Data Figure 7f).
Statins are cholesterol-lowering drugs that inhibit the mevalonate pathway (Extended Data Figure 8a)16. Treating C. elegans with statins also abrogates their ability to sense mitochondrial damage and activate protective programs (Extended Data Figure 7g and 8b). A common side effect of statin therapy is muscle toxicity17. By inhibiting the mevalonate pathway, statins inhibit ubiquinone synthesis, a key component of the electron transport chain (Extended Data Figure 8a). Our data suggests that a combined inhibition of ubiquinone synthesis and mitochondrial surveillance may contribute to the muscle toxicity of statins. In support of a role for mitochondrial surveillance in statin action, a gain-of-function mutation in C. elegans atfs-1 confers statin resistance18. Treating human embryonic kidney HEK293T cells with statin impaired the mitochondrial network and morphology (Extended Data Figure 7h and 9). Statin treatment also decreased ATP production in mouse C2C12 myotubes (Extended Data Figure 7i).
Eukaryotic mitochondrial surveillance pathway components are likely to be targets of microbial toxins and virulence factors. If the animal surveillance of the mitochondria is disabled by a bacterial toxin or virulence factor, other anti-mitochondrial toxins, siderophores, or virulence factors would be rendered more effective. To detect such bacterial anti-surveillance activities, we screened our collection of C. elegans flora for bacterial species which when co-cultured with C. elegans could disrupt the induction of hsp-6p::gfp by antimycin. Six wild bacterial strains of the genus Pseudomonas from three species (vranovensis, brenneri and asplenii) disrupt mitochondrial surveillance (Figure 2d and 2e). The dozens of C. elegans genes we have identified in the mitochondrial surveillance pathway are candidate targets for toxins or virulence factors from these Pseudomonas strains.
Our studies have revealed roles of ceramide and mevalonate in mitochondrial surveillance. The products of these pathways either constitute signals that are transferred within or between cells, or structures within those cells that are necessary for other signals to emerge. We favor that these constitute signals because the expression of these biosynthetic pathways are induced by the mitochondrial insults (Extended Data Figure 4a) and because these molecules are localized to the site of injury (Figure 4d). Ceramides are upstream elements of the stress response of plasma membrane, ER and mitochondria in multiple species19,20, and can act as hormonal signals21. But it is also possible that these constitute membrane elements required for other signals. Variation in the animal pathways for mitochondrial surveillance that we have identified, many of which encode conserved proteins, may underlie variation in the symptoms causes by the same mitochondrial mutations or variation in response to mitochondrial toxins such as the statins. Microbial secondary metabolites that render animals unresponsive to their mitochondrial dysfunctions (Figure 2d and 2e), may have therapeutic potential in the treatment of a predicted up-regulation of detoxification and anti-bacterial pathways which may contribute to the devastating symptoms of some mitochondrial diseases. The anti-surveillance activities of such natural products may also treat other disorders of dysregulated detoxification and innate immunity, such as autoimmunity.
Methods
C. elegans strains
hsp-6p::gfp(zcIs13)V, dpy-5(e907)I; ugt-61p::gfp(sEX11571), hsp-4p::gfp (zcIs4)V, myo-3p::GFPmt(zcIs14), isp-1(qm150), sptl-1(ok1693)II, tag-38(ok490)V, lagr-1(gk327) I; hyl-1(ok976) IV, sphk-1(ok1097) II, hyl-2(ok1766) X, hyl-2(gnv1) X, and asm-3(ok1744) IV were obtained from the Caenorhabditis Genetics Center (CGC). irg-1p::gfp was provided by Fred Ausubel. isp-1; hsp-6p::gfp was generated by crossing hsp-6p::gfp into isp-1(qm150). atfs-1; hsp-60p::gfp, atfs-1(tm4525); hsp-16p::atfs-1Δ1-32.myc::gfp and atfs-1(tm4525); hsp-16p::ATFS-1Δ1-32.myc; hsp-60p::gfp were provided by Cole Haynes. Hermaphrodites were used throughout of the study.
Activation of GFP reporter by wild microbes
Wild microbes isolated from various habitats harboring wild C. elegans populations10 (Samuel BS, Félix MA and Ruvkun G, unpublished) were grown in LB at 18°C shaking for 16–18hrs, concentrated 3X and seeded (30ul) into 24-well NGM plates (no antibiotics) in duplicate. Plates were dried and allowed to grow overnight at room temperature before ~40 synchronized L1 worms were added to the wells. Animals were scored after 48hrs at 20°C. Genera with more than 3 independent isolates that induced hsp-6p::gfp expression are noted along with the percentage of total microbes tested (Figure 2a). Other genera that strongly activate hsp-6p::gfp expression, include Achromobacter, Curtobacterium, Enterobacter, Leucobacter, Mycetocola, Myroides, Raoultella and Rhodococcus.
Activation of GFP reporter by RNAi
RNAi clones were grown in LB containing 50ug/ml carbenicillin at 37 °C overnight and seeded 100ul/well to 24-well worm plates with 5mM IPTG. Dried plates were kept at room temperature overnight to allow IPTG induction of dsRNA expression. Synchronized L1 worms (~40 worms/well) were raised on the RNAi plate at 20 °C. Fluorescence was assayed at 48 hours.
Activation of GFP reporter by drugs
Synchronized L1 worms (~40 worms/well) were raised on a 24-well plate at 20 °C for 48 hours. At this time, each well was treated with 0.5ug antimycin (total volume 20ul) in M9 buffer (3g KH2PO4, 6g Na2HPO4, 5g NaCl, 1ml 1M MgSO4, H2O to 1L), or 1.5ug tunicamycin in DMSO (total volume 20ul). GFP expression was assayed after 24 hours (antimycin), or 7 hours (tunicamycin).
Microscopy
Comparable GFP reporter images were obtained by a Zeiss AxioImager Z1 using the same exposure time. Mitochondrial morphology of C. elegans or HEK293T cells was visualized under an Olympus Fluoview 1000 confocal microscope.
Genome-wide RNAi screen
Primary screen was performed by seeding individual bacterial clones each bearing a distinct C. elegans dsRNA to initiate RNAi onto 24-well RNAi plates. Dried plates were kept at room temperature overnight to induce the expression of dsRNAs. Synchronized L1 hsp-6p::gfp worms (~40 animals/well) were raised on the 24-well plate at 20 °C for 48 hours. At this time, each well was treated with 0.5ug antimycin in M9 buffer (total volume 20ul). GFP expression was assayed after 24 hours. Scores were recorded from 0 (no inhibition of GFP expression) to 4 (strong inhibition of GFP expression).
Positive clones from the primary screen were picked and re-screened in multiple parallel secondary screens. For one screen, synchronized L1 hsp-6p::gfp worms (~40 animals/well) were raised on 24-well plate at 20 °C for 36 hours. At this time, each well was treated with 20-fold concentrated E. coli expressing atp-2 dsRNA (total volume 20ul), to inactivate the mitochondria using RNAi rather than using the antimycin of the primary screen. GFP expression was assayed after 36 hours. In a third test, synchronized L1 isp-1(qm150); hsp-6p::gfp (2 animals/well) were dropped onto 24-well plate using a worm sorter, and raised at 20 °C. This strain carries a mitochondrial mutation that activates hsp-6p::gfp expression in untreated animals as well as in most wells. Fluorescence were scored when they reached L4 stage and again when their progeny (if the strains were fertile) developed to the L4 stage. In a fourth parallel screen, synchronized L1 ugt-61p::gfp worms (~40 animals/well) were raised on 24-well plate at 20 °C for 48 hours. At this time, each well was treated with 0.5ug antimycin in M9 (total volume 20ul). GFP expression was assayed after 24 hours.
Western blotting
Synchronized worms were raised on plates under described conditions. Worms were then washed off plates, resuspended with NuPAGE® LDS Sample Buffer (Invitrogen) and boiled at 95°C for 5 min. Lysates containing the same amount of protein were loaded onto SDS-PAGE and transferred onto Nitrocellulose membrane (Invitrogen). After blocked with 5% nonfat milk, the membrane was probed with the designated first and second antibodies and developed with the enhanced chemiluminescence method (Perkin Elmer) and visualized by X-ray film.
RNA isolation and quantitative RT- PCR
Synchronized L1 wild-type N2 and mutant worms were raised on control or spg-7(RNAi) at 20 °C, and harvested at the L4 stage. Worms were then washed, resuspended with Trizol reagent (Invitrogen), frozen and homogenized by grinding. Total RNA was isolated by chloroform extraction, followed by ethanol precipitation and DNase treatment (Ambion, Turbo DNA free kit). cDNA was then synthesized by reverse transcription (Invitrogen, SuperScript® III First-Strand Synthesis System). Quantitative real-time PCR was carried out using SYBR GREEN PCR Master Mix (Bio-Rad). Quantification of transcripts was normalized to rpl-32 and relative expression levels were calculated as previously described22.
Food avoidance assays
Each gene inactivation that disrupted the response to mitochondrial dysfunction was grown in 1ml LB containing 50ug/ml carbenicillin at 37 °C overnight, pelleted, resuspended with 50ul LB and dropped in the center of a well of 6-well plates containing 5mM IPTG. Dried plates were kept at room temperature overnight to allow IPTG induction of dsRNA expression. Only circular lawns of uniform size and density were used for food avoidance assays. Synchronized L1 worms (~100 animals/well) were dropped in the center of each bacterial lawn. Food avoidance phenotypes were scored at 48 hours. For antimycin treatment, synchronized L1 worms (~100 animals/well) were dropped in the center of each bacterial lawn, and grown at 20 °C for 48 hours. At this time, 50ug antimycin (total volume 80ul) was added directly to the bacteria lawn. Food avoidance was score 8 hours after drug treatment.
Synthetic growth defect on sptl-1(RNAi)
Wild type N2 or mitochondrial mutant allele isp-1 animals were synchronized at the L1 stage. Animals were then raised on control or sptl-1(RNAi). isp-1 animals were grown on control RNAi for 3 days vs. on sptl-1(RNAi) for 5 days to match the developmental stage of wild type N2 animals.
Ceramide biosynthetic mutant analyses
To dissect the pathway of sptl-1 function in mitochondrial surveillance, we tested other mutations in the sphingolipid biosynthetic pathway (Extended Data Figure 4 and 5a). Inactivation of another serine palmitoyl transferase gene sptl-3 had no effect on the induction of hsp-6 upon mitochondrial damage (Extended Data Figure 4, 5a and 5b). A double mutation of hyl-1 and lagr-1, which encode ceramide synthases, reduced the induction of hsp-6 upon mitochondrial damage (Extended Data Figure 5b), whereas hyl-1 or lagr-1 single mutants were less defective (Extended Data Figure 5c). Only the C24 ceramide, synthesized by HYL-1, rescued the deficiency of mitochondrial surveillance caused by sptl-1(RNAi). This is consistent with the defect in hsp-6 induction shown in the hyl-1; lagr-1 double mutant (Extended Data Figure 5b).
Immunostaining
Immunostaining of dissected animals was carried out according to Philips et al23 with minor modifications. Specifically, dissected animals were blocked with 0.5% BSA in PBST for 1 hour at room temperature, and stained with anti-ceramide antibody [MID15B4, Alexis Biochemicals diluted 1:60 (v/v)] in 0.2% BSA in PBST and anti-human OxPhos Complex IV antibody [Invitrogen diluted 1:30 (v/v)] at room temperature for half hour and 4°C overnight. Followed three washes with PBST (10 min each time), slides were then incubated with Cy-3 anti-mouse secondary antibody [diluted 1:200 (v/v)] for 1 hour at room temperature. After three washes again with PBST (10 min each time), slides were mounted and visualized under an Olympus Fluoview 1000 confocal microscope.
HEK293T cells (ATCC) were washed twice with ice cold PBS and fixed with 4% formaldehyde at 4°C for 1 hour. After fixation, cells were permeabilized by 0.1% Triton in PBS at room temperature for 8 minutes, and then blocked with 0.5% BSA in PBST for 1 hour at room temperature, and stained with anti-human OxPhos Complex IV antibody [Invitrogen diluted 1:30 (v/v)] at room temperature for half hour and 4°C overnight. Following three washes with PBST (10 min each time), slides were mounted and visualized under an Olympus Fluoview 1000 confocal microscope.
ATP levels
C2C12 myoblasts (ATCC) were grown in Dulbecco’s Modified Eagle Medium supplemented with 10% (v/v) fetal bovine serum and antibiotics (100 mg/mL penicillin/streptomycin mix) at 37 °C. Differentiation into myotubes was induced at ~90% density by changing the medium to DMEM supplemented with 2% (v/v) horse serum. Differentiation occurred after 5 days. Myotubes were then treated with DMSO, 10 μM simvastatin or 10uM mevastatin. After incubation for 48 h, CellTiter-Glo reagent (Promega) was added to cell-culture medium, and luminescence was measured after 10 min incubation.
Primers for quantitative RT-PCR
| Detects | Forward primer, Reverse primer |
|---|---|
| hsp-6 | CAAACTCCTGTGTCAGTATCATGGAAGG, GCTGGCTTTGACAATCTTGTATGGAACG |
| cyp-14A3 | CAGTTTCCCGCCGAAAACATCCATTTG, CAATGCCGTTCTTCTTTGAAGCCTCCAG |
| ugt-61 | GCAATTGGAGGTCATGACGTAACTATG, GCGAAGAATGATTCGGCATCCATCTTG |
| rpl-32 | AGGGAATTGATAACCGTGTCCGCA, TGTAGGACTGCATGAGGAGCATGT |
| sptl-1 | CTGAAAGGCAGAAAGATGAATTAATTGC, GAATCCACGTGGCCCGCACGATCCTACG |
Primers for genotyping
| gene name | Forward primer, Reverse primer |
|---|---|
| sptl-1 | AGCCCAAGCCAATTATCCTT, AACACGAACTTTGAATCGCC |
| sptl-3 | CTTGGTGTCCCTTTCGTGTT, AGGGCAAGAATTGGGGTAAT |
| F27E5.1 | TGAAAGACAACTTGCTCGGA, TGTCTTTTCAGCAGTCACCG |
| T10B11.2 | TCATTCCGACGGTACCATTT, TGAAGCTTGAAATGCAGTGG |
| asm-3 | CTTGCACTCCTCCTTTCCAC, GGTGACAGAATGCGAGGAAT |
| hyl-1 | GCCCCGTAAATAAGCACAAA, TCGTGTTCTTTCACGTCTCG |
| hyl-2 | GGGGGAGTGATGGAAGAAAT, TTGCAAACCAATTGCAAGAA |
| lagr-1 | ATGCTTGGACCTGAATAC, TTACACGTTCTTCGGTTTAAG |
| sphk-1 | ATGTTCATAGTAGTGGTAAC, CTAGGCAGTTGATGAGAAAACG |
Extended Data
Extended Data Figure 1.

Mitochondrial dysfunction activates homeostatic, detoxification and pathogen responses. a, Drug-induced food avoidance phenotypes on live or dead bacteria. Dead bacteria were obtained by heating bacteria at 90 °C for 30 min. Avoidance behavior was also observed when antimycin was added to dead bacteria, showing that the drug acts directly on C. elegans and is not transformed by the bacteria. b, Quantification of the food avoidance phenotypes of Extended Data Figure 1a (n=4). Error bars represent s.d. c, A dose response of hsp-6p::gfp induction with the addition of antimycin.
Extended Data Figure 2.

Diagram of the genome-wide RNAi screen workflow. For the detailed experimental procedure, see Methods.
Extended Data Figure 3.

Diagram of the sphingolipid metabolism pathway and the corresponding genes.
Extended Data Figure 4.

sptl-1 is required for mitochondrial surveillance. a, A graph showing the fold change in C. elegans sptl-1 transcript level compared to control (n=3). Error bars represent s.d. b, hsp-6p::gfp worms raised on control or sptl-1(RNAi) (n=40). c, Body wall muscle animals expressing a mitochondrially localized GFP reporter. The animals were subjected to sptl-1(RNAi) for 36 hours and transferred onto the second RNAi (control, fzo-1, fis-1 or drp-1). The hyperfusion of mitochondria observed under sptl-1(RNAi) is dependent on the fusion machinery as disruption of mitochondrial fusion by inactivating fzo-1 or fis-1 partially restored the tubular structure. In contrast, inhibition of the gene drp-1 that governs fission led to mitochondrial hyperfusion. The images were taken 36 hours after they were placed on the second RNAi. d, A graph showing the percentage of worms which avoid the spg-7(RNAi) bacteria lawn 48 hours after they were initially placed on the plates (n=4). Error bars represent s.d. e, A graph showing the percentage of worms which avoid the bacteria lawn 8 hours after the addition of antimycin (n=4). Error bars represent s.d. f, wild type N2 or sptl-1(ok1693) mutant animals raised in the presence or absence of 0.8μM antimycin. Photos were taken 4 days after the synchronized L1 worms were placed on the plates. g, hsp-6p::gfp animals were raised on sptl-1(RNAi) for 36 hours and transferred onto a subset of wild microbes. The images were taken after 2 days.
Extended Data Figure 5.

Ceramide biosynthesis is required for mitochondrial surveillance. a, Genotyping of the sphingolipid metabolism pathway mutant alleles. b-c, Fold difference in hsp-6 transcript levels in wild type or sphingolipid metabolism pathway mutants (b) and hyl-1 or lagr-1 mutants (c) (n=3). Error bars represent s.d. d, hsp-6p::gfp worms raised on sptl-1(RNAi) in the presence of increasing amounts of ceramide. e, hsp-6p::gfp in the presence or absence of ceramide.
Extended Data Figure 6.

Ceramide biogenesis is required for mitochondrial surveillance. a, The percentage of worms that avoid the bacterial lawn 8 hours after the addition of antimycin. Animals were pre-treated with control RNAi, control RNAi with ceramide, sptl-1 RNAi or sptl-1 RNAi with ceramide (n=4). Error bars represent s.d. b, Time course experiment for the induction of hsp-6p::gfp with antimycin. c, Dissected young adults after 4 hours antimycin treatment were stained with anti-COX-IV antibody (red) and anti-ceramide antibody (green). d, Nomarski (upper panel) and fluorescent (lower panel) images of intestinal cells in atfs-1; hsp-16p::atfs-1Δ1-32.myc::gfp transgenic animals. e, hsp-6p::gfp worms raised on indicated RNAi in the presence or absence of ceramide.
Extended Data Figure 7.

Inhibition of the mevalonate pathway disrupts mitochondrial surveillance. a, hsp-6p::gfp animals raised on control or hmgs-1(RNAi) in the presence of antimycin. b, Immunoblotting of GFP expressed by hsp-6p::gfp animals, with or without antimycin. c, Antimycin induced food avoidance in control or hmgs-1(RNAi) animals. d, Quantification of food avoidance (n=4). Error bars represent s.d. e, Body wall muscle of control or hmgs-1(RNAi) animals expressing a mitochondrially localized GFP reporter. f, hsp-6p::gfp animals raised on hmgs-1(RNAi), or hmgs-1(RNAi) with addition of mevalonate exposed to antimycin. g, hsp-6p::gfp animals treated with antimycin after pre-treatment with simvastatin or mevastatin. h, Mitochondrial immunostaining in HEK293T cells. i, ATP levels in C2C12 myotubes after treating with simvastatin or mevastatin (n=3). Error bars represent s.d.
Extended Data Figure 8.

a, Diagram of the mevalonate pathway for the biosynthesis of cholesterol, ubiquinone and heme A, and protein N-glycosylation and prenylation. b, hsp-6p::gfp animals treated with antimycin after pre-treatment with increasing concentration of simvastatin. c, hsp-6p::gfp animals with mock, 80ug/ml simvastatin or 80ug/ml mevastatin treatment. d, hsp-6p::gfp animals raised on control RNAi, hmgs-1 RNAi, or hmgs-1 RNAi with the addition of geranylgeranyl pyrophoshate. The animals were then treated with antimycin to induce mitochondrial damage. Statin toxicity has been proposed to be caused by the inhibition of Rab prenylation24. Geranylgeranyl pyrophosphate (GGPP), a precursor of protein prenylation rescued the statin side effect in cell culture24. GGPP also partially rescued the deficiency of mitochondrial surveillance and activated hsp-6p::gfp in antimycin-treated hmgs-1(RNAi) animals.
Extended Data Figure 9.

Mitochondrial immunostaining in HEK293T cells. The cells were treated with DMSO, 10uM simvastatin or 10uM mevastatin for 2 days.
Extended Data Table 1.
Full list of genes identified in the genome-wide RNAi screen for mitochondrial surveillance. The table was sorted by the severity in the defect of the reporter gene induction. The intensity of blue denotes the severity of the defect in mitochondrial inactivation-induced gene response for each gene-inactivation, with darker blue showing the most severe failure to upregulate the response genes. Yellow color indicates previously reported genes for mitochondria unfolded response. N/A means the gene inactivations render the isp-1; hsp-6::gfp animals sick or lethal, which prevented assessment of GFP levels. The synthetic lethality of these mitochondrial surveillance defective gene inactivations with a mitochondrial defect in isp-1(qm150) endorses their role in the homeostatic response that may allow the isp-1(qm150) mutant to be viable.
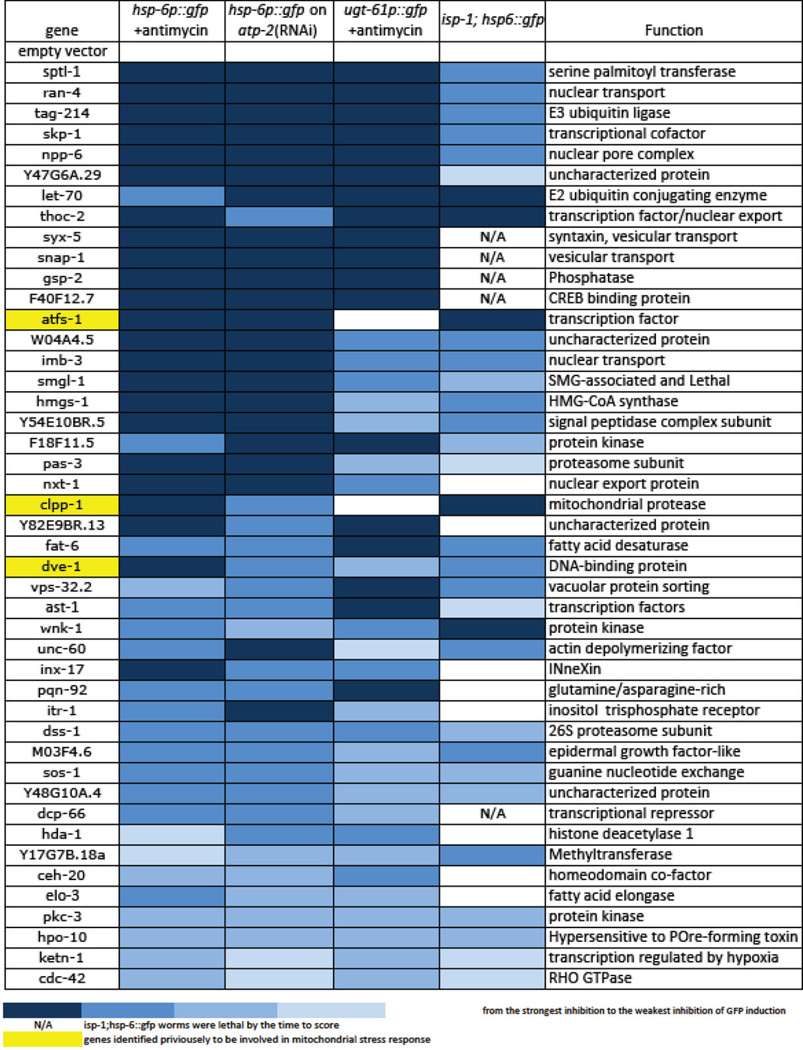 |
Supplementary Material
Acknowledgements
We thank Drs. C. Haynes (Memorial Sloan-Kettering Cancer Center), F. Ausubel (Massachusetts General Hospital), and the Caenorhabditis Genetics Center for providing strains; and V. Mootha lab (Massachusetts General Hospital) for providing the C2C12 cells. We thank J. Larkins-Ford, C. Phillips, S. Garcia, and M. Pellegrino for technical support, as well as MA. Félix for bacterial strains and for guidance in the collection of microbial strains from C. elegans natural habitats. Y.L. is supported by the Helen Hay Whitney research fellowship; B.S.S is supported by the Charles King Trust postdoctoral fellowship. The work is supported by a grant from the National Institute of Health awarded to G.R. (NIH AG043184-16).
Footnotes
Author Contributions Y.L. and G.R. designed experiments; Y.L., B.S.S. and P.C.B. carried out experiments. Y.L., B.S.S. and G.R. wrote the paper. G.R. supervised the project.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper. Correspondence and requests for materials should be addressed to G.R. (ruvkun@molbio.mgh.harvard.edu).
References and notes
- 1.Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149:452–466. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Runkel ED, Liu S, Baumeister R, Schulze E. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS genetics. 2013;9:e1003346. doi: 10.1371/journal.pgen.1003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 5.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chainmediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochimica et biophysica acta. 2013;1833:410–416. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estes KA, Dunbar TL, Powell JR, Ausubel FM, Troemel ER. bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2153–2158. doi: 10.1073/pnas.0914643107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felix MA, Duveau F. Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC biology. 2012;10:59. doi: 10.1186/1741-7007-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SS, et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nature genetics. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 12.Menuz V, et al. Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science. 2009;324:381–384. doi: 10.1126/science.1168532. [DOI] [PubMed] [Google Scholar]
- 13.Deng X, et al. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science. 2008;322:110–115. doi: 10.1126/science.1158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sentelle RD, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nature chemical biology. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colombini M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochimica et biophysica acta. 2010;1797:1239–1244. doi: 10.1016/j.bbabio.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Wenner Moyer M. The search beyond statins. Nature medicine. 2010;16:150–153. doi: 10.1038/nm0210-150. [DOI] [PubMed] [Google Scholar]
- 17.Graham DJ, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipidlowering drugs. JAMA : the journal of the American Medical Association. 2004;292:2585–2590. doi: 10.1001/jama.292.21.2585. [DOI] [PubMed] [Google Scholar]
- 18.Rauthan M, Ranji P, Aguilera Pradenas N, Pitot C, Pilon M. The mitochondrial unfolded protein response activator ATFS-1 protects cells from inhibition of the mevalonate pathway. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5981–5986. doi: 10.1073/pnas.1218778110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hage-Sleiman R, Esmerian MO, Kobeissy H, Dbaibo G. p53 and Ceramide as Collaborators in the Stress Response. International journal of molecular sciences. 2013;14:4982–5012. doi: 10.3390/ijms14034982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senkal CE, et al. Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. The Journal of biological chemistry. 2011;286:42446–42458. doi: 10.1074/jbc.M111.287383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breslow DK, Weissman JS. Membranes in balance: mechanisms of sphingolipid homeostasis. Molecular cell. 2010;40:267–279. doi: 10.1016/j.molcel.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nuclear receptor signaling. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips CM, McDonald KL, Dernburg AF. Cytological analysis of meiosis in Caenorhabditis elegans. Methods in molecular biology. 2009;558:171–195. doi: 10.1007/978-1-60761-103-5_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner BK, et al. A small-molecule screening strategy to identify suppressors of statin myopathy. ACS chemical biology. 2011;6:900–904. doi: 10.1021/cb200206w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.