

Abstract
In recent years, inflammation has become implicated as a major pathogenic factor in the onset and progression of Parkinson's disease. Understanding the precise role for inflammation in PD will likely lead to understanding of how sporadic disease arises. In vivo evidence for inflammation in PD includes microglial activation, increased expression of inflammatory genes in the periphery and in the central nervous system (CNS), infiltration of peripheral immune cells into the CNS, and altered composition and phenotype of peripheral immune cells. These findings are recapitulated in various animal models of PD and are reviewed herein. Furthermore, we examine the potential relevance of PD-linked genetic mutations to altered immune function and the extent to which environmental exposures that recapitulate these phenotypes, which may lead to sporadic PD through similar mechanisms. Given the implications of immune system involvement on disease progression, we conclude by reviewing the evidence supporting the potential efficacy of immunomodulatory therapies in PD prevention or treatment. There is a clear need for additional research to clarify the role of immunity and inflammation in this chronic, neurodegenerative disease.
Keywords: Immune System, Parkinson's Disease, lymphocytes, inflammation, immunomodulation, adaptive immunity
Introduction
A great need exists in the PD field for disease-modifying therapies, for better comprehension of disease pathogenesis, and for identification of relevant biomarkers [1]. In recent years, inflammation has been implicated as an important mediator of disease pathogenesis. Clarifying the role of inflammation in PD will better enable investigators to address the field's needs. Functional profiling of peripheral immune cells could provide important disease biomarkers while immunomodulatory therapeutics may be able to delay or attenuate disease [2]. Even though the vast majority of research in PD has focused on neuronal and glial dysfunction, this review focuses on synthesizing evidence for the role of inflammation and adaptive immunity in promoting disease susceptibility and progression.
Parkinson's Disease
Clinical diagnosis of PD is established through physical examination and rating of motor symptoms including bradykinesia, resting tremor, and muscle rigidity but it is now recognized that REM-behavior disorder, constipation, depression, and anosmia precede manifestation of motor symptoms by years to decades [3]. Hereditary forms of PD, caused by specific gene mutations, only comprise 5-10% of PD cases [4]. These genetic forms of PD typically have an earlier age of onset and provide insight into the potential role of ubiquitously expressed proteins in neuronal and immune dysfunction in sporadic PD [5]. Other non-motor symptoms develop later in disease progression such as psychosis, autonomic dysfunction, and cognitive abnormalities [4, 6]. The progressive, multi-system pattern of PD symptomatology is consistent with a prominent role for a chronic inflammatory process.
PD pathology is characterized by chronic neuroinflammation, Lewy body inclusions, and loss of dopamine-producing (DA) neurons in the substantia nigra parts compacta (SNpc) of the midbrain [4, 6]. The Braak hypothesis (Fig 1) proposes a PD-specific pattern for progression of Lewy body pathology, intraneuronal aggregate of various proteins, including α-synuclein, tau, and ubiquitin. Lewy bodies are hypothesized to initially form in the olfactory bulb or the gastrointestinal tract (areas that are constantly exposed to the environment). As disease progresses, it is thought that pathological hallmarks then appear in the vagus or olfactory nerve and subsequently in the brainstem. When patients present clinically with motor symptoms, over 70% of DA neurons in the SNpc have died and significant Lewy body pathology as well as inflammation is present in that region. In final stages of the disease, pathology also occurs in the forebrain and cortical regions. An attractive feature of this hypothesis is that it may explain the presence and timing of non-motor and motor symptoms, as well as the multi-systemic nature of the disease that could me mediated in part by the spread of inflammation. Efforts to use non-motor symptom-based screening to identify people “at-risk” for PD are underway and will be important in clarifying underlying pathological mechanisms [7].
Fig 1. The Braak Hypothesis proposes a pattern for progression of disease pathology.
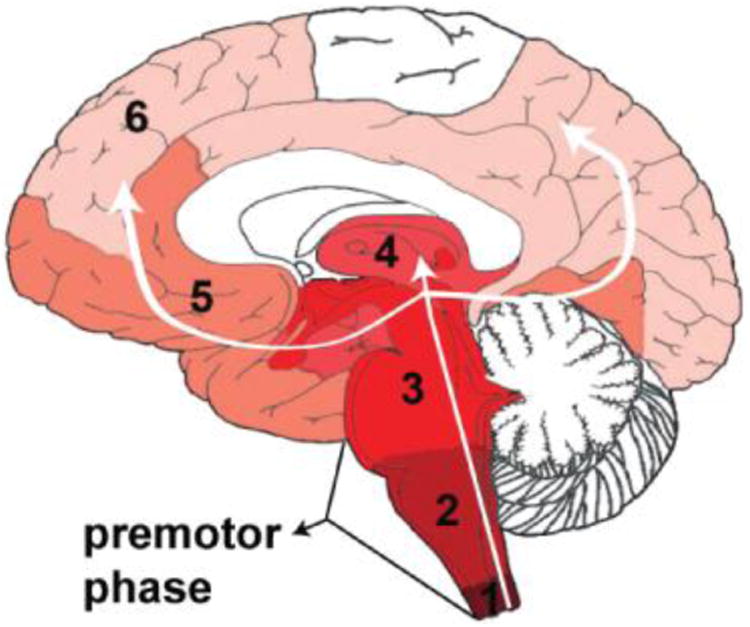
Pathology initiates in the periphery and progresses into the CNS via the vagus nerve and/or olfactory nerve in a predictable, progressive manner. Numbers indicate proposed sequence of progression into various brain regions.
Etiology of Sporadic Parkinson's Disease
The etiology of sporadic PD is undetermined because significant loss of DA neurons (∼70%) has already occurred by the time of clinical presentation [8]. Nevertheless, it is generally accepted that both genetic and environmental factors contribute. Multiple genetic polymorphisms have been associated with sporadic PD through genome-wide association studies (GWAS) (Table 1) [9]. The results of these GWAS identify specific genetic loci that contribute to disease susceptibility. Such loci include genes for α-synuclein, glucocerebrosidase, and tau. Thus, genetic background combined with specific environmental exposures could promote the manifestation of the pathologic process underlying PD.
Table 1. Genetic polymorphisms associated with sporadic forms of PD.
| Genome Wide Association-Based Loci | |||
|---|---|---|---|
|
| |||
| Chromosome | RefSNP # | Gene | Odds Ratio |
| 1q22 | N370S | GBA | 3.08 |
| 4p16 | rs11248060 | DGKQ | 1.69 |
| 4q22 | rs356229 | SNCA | 1.35 |
| 6p21 | rs3129882 | HLA-DRA | 1.31 |
| 21q21 | rs2823357 | USP25 | 1.15 |
| 12q12 | rs1491942 | LRRK2 | 1.19 |
| 17q21 | rs1724425 | MAPT | 0.68 |
| 4p15 | rs4538475 | BST1 | 1.25 |
| 3q27 | rs11711441 | LAMP3 | 0.82 |
Adapted from Ross OA, Curr Genet Med Rep, (2013), 1:52-57.
Environmental exposures also modify the risk for PD. Indeed, the identification of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was critical in recognizing the loss of these neurons as the cause of motor symptoms in disease [10, 11]. Upon absorption into the bloodstream, the highly lipophilic MPTP molecule quickly enters the CNS, where it is metabolized by monoamine oxidase-B into 1-methyl-4-phenylpridinium (MPP+). MPP+ inhibits cellular respiration by direct inhibition of the mitochondrial transport chain leading to neuronal injury and death [12]. Unlike MPTP, the mechanisms through which other environmental exposures modulate risk of PD are unclear. PD risk is increased by pesticide/organophosphate exposure and head trauma while it is decreased by moderate amounts of cigarette use and caffeine consumption [13-15]. Positive associations of PD with influenza, toxoplasmosis, and autoimmunity have been reported while non-steroidal anti-inflammatory drugs decrease risk for PD [16-24]. Genetic factors, as well as environmental factors clearly contribute to the development of PD; however, neither is believed to be independently sufficient to cause sporadic disease. Thus, it is generally accepted that the complex interplay between genetic and environmental factors predisposes people to the development of sporadic PD.
The immune system is in constant dialogue with our environment, tolerating certain exposures or antigens while responding to others. Dysregulated immune responses lead to disease. An inadequate response to an infection leads to bacteremia while hyper-reactivity to a harmless antigen can cause allergy. The innate immune system plays a key role in initiating inflammation in response to conserved antigen or cell death signals such as bacterial flagella, single-stranded viral DNA, or release of inflammatory cytokines from injured cells. The activated innate immune system will then induce an adaptive immune response, which are specific, targeted, and highly potent against the antigens present in the milieu of the inflamed region. Since adaptive immune cells can promote both anti-inflammatory and pro-inflammatory reactions in response to specific antigens, they are uniquely suited to modulate inflammation in the context of certain environmental exposures and neuronal dysfunction [25, 26]. In addition, complex immunoregulatory processes that require intricate crosstalk between the adaptive and innate immune system occur at mucosal surfaces that are constantly exposed to antigens [27]. Interestingly, the Braak hypothesis suggests that these mucosal sites (nasal and intestinal mucosa) may be where PD pathology is initiated. Thus, the immune system is poised to determine the body's response to various environmental exposures in predisposing to or protecting against PD. Furthermore, since PD is a multi-system progressive disease, the immune system could play a role in propagating neuronal dysfunction and pathology within the central nervous system (CNS). Nevertheless, contextualizing the role of the immune system will be difficult given contributions from genetic background, neuronal dysfunction, environmental exposure, and immunologic memory. A survey of our knowledge of the role of the immune system in PD is presented below.
Evidence for Inflammation and Innate Immunity in PD
The interplay between inflammation and neuronal dysfunction is complex. Neuronal death can induce inflammation by releasing apoptotic and necrotic cellular factors that are recognized by innate immune cells. The activation of innate immune cells by these general factors could then induce an adaptive immune response that would lead to specific and selective patterns of neuronal injury. Conversely, inflammation can induce neuronal death through the production of neurotoxic cytokines or direct neuronal injury. Many mechanisms of neuronal dysfunction have been implicated in PD pathogenesis, including proteasome-mediated protein degradation, mitochondrial dysfunction, and oxidative stress [28]. Inflammation-mediated neuronal dysfunction has also been well established. Specifically in PD, midbrain DA neurons are very sensitive to cytokines such as TNF-α and IFNγ [29-32]. Microglia, CNS-resident innate immune cells, densely populate the SN, and therefore could contribute to the enhanced sensitivity of this region to inflammatory stimuli [33]. The bacterial endotoxin, lipopolysaccharide (LPS), directly activates TLR4 receptors on microglia (and other innate immune cells) to initiate an inflammatory response. Within a week after direct LPS injection into the SN of mice, significant decreases in dopamine and dopamine metabolite levels (>60%) in the striatum and in tyrosine hydroxylase immunostain-positive (TH+) neuron number in the SN are detectable. These changes are detectable even a year after the injection [34, 35]. Chronic infusion of a low dose of LPS into the rat SN leads to peak microglial activation in two weeks followed by delayed and selective loss of DA neurons. Studies in vitro with neuron-enriched versus mixed neuron/glia cultures suggested this was due to microglial-specific activation because dopaminergic neurons do not express TLR4 [36].
Abundant evidence in humans also demonstrates a role for chronic inflammation and innate immune activation in PD. Increased levels of cytokines (including IL-1β, TGF-β, IFNγ, and IL-6) as measured post-mortem were found in the CSF and nigrostriatal regions of individuals with PD relative to age-matched healthy controls [31, 37-39]. Furthermore, proteins of the complement system, a serum-mediated mechanism designed to clear antibody and various immune targets, are found in extraneuronal Lewy bodies postmortem. This finding suggests that innate immune activation occurred in association with or in response to Lewy body formation [40]. Serum levels of TNFα are elevated in PD patients and the serum levels of IL-6 correlate with Hoehn and Yahr staging [41]. Taken together, this evidence indicates that an active inflammatory process with definite innate immune involvement is ongoing in the CNS of PD patients.
Until we can identify people with PD earlier in the course of the disease, we will be unable to definitely establish whether innate immune activation is an etiologic or resultant process in the pathophysiology of disease. However, inflammation and innate immunity are also known to play an important role in various animal models of PD. In these models, it is often present before detectable neuronal dysfunction or death occurs. In the MPTP mouse model, activation of microglia, increased endothelial expression of adhesion molecules, and infiltration of T lymphocytes can be dampened by dexamethasone treatment. Dexamethasone is a potent corticosteroid that globally suppresses immune responses. The anti-inflammatory effects of dexamethasone in the MPTP mouse model protect against DA neuron loss [42, 43]. The MPTP model does not display significant blood brain barrier (BBB) disruption and thus, may not completely reflect the disease process in that respect [42]. People with PD have significant BBB dysfunction, which is discussed below. The use of anti-inflammatory drugs in neurotoxin animal models such as MPTP and 6-hydroxydopamine (6-OHDA) also attenuates DA neuron loss [44]. The direct LPS intranigral injection model is useful in understanding how direct engagement of receptors on innate immune cells can initiate a neurotoxic inflammatory response. On the other hand, direct toxin models may better reflect how neuronal death induces inflammation, which then propagates further neuronal injury [34]. Viral overexpression of human α-synuclein in the mouse SN induces inflammation and microglial activation that is followed months later by progressive death of TH+ neurons [45]. Innate immune activation is also observed in various transgenic mouse models of α-synuclein overexpression. In a mouse model, expression of wild-type α-synuclein under the direction of the Thy1 promoter resulted in microglial activation and TNFα expression in the striatum at 1 month of age and progression to the SN at 5-6 months of age [46]. These changes can persist up to 14 months of age. Other various models of transgenic mutant and wild type α-synuclein overexpression also display chronic microgliosis [47-50]. In summary, direct neurotoxin models (MPTP, 6-OHDA) typically involve necrosis of DA neurons that initiate an inflammatory response through innate immune activation, whereas in LPS and α-synuclein over-expression models, an initial inflammatory reaction precipitates neuronal dysfunction and death. It is unclear which of these two pathogenic processes predominates in PD since we are only able to detect human disease at late stages. Investigation of inflammation in the context of multiple animal models remains extremely important because they allow investigators to clarify the interactions between neuronal dysfunction, innate immune activation, and inflammation.
Inflammation can also alter the permeability of the BBB allowing increased flux of inflammatory factors and immune cells from the periphery. Innate immune activation leads to release of proinflammatory factors that mediate this increased BBB permeability. Adaptive immune cells normally survey the CNS. Increased BBB permeability could allow for increased influx of peripheral adaptive immune cells which could then be activated by inflamed microglia as they are the primary innate immune cell of the CNS. These adaptive immune cells are activated in response to specific antigens and would be able to promote inflammation in other CNS regions that contain these same antigens. This inflammatory response could induce neuronal injury (if not already present) that in turn could propagate a positive-feedback cycle of neuronal injury and inflammation in the development of PD [32]. Thus, increased BBB permeability could be a catalytic factor in the development of PD.
In vivo evidence for increased blood brain permeability has been revealed through PET measurement of ligand uptake by the molecular efflux pump, P-glycoprotein, and increased albumin in the CSF of PD patients [51, 52]. Possible mechanisms for this BBB leakiness include cerebral capillary basement membrane thickening and collagen accumulation [53]. Increased proliferation of blood vessels or endothelial cells also occurs in PD [54]. Upregulation of ICAM-1, an important adhesion molecule for immune cell diapedesis, has been reported on blood vessels near the SN in postmortem brain tissue from PD patients. Endothelial upregulation of ICAM-1 is also present in MPTP-induced rodent and non-human primate PD models [55, 56]. Inflammatory responses that promote BBB leakiness may promote an efflux of antigen from the CNS to lymphoid tissues where adaptive immunity can be engaged. An activated endothelium and BBB could also permit greater influx of peripheral immune cells that further propagate inflammation. Understanding the process of how innate immune cells present neuronal antigens to adaptive immune cells and then how these adaptive immune cells are recruited to the CNS in PD will be important for developing a coherent model of disease pathogenesis. Understanding the role of inflammation in this disease will aid development of therapeutic approaches to promote neuroprotective immune responses.
Overall, it is clear that DA neurons are especially susceptible to damage from inflammatory stimuli. Furthermore, inflammation is a key pathogenic process in models of PD that often precedes detectable neuronal injury and degeneration. It will be difficult in humans to determine whether inflammation precedes neuronal injury until earlier diagnosis or biomarkers are established. Nevertheless, the brains of PD patients display chronic neuroinflammation that could be targeted to modify disease progression. The selective loss of certain neuronal populations while others are spared could point to a role for adaptive immunity in mediating this inflammatory process.
Engagement of Adaptive Immunity: Microglial Activation and MHC in PD
Some of the earliest evidence for inflammation in PD is from identification of chronic microglial activation in post-mortem brains. Microglia are CNS-resident immune cells that play an important role in neurologic development and maintenance. They are critical for recruitment of peripheral immune cells through secretion of chemokines and thus, amplify inflammatory signals in the CNS. These inflammatory signals could originate from neuronal dysfunction or external insults (trauma, toxicants, etc.). Microglia respond to numerous signals such as bacterial and viral products, α-synuclein, complement, antibodies, and cytokines [57-60]. Neuronal death may trigger microglial activation through loss of inhibitory CD200:CD200R signaling, ligation of microglial receptors that recognize factors released during apoptosis, release of α-synuclein, and/or through binding of complement or antibodies bound to neurons [58, 59]. In response to these stimuli, microglia produce cytokines [59, 61, 62], reactive oxygen species [63, 64], prostanoids that have immunomodulatory functions [65], and chemokines that recruit peripheral immune cells [66, 67]. Neuronal death can occur in response to cytokines such as TNFα, to ligation of death receptors such as Fas, and to toxicity of reactive oxygen species, as well as phagocytosis [59, 66]. Like other cells with antigen-presentation functions (B lymphocytes, macrophages, monocytes, dendritic cells), activated microglia will express Major Histocompability Class II (MHC-II) molecules that present endocytosed or lysosomal peptides to CD4 T lymphocytes (Fig 2). MHC-II molecules are α/β heterodimers that require binding of a peptide approximately 8-10 amino acids in length to be stably expressed on the surface of activated antigen-presenting cells. In humans, MHC-II molecules include HLA-DR, HLA-DQ, and HLA-DP. The majority of these peptides are derived from the processing of endocytosed proteins or from the typical protein constituents of endolysosomes. MHC-II genes are commonly known for their use in tissue typing for organ donation. Recognition of a specific peptide:MHC-II complex by a cognate CD4+ T cell through its unique receptor will activate the T cell to allow it to perform its effector functions: proliferation, cytokine secretion, assisting antibody-secreting B cells to produce higher affinity antibodies through a process called hyperaffinity maturation, and allow for proper activation and maintenance of cytotoxic CD8 T cells [68-71]. Presentation of antigens on MHC on the surface of activated microglia or other infiltrating innate immune cells is the key to engagement of adaptive immunity, which in turn can propagate the inflammatory process. MHC expression has been well-documented in PD.
Fig 2. Biosynthesis of peptide-MHC complexes.
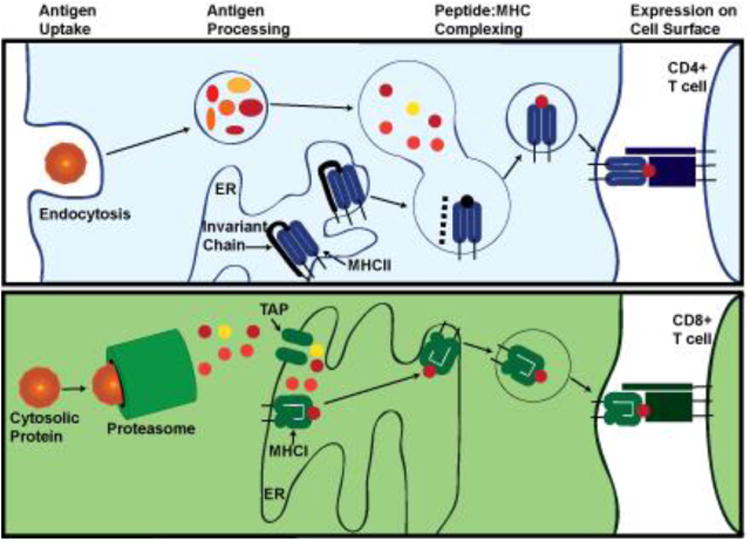
MHC-II peptides are derived from endocytosed antigens while MHC-I proteins are derived from proteasome-processed cytosolic proteins. MHC-II presents peptides to CD4 T cells while MHC-I presents peptides to CD8 T cells.
Microglial expression of MHC-II molecules has been reported in states of chronic neuroinflammation, including neurodegenerative diseases. In the healthy CNS, microglial MHC-II expression is difficult to detect. In PD brains, HLA-DR immunostain-positive microglia are found throughout the nigrostriatal tract and other parts of the CNS, including the hippocampus, entorhinal cortex, and cingulated cortex [72-74]. In those individuals exposed to MPTP through the use of contaminated heroin, microgliosis is present even years after exposure [75]. Chronic microglial activation is also seen years after initial MPTP administration in primate models of PD irrespective of acute or chronic dosing [76-78]. Positive correlations between disease duration and the microglial/macrophage activation marker CD68 and between MHC-II expression and amount of α-synuclein deposition in the SN of post-mortem human brain sections have been reported [57]. These activated microglia are associated with damaged neurons and Lewy body pathology [74]. In vivo PET imaging of PD patients demonstrates that microglial activation is concentrated in the SN and also occurs throughout the diseased CNS. Microglial activation does not seem to progress over the course of two years, suggesting that it is sustained at a constant level during clinically detectable disease and likely plays a role early in disease [79]. PET studies have also correlated microglial activation in the human midbrain with increased motor score on the Unified Parkinson's Disease Rating Scale (UPDRS) [80]. Microglial activation is prevalent in PD and occurs early in the disease process, suggesting that inflammatory processes and microglial effector functions play a prominent role in promoting disease development. Microglial effector functions could engage adaptive immunity in the context of antigen presentation on MHC to amplify and propagate inflammation.
Particularly relevant for adaptive immunity, single nucleotide polymorphisms (SNPs) in the MHC-II locus have been associated with increased risk of developing PD. In particular, people homozygous for a risk-conferring SNP, r3129882, in the first intron of HLA-DRA have a 1.7 fold higher risk for PD than individuals homozygous for the non-risk conferring allele [81, 82]. Other SNPs in the MHC-II locus associated with PD have also been reported in the HLA-DRB1 and -DRB5 genes. Another study has shown an association with the HLA-DQB1*06 allele in a German cohort [83-87]. The up-regulation of MHC-II mRNA (HLA-DRA, HLA-DPA1, HLA-DQA1) and increased expression of HLA-DR on monocytes in cerebrospinal fluid (CSF) has been reported [88, 89]. Further study is needed to assess whether changes in MHC expression occurs in PD and in particular in association with the rs3129882 SNP. These associations between PD and the MHC-II locus suggest that certain antigens or patterns of MHC-II expression could promote the development of PD. The quality of the interaction between CD4 T cell receptors and peptide:MHC-II impacts CD4 T cell expansion and differentiation [69, 71] (Fig 3). Thus, MHC-II expression could modulate the overall quality of the inflammatory response via activation of the adaptive immune system. Individuals with a certain MHC-II expression pattern or haplotype could propagate inflammation initiated by neuronal dysfunction or death more readily. Activation of the adaptive immune system by innate immune cells could allow for propagation of chronic inflammation in response to particular stimuli and antigens. Such a mechanism would promote or exacerbate PD progression. The evidence for involvement of T cells, B cells, and antibodies in promoting inflammation in PD is discussed below.
Fig 3. Model of MHCII mediated antigen presentation to CD4 T cells.
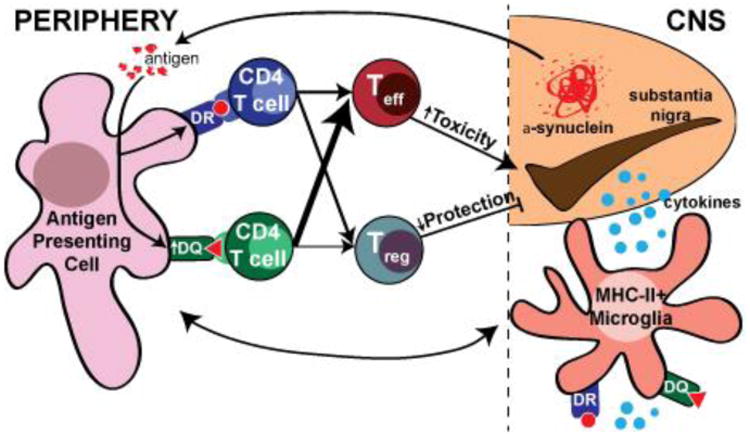
Antigen (potentially α-synuclein) from the CNS is presented by either HLA-DR or -DQ molecules in secondary lymphoid tissues to activate and differentiate CD4 T cells. Failure to induce the appropriate regulatory (Treg) or effector (Teff) T cells could contribute to dopaminergic neurondegeneration in PD.
Certain antigens or environmental exposures could synergize with a person's MHCII genome to put them at risk for developing PD. In this manner, the immune system would be an etiologic factor. Peptides presented on MHCII would activate T cells to influence neuronal function through cytokine secretion, which may promote cytotoxicity. This idea is supported by the association between PD and autoimmune diseases, such as bullous pemphigoid, systemic lupus erythematosus, and Sjogren's syndrome [16, 17, 20, 22]. Gut inflammation is hypothesized to play a role in the development of sporadic PD. Overrepresentation of CARD15 mutations associated with Crohn's disease and Helicobacter pylori seropositivity before age 75 in people with sporadic PD supports this hypothesis [90, 91]. A GWAS identified polymorphism in the LRRK2 gene is associated with Crohn's disease. Mutations in this gene give rise to an autosomal dominant form of PD (see below). Given the extensive neuronal network of the GI tract, its constant interaction with the environment, and its extensive, unique immunological repertoire, the GI tract represents a potentially interesting site for initiation of PD pathogenesis because of the complex, continuous interplay between the innate and adaptive immune system through antigen presentation.
Immune responses to infections are also implicated in PD through association with influenza, toxoplasmosis, and Epstein-Barr Virus (EBV) [21, 23, 92]. Post-encephalitic parkinsonism after the 1918 Spanish Flu pandemic and a cross-reactive antibody between EBV and α-synuclein suggest that adaptive immune responses to certain viruses may predispose an individual to PD [21, 92]. Infection of mice with the A/VN/1203/04 strain of the H5N1 influenza virus demonstrated viral replication in the CNS, microglial activation and increased proinflammatory cytokine and chemokine expression lasting 90 days after viral clearance. A transient decrease in tyrosine hydroxylase (TH) expression in the SN was also observed [93]. This virus can also induce α-synuclein phosphorylation and aggregation [94]. Japanese Encephalitis Virus (JEV) has also induced a post-encephalitic parkinsonism with similar neuropathologic features and locomotor dysfunction as that seen in people with sporadic PD [95]. JEV infection in rats results in hypokinesia attributable to depletion of catecholamines in the CNS [96]. These studies provide provocative evidence that an adaptive immune response could induce PD-related neurologic dysfunction initiated solely by inflammation. MHC expression on stimulated innate immune cells would play a critical role in determining which antigens are presented to the adaptive immune system. Activated adaptive immune cells would then propagate the inflammatory reaction. The use of animal models of PD such as the human α-synuclein-expressing adenoassociated virus (AAV-Syn) model [45] that more directly engages adaptive immunity could help address the validity of these associations. Global loss of MHC-II in a mouse model is protective against neurodegeneration, microglial activation, and T cell infiltration when human α-synuclein is virally overexpressed in the SN. While this model lacks CD4 T cells and has impaired antibody production, it does provide interesting evidence for a role of CD4 T cells and MHC-II expression in PD pathogenesis [97]. Other neurotoxin models employing MPTP or 6-hydroxydopamine (6-OHDA) may be less likely to engage adaptive immune mechanisms because they are mainly driven by neuronal toxicity rather than a more direct inflammatory stimulus. Clarifying whether the immune system acts as an etiologic factor in PD or simply propagates inflammation initiated by neuronal dysfunction will be an important part of determining PD etiology and pathogenesis. Better understanding of the immune system's role will certainly have therapeutic implications.
Adaptive Immunity (I): T Lymphocytes in PD
Given evidence for homeostatic surveillance of the CNS by naïve and memory T cells, these lymphocytes could be involved in both initiating and propagating steps of PD pathogenesis [98, 99]. In both PD patients and animal models, infiltration of T cells into the SN has been demonstrated [55, 72, 73]. In conjunction with HLA-DR expression on microglia, T cell infiltration has been shown in post-mortem brain sections from PD patients. This evidence suggests potential direct interaction with antigen-presenting functions of microglia [72, 73]. Furthermore, levels of B2 microglobulin, a protein required for stability of MHC-I molecules, are increased in the striatum of PD patients [38]. MHC-I molecules activate CD8 T cells by presenting cytosolic peptides processed through the proteasome pathway (Fig 2). Unlike MHC-II, MHC-I molecules are ubiquitously expressed on all cells (including neurons) except erythrocytes. Upon engagement of peptide:MHCI, CD8 T cells can directly kill cells by engagement of death receptors via Fas or TNF-α or by direct lysis through release of granzymes and perforin. In addition to CD4 T cell-mediated toxicity, direct neuronal injury may be caused by CD8 T cells that are reactive against neuronal antigens presented on MHCI molecules [100]. Thus, T-cell mediated neurotoxcity may be driven by direct cell lysis, engagement of cell death receptors, and cytokine secretion through recognition of peptide:MHC molecules on innate immune cells.
To better understand if adaptive immune responses are altered in PD patients, a handful of studies have examined the composition of T-cell subsets in the peripheral blood. PD patients are reported to have decreased overall numbers of lymphocytes without a change in frequency [101, 102]. Compared to people with other neurologic diseases, individuals with PD have increased memory T cells but decreased naïve T cells [88]. Memory T cells respond faster and with greater magnitude than activated naïve T cells. A more activated, cytotoxic T cell response is suggested by decreased CD4:CD8 ratios and a shift to more IFNγ- versus IL-4-producing T cells in PD patients [101-103]. Within the CD4 T cell compartment, PD patients have an increase in CD45RO+ T cells, which represent an activated and/or memory T cell population [104]. CD45RO+ expression also positively correlates with UPDRS motor score. Naïve CD4 T cells have increased Fas expression, which may explain why their frequency is decreased [102]. Fas expression is normally upregulated after T-cell activation and ligation by FasL induces apoptosis. This process is used on T lymphocytes to maintain immune privilege or initiate contraction after an immune response [105]. Increased memory and activated CD4 T cells in conjunction with a relative increase in CD8 T cells could suggest an active inflammatory process. Conflicting reports of changes in relative frequencies of CD4+CD25+ T cells in peripheral blood exist but without further characterization of this subpopulation, the biological significance of this is unclear [101-103, 106]. CD25 is increased on activated or memory T cells, as well as regulatory T (Treg) cells. One study that demonstrated that CD4+CD25+CD127- regulatory T cells from PD patients had less suppressive capacity compared to cells from healthy controls [104]. The relative number of effector to regulatory T cell responses is thought to regulate immune responses. Decreased effectiveness of regulatory T cells could promote a chronic neuroinflammatory state or allow for breaking of tolerance to neuronal antigens that would lead to abnormal immune responses against CNS proteins. Investigation of why these regulatory T cells have reduced suppressive capacity could also provide insight into PD pathogenesis and disease progression.
Despite information about changes in relative frequency of T cell subsets, little is known about the identity of CNS antigens to which activated and memory T cells in people with PD are responding. One study looked at whether specific T cell receptor genes are preferentially expressed in PD patients and reported that CD8 T cells have a lower frequency of Vβ8 expressing cells [107]. The use of deep sequencing techniques to identify patterns of T cell receptor usage in PD patients could provide evidence for response to specific antigens and help to identify those antigens. Identification of such patterns could also be a useful biomarker. T cell biomarkers have been investigated and some candidate proteins have been identified within these cells such as β-fibrinogen and transaldolase [108]. The pathogenic relevance of these changes is not yet understood.
Other overall pathogenic changes in peripheral blood lymphocytes from PD patients have been reported. Lymphocytes from PD patients display an increased incidence of micronuclei, single strand DNA breaks, and oxidized purine bases [109]. Interestingly, levodopa treatment seems to reduce this DNA damage in peripheral blood lymphocytes [110]. Markers of apoptosis, caspase-3, and Cu/Zn superoxide dismutase activity are increased in lymphocytes from people with PD [111]. This DNA damage and increased level of apoptosis could be representative of a systemic, pathogenic process involving oxidative stress, specific immune responses, and/or intrinsic, genetic factors. The use of animal models to assess the contribution of lymphocytes will inform us of how these in vivo human changes may contribute to disease etiology and progression.
The contribution of T cells to PD-like pathology in animal models has been assessed but mainly in direct toxin models. CD8 T cells are seen in greater amounts than CD4 T cells in the acute MPTP neurotoxin model. In this model, these lymphocytes have increased expression of LFA-1, an integrin that binds endothelial adhesion molecules, to allow for diapedesis across the BBB [42]. DA neuron loss and behavioral deficits caused by both chronic and acute MPTP administration are attenuated in RAG2 knockout mice that lack both T and B cells [55, 112]. Mice in which there is a global loss of αβ-T cells through knockout of the T cell receptor β-chain, as well as mice lacking CD4 T cells (CD4-/-) are also protected in the MPTP model [55]. The loss of CD8 T cells was not protective. Reconstitution of Rag1-/- mice with FasL-mutant splenocytes attenuated DA neuron cell death while reconstitution with IFNγ-/- splenocytes did not. These findings suggest that Fas-FasL interactions involving CD4 T cells play an important role in promoting MPTP-induced neurodegeneration. As discussed above, cytotoxic T cells use FasL to induce apoptosis in target cells but Fas receptor is also important in T cell homeostasis and contraction. Impaired FasL-Fas interactions result in unchecked T cell activation and proliferation. An extreme example manifests itself in autoimmune lymphoproliferative syndrome caused by unchecked T cell proliferation [113]. Dysregulation of this pathway may also leave chronically activated macrophages or microglia unchecked allowing for propagation of the inflammatory response. Fas-FasL signaling may also indirectly be contributing to neuronal damage by inducing Fas-induced apoptosis-resistant astrocytes to produce proinflammatory cytokines that damage neurons [114]. In the intranigral AAV-human-α-synuclein overexpression mouse model, B and T lymphocyte infiltration in the SN persists after peak of microglial activation suggesting that adaptive immune cells propagate inflammation in this model as well [45]. One study with MPTP gives a potential model of adaptive immune engagement where nitrated α-synuclein drains from the CNS into cervical lymph nodes. Robust T cell responses were initiated when mice were immunized with the C-terminus of the nitrated α-synuclein. Transfer of T cells from immunized mice into MPTP-treated mice enhanced neuroinflammation to a slight but significant degree [115]. T cells from mice immunized with nitrated α-synuclein produced mostly IL-17 and TNFα after ex vivo restimulation. Adoptive transfer of Th17-polarized T cells from nitrated α-synuclein-immunized mice into wild-type mice greatly exacerbated MPTP-induced neurodegeneration. Neurodegeneration was not exacerbated by adoptive transfer of T cells polarized to Th1 (IFNγ-producing) or Th2 (IL-4-producing) phenotypes. Adoptive transfer of ex vivo differentiated regulatory T cells into MPTP-induced mice attenuated neurodegeneration [67]. The novelty of this study is that it demonstrates that modulation of the adaptive immune response can indeed determine the outcome of neurodegeneration in a model of PD. Given this evidence, T cell responses to modified α-synuclein or other modified antigens from DA neurons could initiate an adaptive immune response that propagates neuronal death. These responses would differ based on the context of antigen presentation by innate immune cells. Various etiologies could allow for the adaptive immune system to be exposed to modified α-synuclein (i.e. toxins) or increased escape of CNS antigen (i.e. head trauma). These mechanisms would break normal tolerance mechanisms by presenting self-antigens that are cross-reactive with cognate peptide:MHC of existing memory T cells. These antigens could also activate naive T cells by presenting self-antigens for which T cells are not normally negatively selected against or tolerized to in the context of an inflammatory response. Further studies to identify specific antigens that T cells respond in animal models and humans will be critical to understanding the role of the adaptive immune system in pathogenesis of PD.
Adaptive Immunity (II): Antibodies and B Lymphocytes in PD
B lymphocytes are the antibody-secreting cells of the immune system. They are the key mediators of humoral immunity in that their secreted immunoglobulins can have effects at sites far from the actual site of secretion [25]. Unique antibodies, as well as increased antibody levels against CNS proteins are present in people with PD [116, 117]. However, infiltration of antibody-producing B lymphocytes into the CNS has not been not reported in brains of PD patients [55, 72, 73]. While infiltration of B lymphocytes is present under some inflammatory conditions, the aggregation of antibodies at a specific site is enough to promote inflammation. Antibodies can precipitate inflammatory reactions through activation of the complement system or effector cells through surface immunoglobulin receptors. B cells can be activated to secrete antibody upon engagement of their surface immunoglobulin receptor in the context of inflammatory cytokines from innate immune cells or direct recognition of stimuli through pattern recognition receptors. These lymphocytes also can re-engineer the antibodies they secrete through a process called affinity maturation that occurs in secondary lymphoid organs such as the spleen or lymph nodes. Particularly for PD, there is some evidence that the meninges can act as a secondary lymphoid organ [118, 119]. Affinity maturation is mediated by activated T cells and occurs through interactions between the T cell receptor and peptide-MHC-II on the surface of the B cell. B cells that have undergone affinity maturation produce antibodies of higher affinity to their target and can become memory cells that are long-lived [25]. B cells in this manner have a unique niche in the adaptive immune system that depends heavily on signals from innate immune cells and CD4 T cells.
A decrease in the number of peripheral blood B cells in PD patients has been reported but the significance of this finding needs to be contextualized [101, 102]. Reduced peripheral blood B cell counts in other autoimmune or inflammatory diseases are due to decreased circulating memory B cells. This reduction in B cell number may be related to active inflammation and cellular activation [120-122]. Increased study is needed to understand the role for B cells in PD outside of their antibody-secreting functions discussed below.
Antibodies against DA neurons have been identified in PD patients that are not present to the same extent in healthy people [116, 117, 123, 124]. At present, the lack of a universal antibody or “PD”-specific antigen refutes an etiologic role for B cells and antibodies but does not rule out either a pathogenic or a protective role for B cells in modulating inflammation in PD. It is also possible that a universal antibody or antigen in PD does not exist because no single antigen is required for disease induction. On the other hand, we may not able to identify one because we only assess antibody production at a late stage of disease. Within human PD brain sections, immunoglobulins are found to colocalize with pigmented DA neurons in close proximity to FcγR+ microglia. The number of IgG+ neurons positively correlated with the number of MHCII+ microglia suggesting a link between antibody-mediated destruction of neurons and antigen presentation. Further supporting this link, these microglia contain pigmented granules suggesting antibody-mediated phagocytosis of pigmented DA neurons [125]. A pathogenic role for anti-neuronal antibodies in PD is further supported by a study using immunoglobulins from PD patients in a rat model. These immunoglobulins were injected into the SN of rats resulting in a loss of TH+ neurons while acetylcholinesterase-positive neurons in the medial septal region were spared [126]. These immunoglobulins induced perivascular inflammation, microgliosis, and loss of TH+ neurons in the SN that was dependent on expression of the Fcγ receptor [127]. Similarly, Fcγ receptor knockout mice are resistant to chronic but not acute MPTP administration [112]. One caveat to these results is that the deletion of Fcγ receptor knockout mice are resistant to chronic but not acute MPTP administration [112]. One caveat to these results is that the deletion of Fcγ receptor in mouse models impacts numerous cell types (monocytes, microglia, neutrophils, basophils, mast cells, NK cells) and reduces the ability to respond to cellular bound IgG through multiple mechanisms. These mice are also resistant to neurodegeneration and microglial activation induced by AAV-induced intranigral expression of human wild type α-synuclein [128]. The AAV- α-synuclein model also leads to significant IgG deposition, suggesting that humoral immunity and/or breakdown of the BBB is playing a role [45]. These findings suggest that with DA neuron injury, the humoral arm of the immune system can play a role in chronic neuroinflammation.
Given the chronic inflammatory nature of PD, humoral immunity could play an important role in the progression of PD while T cell immunity may be more important for disease onset. Intuitively, this idea makes sense because CD4 T cell activation is essential for potent engagement of humoral immunity. In contrast, antibodies may also play a protective role. The use of a mouse antibody against α-synuclein showed that microglia can take up antibody-bound α-synuclein in vivo and prevent neuron to astrocyte transmission of α-synuclein [129]. It is unknown whether this phenomenon can happen in the PD brain but it raises the distinct possibility that certain sets of antibodies could play a protective role. Pathogenic antibodies could promote inflammatory reactions or propagate α-synuclein spread through uptake by Fc receptor-expressing cells. Other antibodies may be protective by aiding in clearance of α-synuclein aggregates.
There have been a few attempts to use antibodies as biomarkers for PD. One study found decreased levels of anti-α-synuclein antibodies in PD patients but not in controls or patients with other neurodegenerative conditions with sensitivity and specificity of 85 and 25%, respectively. These antibodies did not correlate with age, duration of disease, or Hoehn and Yahr staging [116]. The presence of a-synuclein antibodies may hint at pathologic mechanisms but are not specific enough to be used diagnostically. Another study demonstrates the ability to use 10 autoantibody biomarkers to differentiate PD from normal aging, Alzheimer's disease, breast cancer, and multiple sclerosis with accuracies over 85% [117]. Another report describes that in people with sporadic PD, the frequency of those with anti-α-synuclein antibodies is not significantly different from healthy controls. However, around 90% of people with familial PD have these antibodies. The authors suggest that the antibodies in familial PD may represent more pathogenic epitopes rather than incidental ones [124]. Studies using antibodies from patients at earlier stages of disease will likely give more insight into the role of antibodies in PD pathogenesis. Such antibodies are less likely to be incidental from the release of CNS antigens as inflammation propagates in disease.
Genetic Mutations in PD and Adaptive Immunity
Many of the genetic causes of familial PD have been identified [130, 131] (Table 2). Most genetic mutations cause early-onset forms of PD. It is generally believed that genetic and sporadic forms of the disease are likely to share common mechanisms. The study of these genetic mutations has provided insight into how perturbation of certain pathways may contribute to the development of sporadic PD. The majority of research efforts to date have investigated the pathogenic roles of these mutations in neurons. Some of these proteins have shown to be involved in the function of immune cells and may contribute to non-autonomous cell death of neurons. Dysfunction of these proteins in both neurons and immune cells would allow for synergy of inflammatory and neuronal death mechanisms that promote the development and/or progression of PD.
Table 2. Genetic loci associated with familial forms of PD.
| Familial Parkinson's Disease Loci | ||||
|---|---|---|---|---|
|
| ||||
| Locus | Region | Gene | Onset | Inheritance |
| PARK 1/4 | 4q21 | SNCA | Late | AD |
| PARK 2 | 6q26 | PRKN | Early | AR |
| PARK 6 | 1p36.12 | PINK1 | Early | AR |
| PARK 7 | 1p36.23 | DJ-1 | Early | AR |
| PARK 8 | 12p12 | LRRK2 | Late | AD |
| PARK 9 | 1p36.13 | ATP13A2 | Early | AR |
| PARK 14 | 22q13.1 | PLA2G6 | Early | AR |
| PARK 15 | 22q12.13 | FBXO7 | Early | AR |
| PARK 17 | 16q11.2 | VPS35 | Late | AD |
Adapted from Ross OA, Cutt Genet Med Rep, (2013), 1:52-57.
The first identified mutation to cause PD was in the SNCA gene, which encodes α-synuclein. Since then, polymorphisms in the promoter region that cause increased expression of α-synuclein have also been identified [9, 132]. Signs of synergy between inflammation and α-synuclein-induced degeneration have been observed in numerous cell culture and animal model studies as discussed previously. This synergy is likely to have important consequences in the brains of humans affected with PD. But it should be noted that SNCA and therefore the α-synuclein protein (as are most of the genes implicated in familial forms of PD) is actually expressed in a wide variety of immune cells including T cells, B cells, NK cells, microglia, and monocytes [133-136]. In microglia, α-synuclein has been shown to have a homeostatic role. Microglia from mice lacking SNCA have a more activated phenotype in terms of morphology and cytokine secretion in addition to decreased phagocytic ability [136, 137]. The level of α-synuclein protein increases after LPS stimulation of monocytes and lymphocytes [138], suggesting it may have some regulatory role in inflammatory responses. CD4 T cells from people with the autoimmune disorder systemic lupus erythematosus have a 5-fold increase in α-synuclein expression that correlates with higher basal levels of autophagy [139]. Authophagy plays a critical role in maintenance, activation, and proliferation of T cells [140]. Therefore, changes in immune cell levels of α-synuclein could modulate cellular function via mechanisms such as altered autophagy. This last point may be especially relevant as altered number or frequency of T and B cells have been reported in PD patients. In mice expressing human wild type α-synuclein under the Thy1 promoter, frequencies of CD4 and CD8 T cells are increased in the peripheral blood [46]. α-synuclein is also over-expressed in the T cells of these mice, thus the role of this protein in immune cells versus neurons will need to be evaluated in greater depth. It is unknown whether the inflammatory phenotype is attributed to primary deficits in immune cells over-expressing α-synuclein versus to secondary activation of immune cells as a result of over-expression of α-synuclein in neurons and neuronal dysfunction. The critical point here is that the functional significance of mutations or polymorphisms in α-synuclein in immune cells has been understudied. In addition, perturbations in the levels of α-synuclein by various stimuli, such as LPS, in the context of genetic polymorphisms inherent in immune gene loci could lead to immune dysfunction in those individuals that may synergize with and exacerbate neuronal dysfunction.
Mutations in LRRK2 (PARK8), a gene encoding a MAPK-kinase-kinase, GTPase, and ankyrin domain-containing protein, cause an autosomal dominant form of PD with a clinical presentation and age of onset similar to sporadic PD [141]. The incomplete penetrance (estimated at 33-67%) of these mutations also suggests a strong environmental and/or immune component that modifies disease onset [142, 143]. Genetic variants in this gene have also been associated with increased susceptibility for sporadic PD [85, 144]. Initial studies looking at LRRK2 function and expression in neurons demonstrate that its expression is low in the SN and greater in areas of the brain that receive dopamine signals [145-148]. In vitro and targeted in vivo overexpression of mutant LRRK2 induces neurotoxicity in a kinase- and GTPase-dependent manner [149-153]. However, germ-line mutant LRRK2 transgenic mice do not completely replicate PD-like phenotypes or show spontaneous DA neuron degeneration [154-157]. However, these mouse models do show behavioral alterations and impaired dopamine release in the nigrostriatal system. Indeed, perturbations in LRRK2 function alone may not be sufficient to induce PD-like pathology, but may require a second insult such as inflammation.
Interestingly, LRRK2 expression occurs in many immune cells: monocytes, microglia, B cells, and T cells [158, 159]; yet the role of LRRK2 in various immune cell populations has only received attention in recent years. The level of expression in many immune cell subsets is significantly greater than in neuronal populations [160]. Mouse bone marrow-derived macrophages (BMDMs) significantly upregulate LRRK2 mRNA expression after stimulation with LPS and lentiviral particles. BMDMs from R144C-LRRK2 mutant mice have reduced LC3-II expression, a marker of autophagy [158]. IFNγ stimulation of immune cells results in upregulation of LRRK2 expression, which was shown to activate the NF-κB pathway, to be important for production of microbicidal reactive oxygen species during phagocytosis, and to colocalize near pathogens during intracellular infection [160]. In microglia, LRRK2 expression can be detected upon in vivo activation with LPS and is upregulated by ex vivo LPS treatment. Furthermore, LRRK2 inhibition and knockdown in microglia attenuated LPS-induced inflammatory signaling as measured by cytokine secretion, microglial chemotaxis, and morphologic remodeling [161]. Polymorphisms in LRRK2 are also associated with leprosy and Crohn's disease [162, 163]. In fact, increased LRRK2 expression is found in the epithelium of Crohn's disease patients in many cell types including macrophages, dendritic cells, and B lymphocytes [160]. LRRK2-/- mice are more susceptible to dextran sodium sulfate-induced colitis [164]. Interestingly for adaptive immunity, LRRK2 inhibits the nuclear translocation of NFAT, a master regulator of transcriptional activation in T cells [164]. Overall, it is clear that LRRK2 is involved in regulating the activation status and effector functions in multiple kinds of immune cells. In summary, the role of pathogenic LRRK2 mutations in various LRRK2 domains needs to be further studied in the immune system.
Loss of function mutations and deletions in the Parkin (PARK2) gene cause autosomal recessive forms of juvenile PD [165]. The Parkin gene encodes a multidomain protein that contains E3 ubiquitin ligase activity that plays a role in the regulation of numerous cellular activities including proteasomal degradation and ubiquitin-mediated signaling. Parkin's physiologic substrates are unknown [166]. Potential roles for this protein are in preventing protein aggregation and/or promoting mitophagy [167-169]. Parkin has also been suggested to function as a transcription factor, regulating p53 expression [170] independent of its ligase activity. Parkin knockout animals do not have loss of DA neurons in the SN and do not have increased vulnerability to MPTP, yet they do show non-motor behavioral changes related to changes in the nigrostriatal pathway [171, 172]. This phenotype suggests compensatory changes in the global Parkin knockout animal that prevent death of DA neurons or that a second insult is required in conjunction with lack of Parkin expression. However, Parkin-deficient animals have interesting inflammation-related phenotypes, suggesting that inflammatory stimuli could be this secondary insult. Chronic peripheral low dose LPS intraperitoneal administration in Parkin-/- mice triggers fine-motor deficits and loss of DA neurons in the SN [173]. In microglia, neurons, and peripheral macrophages, LPS or TNFα stimulation transiently reduces Parkin expression in a dose-dependent manner. This decrease was found to be due to a NF-kB signaling response element in the Parkin promoter. Thus, chronic inflammation may down regulate Parkin and phenocopy loss of function Parkin mutations [174]. Interestingly, Parkin has also been implicated as a susceptibility locus for leprosy and typhoid fever [175, 176]. Furthermore, genetic loss of Parkin in mice and Drosophila models has been shown to increase susceptibility to infection. This effect on immune function was shown to be related to Parkin's role in autophagy [177]. The role of Parkin in inflammation and immune cells needs to be more thoroughly evaluated. Current evidence demonstrates that inflammation regulates Parkin expression and that Parkin is involved in PD-implicated pathways in neurons.
Identification of the role of proteins in which pathogenic mutations cause genetic forms of PD will be critical in identifying pathogenic processes. Much research has focused on the role of these proteins in neurons and CNS pathways despite their expression in many other cell types. Immune cells in particular are poised to interact with the environment and respond in a coordinated manner. In genetic forms of PD, dysfunction of these proteins could propagate and promote neuronal dysfunction. In sporadic PD, the way in which these proteins are altered by environmental factors in immune cells could then allow these cells to modify neuronal function and promote the development of PD. Further study of these proteins in immune cells is well-justified.
Immunomodulatory Therapies in PD
Given the chronic, progressive nature of PD and the large amount of neurodegeneration that has already occurred before clinical presentation, it is unlikely that disease-modifying therapies will have a significant impact in altering the course of disease. Preclinical diagnosis and timely treatment will be necessary to significantly impact disease. Nevertheless, because intense investigation into biomarkers of early disease is in progress, it is critical that we continue identifying immunomodulatory therapies that could be translated to the clinic. To this end, there are a few therapies that target immune pathways have shown therapeutic benefit in animal models of PD.
One such example is that adoptive transfer of T cells from a copaxone-immunized animal showed protection against neurodegeneration in an MPTP model [178]. Copaxone, also known as glatiramer acetate, is used in the treatment of multiple sclerosis. It is a random polymer of amino acids from myelin basic protein. This mixture is thought to mimic the antigenic properties of myelin basic protein but alter the T cell response to a more anti-inflammatory phenotype [179]. This study suggests that T cell skewing to dampen the adaptive immune response could potentially have a disease-modifying effect. The adoptively transferred T cells accumulate in the SN, suppress microglial activation, and are correlated with an increase in neurotrophic factors [180]. Adoptive transfer of Tregs also attenuated neurodegeneration in an MPTP model [67]. Decreased effectiveness of regulatory T cells in their suppressive capacities and changes in the T cell compartment suggest that correcting phenotypic T cell changes in PD patients may provide a beneficial disease-modifying effect [104]. Indeed, Howard Gendelman is initiating a clinical trial to assess the efficacy of Sargramostim, a granulocyte-monocyte colony stimulating factor analog thought to promote the development of Tregs, in the treatment of PD. If successful, this trial could demonstrate the usefulness of immunomodulatory therapies in treating this disease [181].
The use of a dominant-negative inhibitor of TNFα (DN-TNF) in PD animal models suggests that anti-TNF biological agents may also be of clinical value to delay or attenuate nigral DA neuron degeneration. Administration of soluble DN-TNF via osmotic pump or lentiviral overexpression in the SN shows protection in 6-OHDA models [182, 183]. Even delayed lentiviral overexpression of DN-TNF in the SN can be protective against progressive DA neuron loss [184]. As a way to dampen overall levels of inflammation, anti-TNF biological tools could be of therapeutic value.
Passive immunization in animal models with anti-α-synuclein antibodies have also been investigated as a potential mechanism for clearing accumulations of α-synuclein. In mice expressing human α-synuclein under the platelet-derived growth factor β promoter, administration of an anti-α-synuclein antibody improves behavioral performance and promoted degradation of accumulated α-synuclein [185, 186]. Clearance of α-synuclein is thought to occur via uptake through Fc-receptor expressing cells such as microglia. Whether clearance of α-synuclein-containing Lewy bodies would ameliorate PD symptoms, is an important question in order to pursue such a therapy. These observations will need to be replicated using other human α-synuclein overexpression models independently to confirm the promising potential of synuclein immunotherapy.
Given some of the pathogenic contributions of adaptive immunity in PD described in this review, a few other strategies could potentially be used. These therapeutics would need validation or testing in animal models of PD. Therapies that block T cell proliferation may be useful in modifying progression or onset of PD. Such therapeutics could include corticosteroids, colchicine, or peripheral T-cell depleting antibodies such as muromonab-CD3. Colchicine blocks mitosis and thereby preventing the proliferation of rapidly dividing cells such as activated immune cells. Other alternatives or modifications of this drug may be needed as there are significant gastrointestinal side effects. In a rotenone rat PD model, colchicine showed benefit in preventing neurodegeneration [187]. Muronomab initially activates CD3+ T cells but then these activated cells die. This antibody is currently used in treatment of transplant rejection [188].
A second avenue is the use of drugs that block T cell activation or recruitment. Abatacept and belatacept block the interaction between CD28 on T cells with CD80/CD86 molecules on antigen presenting cells [188, 189]. These drugs have been used in transplant rejection and rheumatoid arthritis with great success. Given the documented changes in activated and memory T cell subsets in people with PD, these drugs could have therapeutic success in limiting development of such T cell populations. The drug, natalizumab has been successful in the treatment of multiple sclerosis [190]. As an antibody against the α4 integrin, this antibody blocks the recruitment of T cells to sites of inflammation. By blocking activation and recruitment of T cells in PD, neuroinflammation could be dampened to prevent subsequent neurodegeneration.
Ex vivo modification of autologous T cells to make them produce anti-inflammatory molecules or have them target effector T cells that are pathogenic in PD might also be another useful therapeutic approach. This approach would require identification of pathogenic T cell populations or the relevant antigens to which pathogenic T cells in PD respond. Such technologies are currently being explored in harnessing immune responses against cancer [191].
Therapies to target humoral immunity do exist. Rituximab is an example of a B cell depleting antibody that has shown some effectiveness in autoimmune diseases where antibodies are known to play a pathogenic role [192]. Intravenous immunoglobulin (IVIG) has been effective in treating diseases that are mediated by pathogenic antibody responses such as erythroblastosis fetalis, autoimmune thrombocytopenia, and chronic inflammatory demyelinating polyneuropathy. IVIG is thought to block proinflammatory antibody-mediated responses through many pathways including modulating Fc-receptor expression, expansion of Tregs, and blocking activating Fc-receptors [193]. IVIG is generally well tolerated with less than 5% of people experiencing headache, fever, or nausea. In a recent Phase III trial by Baxter, IVIG did not show efficacy in the treatment of mild to moderate Alzheimer's disease that may not reflect on its potential in PD since underlying pathogenic mechanisms are different. However, this study demonstrated that IVIG was safe in the elderly with neurodegenerative disease. Thus, IVIG could be a safe immunomodulatory therapy potentially useable for PD.
As with any immunomodulatory therapy, immunosuppression or off-target effects are of great concern, especially in an elderly population that is at increased susceptibility to infections. But as learned in the transplant field, timing of these and any other therapeutic intervention is also critical [188]. Use of these therapeutics at the time where inflammation is beginning or is primed could have significant impact on the course of disease while having minimal effects much later in disease. Further understanding of the role of the adaptive immune system will help determine which avenues of immunomodulatory therapies are most likely to be effective.
Conclusions
The coordinated role of the innate and adaptive immune systems in the pathogenesis and pathophysiology of PD is becoming more clearly understood (Fig 4). The adaptive immune system normally responds to specific pathogens or antigens and has various effector mechanisms to clear them, i.e. direct cytotoxicity, cytokine production, and antibody production. The adaptive immune system must receive the appropriate signals from innate immune cells to be activated in order to respond to specific antigens as recognized by specific, unique receptors. T cells recognize peptides presented on MHC molecules while B cells recognize antigens through their cell surface receptors. Abundant evidence exists for the role of inflammation in human PD as well as propagating neurodegeneration in animal models. Further investigation in delineating a role for the adaptive immune system in PD pathology and the specific peptides that are being presented is required. Environmental and genetic factors synergize to create susceptibility to PD. It remains to be seen whether these factors create susceptibility primarily through inducing neuronal dysfunction or through direct activation of the immune system that then propagates neuronal dysfunction (Fig 4). Regardless, activation of the innate immune system to present antigens to the adaptive immune system could further precipitate neuronal dysfunction in a specific manner, i.e. from antigens derived from DA neurons. Modification of these antigens by inflammation and environmental factors may allow for an adaptive immune response in bypassing normal tolerance mechanisms. Nitrated α-synuclein is one potential antigen in this case. From the initiation site of this inflammation by the innate immune system, a chronic adaptive immune reaction could spread neuronal damage and dysfunction to other CNS pathways implicated in sporadic PD. The adaptive immune system may prove to be the missing link between environment, genetics, and the spread of pathology put forth by the Braak hypothesis.
Fig 4. Overall Paradigm for Engagement of Adaptive Immunity in PD.
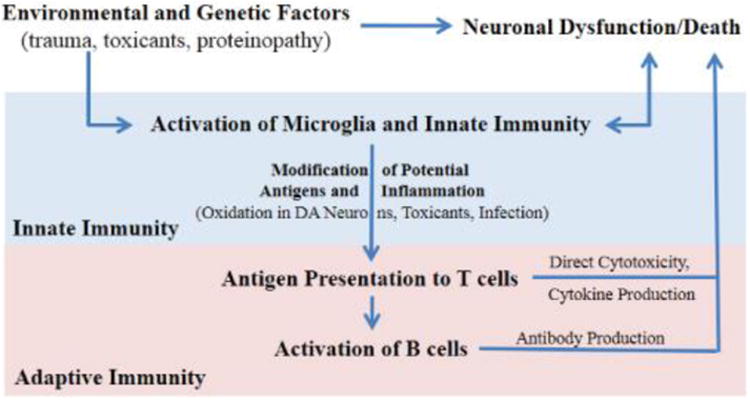
Environmental/genetic factors may initiate 1) neuronal dysfunction or 2) activation of microglia and/or other innate immune mechanisms which would amplify one another. Antigen presentation by microglia would activate T cells which in turn would activate B cells to produce antibodies. Continued release and presentation of antigens from dying neurons or modified antigens would allow for propagation of a specific, chronic inflammatory response mediated by the adaptive immune system towards the neurons that degenerate in PD.
In this review, the evidence for activation of the adaptive immune system in human PD has been presented, as well as how effector functions of adaptive immune cells damage DA neurons in animal models. Certain immune functions of genes known to be mutated in genetic forms of PD have also been presented. Taken together, exciting clues for an unrecognized role of the adaptive immune system exist but much work remains to be done. Closer examination of this system may provide a paradigm-shifting understanding of PD as well as an opportunity for identification of biomarkers and disease-modifying therapies.
Acknowledgments
Financial support for the authors of this review includes grants from The Michael J. Fox Foundation for Parkinson's Research Target Validation program (JMB), NIH/NINDS R01NS072467-04 (MGT), NIH/NINDS F31 NS081830-01 (GTK) and NIH/NIGMS RO1 GM47310-16 (JMB).
MGT is an ex-employee of Xencor Inc., a biotherapeutics company developing anti-TNF biologics for neurological disorders and co-inventor on several patents held by Xencor Inc. JMB and GTK have no conflicts to declare.
References
- 1.Chen JJ. Parkinson's Disease: Health-Related Quality of Life, Economic Cost, and Implications of Early Treatment. AJMC. 2010:S87–S93. [PubMed] [Google Scholar]
- 2.Henchcliffe C, Severt WL. Disease modification in Parkinson's disease. Drugs Aging. 2011;28:605–615. doi: 10.2165/11591320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Factor S, Weiner W. Parkinson's Disease: Diagnosis and Clinical Management. Demos Medical Publishing; [Google Scholar]
- 4.Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet. 2004;363:1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- 5.Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 6.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56:33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 7.Hawkes CH, Del Tredici K, Braak H. A timeline for Parkinson's disease. Parkinsonism Relat Disord. 2010;16:79–84. doi: 10.1016/j.parkreldis.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Brooks DJ. The early diagnosis of Parkinson's disease. Ann Neurol. 1998;44:S10–18. doi: 10.1002/ana.410440704. [DOI] [PubMed] [Google Scholar]
- 9.Ross OA. A prognostic view on the application of individualized genomics in Parkinson's disease. Curr Genet Med Rep. 2013;1:52–57. doi: 10.1007/s40142-012-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis GC, Williams AC, Markey SP, Ebert MH, Caine ED, Reichert CM, Kopin IJ. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979;1:249–254. doi: 10.1016/0165-1781(79)90006-4. [DOI] [PubMed] [Google Scholar]
- 11.Ballard PA, Tetrud JW, Langston JW. Permanent human parkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): seven cases. Neurology. 1985;35:949–956. doi: 10.1212/wnl.35.7.949. [DOI] [PubMed] [Google Scholar]
- 12.Bove J, Perier C. Neurotoxin-based models of Parkinson's disease. Neuroscience. 2012;211:51–76. doi: 10.1016/j.neuroscience.2011.10.057. [DOI] [PubMed] [Google Scholar]
- 13.Bower JH, Maraganore DM, Peterson BJ, McDonnell SK, Ahlskog JE, Rocca WA. Head trauma preceding PD: a case-control study. Neurology. 2003;60:1610–1615. doi: 10.1212/01.wnl.0000068008.78394.2c. [DOI] [PubMed] [Google Scholar]
- 14.Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Huang X, Guo X, Mailman RB, Park Y, Kamel F, Umbach DM, Xu Q, Hollenbeck A, Schatzkin A, Blair A. Smoking duration, intensity, and risk of Parkinson disease. Neurology. 2010;74:878–884. doi: 10.1212/WNL.0b013e3181d55f38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chemaly RE, Moussalli AS. Parkinsonian syndrome as a complication of systemic lupus erythematosus. Report of a case and review of the literature. J Med Liban. 2012;60:103–105. [PubMed] [Google Scholar]
- 17.Li X, Sundquist J, Sundquist K. Subsequent risks of Parkinson disease in patients with autoimmune and related disorders: a nationwide epidemiological study from Sweden. Neurodgener Dis. 2012;10:277–284. doi: 10.1159/000333222. [DOI] [PubMed] [Google Scholar]
- 18.Rugbjerg K, Friis S, Ritz B, Schernhammer ES, Korbo L, Olsen JH. Autoimmune disease and risk for Parkinson disease: a population-based case-control study. Neurology. 2009;73:1462–1468. doi: 10.1212/WNL.0b013e3181c06635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samii A, Etminan M, Wiens MO, Jafari S. NSAID use and the risk of Parkinson's disease: systematic review and meta-analysis of observational studies. Drugs Aging. 2009;26:769–779. doi: 10.2165/11316780-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Walker RH, Spiera H, Brin MF, Olanow CW. Parkinsonism associated with Sjögren's syndrome: three cases and a review of the literature. Mov Disord. 1999;14:262–268. doi: 10.1002/1531-8257(199903)14:2<262::aid-mds1011>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 21.Ravenholt RT, Foege WH. 1918 influenza, encephalitis lethargica, parkinsonism. Lancet. 1982;2:860–864. doi: 10.1016/s0140-6736(82)90820-0. [DOI] [PubMed] [Google Scholar]
- 22.Langan SM, Groves RW, West J. The relationship between neurological disease and bullous pemphigoid: a population-based case-control study. J Invest Dermatol. 2011;131:631–636. doi: 10.1038/jid.2010.357. [DOI] [PubMed] [Google Scholar]
- 23.Miman O, Kusbeci OY, Aktepe OC, Cetinkaya Z. The probable relation between Toxoplasma gondii and Parkinson's disease. Neurosci Lett. 2010;475:129–131. doi: 10.1016/j.neulet.2010.03.057. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol. 2003;60:1059–1064. doi: 10.1001/archneur.60.8.1059. [DOI] [PubMed] [Google Scholar]
- 25.Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247:52–63. doi: 10.1111/j.1600-065X.2012.01124.x. [DOI] [PubMed] [Google Scholar]
- 26.Salmond RJ, Zamoyska R. The influence of mTOR on T helper cell differentiation and dendritic cell function. Eur J Immunol. 2011;41:2137–2141. doi: 10.1002/eji.201141523. [DOI] [PubMed] [Google Scholar]
- 27.Sommer F, Backhed F. The gut microbiota--masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 28.Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 29.McGuire SO, Ling ZD, Lipton JW, Sortwell CE, Collier TJ, Carvey PM. Tumor necrosis factor alpha is toxic to embryonic mesencephalic dopamine neurons. Exp Neurol. 2001;169:219–230. doi: 10.1006/exnr.2001.7688. [DOI] [PubMed] [Google Scholar]
- 30.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 31.Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, Anisman H, Hayley S, Park DS. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. 2007;27:3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tansey MG, Goldberg MS. Neuroinflammation in Parkinson's disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 34.Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7:429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- 35.Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70:1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- 36.Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. 2002;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- 37.Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Letter. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- 38.Mogi M, Harada M, Kondo T, Riederer P, Nagatsu T. Brain beta 2-microglobulin levels are elevated in the striatum in Parkinson's disease. J Neural Transm Park Dis Dement Sect. 1995;9:87–92. doi: 10.1007/BF02252965. [DOI] [PubMed] [Google Scholar]
- 39.Blum-Degen D, Müller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer's and de novo Parkinson's disease patients. Neurosci Letter. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- 40.Yamada T, McGeer PL, McGeer EG. Lewy bodies in Parkinson's disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992;84:100–104. doi: 10.1007/BF00427222. [DOI] [PubMed] [Google Scholar]
- 41.Koziorowski D, Tomasiuk R, Szlufik S, Friedman A. Inflammatory cytokines and NT-proCNP in Parkinson's disease patients. Cytokine. 2012;60:762–766. doi: 10.1016/j.cyto.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 42.Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- 43.Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration. 1996;5:137–143. doi: 10.1006/neur.1996.0020. [DOI] [PubMed] [Google Scholar]
- 44.Mogi M, Togari A, Tanaka K, Ogawa N, Ichinose H, Nagatsu T. Increase in level of tumor necrosis factor-alpha in 6-hydroxydopamine-lesioned striatum in rats is suppressed by immunosuppressant FK506. Neurosci Lett. 2000;289:165–168. doi: 10.1016/s0304-3940(00)01275-1. [DOI] [PubMed] [Google Scholar]
- 45.Theodore S, Cao S, McLean PJ, Standaert DG. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2008;67:1149–1158. doi: 10.1097/NEN.0b013e31818e5e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E, Effros RB, Chesselet MF. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol. 2012;237:318–334. doi: 10.1016/j.expneurol.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tofaris GK, Garcia Reitbock P, Humby T, Lambourne SL, O'Connell M, Ghetti B, Gossage H, Emson PC, Wilkinson LS, Goedert M, Spillantini MG. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emmer KL, Waxman EA, Covy JP, Giasson BI. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem. 2011;286:35104–35118. doi: 10.1074/jbc.M111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller RM, Kiser GL, Kaysser-Kranich T, Casaceli C, Colla E, Lee MK, Palaniappan C, Federoff HJ. Wild-type and mutant alpha-synuclein induce a multi-component gene expression profile consistent with shared pathophysiology in different transgenic mouse models of PD. Exp Neurol. 2007;204:421–432. doi: 10.1016/j.expneurol.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 51.Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 52.Pisani V, Stefani A, Pierantozzi M, Natoli S, Stanzione P, Franciotta D, Pisani A. Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson's disease. J Neuroinflammation. 2012;9:188. doi: 10.1186/1742-2094-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farkas E, De Jong GI, de Vos RA, Jansen Steur EN, Luiten PG. Pathological features of cerebral cortical capillaries are doubled in Alzheimer's disease and Parkinson's disease. Acta Neuropathol. 2000;100:395–402. doi: 10.1007/s004010000195. [DOI] [PubMed] [Google Scholar]
- 54.Faucheux BA, Bonnet AM, Agid Y, Hirsch EC. Blood vessels change in the mesencephalon of patients with Parkinson's disease. The Lancet. 1999;353:981–982. doi: 10.1016/S0140-6736(99)00641-8. [DOI] [PubMed] [Google Scholar]
- 55.Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miklossy J, Doudet DD, Schwab C, Yu S, McGeer EG, McGeer PL. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp Neurol. 2006;197:275–283. doi: 10.1016/j.expneurol.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 57.Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation. 2005;2:14. doi: 10.1186/1742-2094-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 59.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 60.Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging. 2008;29:739–752. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 61.Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging. 2008;29:1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su X, Federoff HJ, Maguire-Zeiss KA. Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox Res. 2009;16:238–254. doi: 10.1007/s12640-009-9053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilms H, Rosenstiel P, Romero-Ramos M, Arlt A, Schäfer H, Seegert D, Kahle PJ, Odoy S, Claasen JH, Holzknecht C, Brandenburg LO, Deuschl G, Schreiber S, Kirik D, Lucius R. Suppression of MAP kinases inhibits microglial activation and attenuates neuronal cell death induced by alpha-synuclein protofibrils. Int J Immunopathol Pharmacol. 2009;22:897–909. doi: 10.1177/039463200902200405. [DOI] [PubMed] [Google Scholar]
- 64.Lee EJ, Woo MS, Moon PG, Baek MC, Choi IY, Kim WK, Junn E, Kim HS. Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J Immunol. 2010;185:615–623. doi: 10.4049/jimmunol.0903480. [DOI] [PubMed] [Google Scholar]
- 65.Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC. The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation. 2011;8:166. doi: 10.1186/1742-2094-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marin-Teva JL, Cuadros MA, Martin-Oliva D, Navascues J. Microglia and neuronal cell death. Neuron Glia Biol. 2011;7:25–40. doi: 10.1017/S1740925X12000014. [DOI] [PubMed] [Google Scholar]
- 67.Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol. 2010;184:2261–2271. doi: 10.4049/jimmunol.0901852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Waldburger JM, Masternak K, Muhlethaler-Mottet A, Villard J, Peretti M, Landmann S, Reith W. Lessons from the bare lymphocyte syndrome: molecular mechanisms regulating MHC class II expression. Immunol Rev. 2000;178:148–165. doi: 10.1034/j.1600-065x.2000.17813.x. [DOI] [PubMed] [Google Scholar]
- 69.Luckheeram RV, Zhou R, Verma AD, Xia B. CD4(+)T cells: differentiation and functions. Clin Dev Immunol. 2012;2012:925135. doi: 10.1155/2012/925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marsh SG. System WHONCfFotH. Nomenclature for factors of the HLA system, update December 2012. Tissue Antigens. 2013;81:253–257. doi: 10.1111/tan.12077. [DOI] [PubMed] [Google Scholar]
- 71.Yamane H, Paul WE. Cytokines of the gamma(c) family control CD4+ T cell differentiation and function. Nat Immunol. 2012;13:1037–1044. doi: 10.1038/ni.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 73.McGeer PL, Itagaki S, McGeer EG. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 1988;76:550–557. doi: 10.1007/BF00689592. [DOI] [PubMed] [Google Scholar]
- 74.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol. 2003;106:518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 75.Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 76.McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- 77.Barcia C, Sanchez Bahillo A, Fernandez-Villalba E, Bautista V, Poza YPM, Fernandez-Barreiro A, Hirsch EC, Herrero MT. Evidence of active microglia in substantia nigra pars compacta of parkinsonian monkeys 1 year after MPTP exposure. Glia. 2004;46:402–409. doi: 10.1002/glia.20015. [DOI] [PubMed] [Google Scholar]
- 78.Vázquez-Claverie M, Garrido-Gil P, San Sebastián W, Izal-Azcárate A, Belzunegui S, Marcilla I, López B, Luquin MR. Acute and chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administrations elicit similar microglial activation in the substantia nigra of monkeys. J Neuropathol Exp Neurol. 2009;68:977–984. doi: 10.1097/NEN.0b013e3181b35e41. [DOI] [PubMed] [Google Scholar]
- 79.Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ. In vivo imaging of microglial activation with 11C.(R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 80.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 81.Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo Y, Deng X, Zheng W, Xu H, Song Z, Liang H, Lei J, Jiang X, Luo Z, Deng H. HLA rs3129882 variant in Chinese Han patients with late-onset sporadic Parkinson disease. Neurosci Lett. 2011;501:185–187. doi: 10.1016/j.neulet.2011.05.245. [DOI] [PubMed] [Google Scholar]
- 83.Ahmed I, Tamouza R, Delord M, Krishnamoorthy R, Tzourio C, Mulot C, Nacfer M, Lambert JC, Beaune P, Laurent-Puig P, Loriot MA, Charron D, Elbaz A. Association between Parkinson's disease and the HLA-DRB1 locus. Mov Disord. 2012;27:1104–1110. doi: 10.1002/mds.25035. [DOI] [PubMed] [Google Scholar]
- 84.Hill-Burns EM, Factor SA, Zabetian CP, Thomson G, Payami H. Evidence for more than one Parkinson's disease-associated variant within the HLA region. PLoS One. 2011;6:e27109. doi: 10.1371/journal.pone.0027109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 86.International Parkinson Disease. Genomics C, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, Stefansson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun C, Wei L, Luo F, Li Y, Li J, Zhu F, Kang P, Xu R, Xiao L, Liu Z, Xu P. HLA-DRB1 alleles are associated with the susceptibility to sporadic Parkinson's disease in Chinese Han population. PLoS One. 2012;7:e48594. doi: 10.1371/journal.pone.0048594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fiszer U, Mix E, Fredrikson S, Kostulas V, Link H. Parkinson's disease and immunological abnormalities: increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol Scand. 1994;90:160–166. doi: 10.1111/j.1600-0404.1994.tb02699.x. [DOI] [PubMed] [Google Scholar]
- 89.Botta-Orfila T, Sanchez-Pla A, Fernandez M, Carmona F, Ezquerra M, Tolosa E. Brain transcriptomic profiling in idiopathic and LRRK2-associated Parkinson's disease. Brain Res. 2012;1466:152–157. doi: 10.1016/j.brainres.2012.05.036. [DOI] [PubMed] [Google Scholar]
- 90.Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Juzwiak S, Kurzawski G, Tan EK, Drozdzik M. CARD15 variants in patients with sporadic Parkinson's disease. Neurosci Res. 2007;57:473–476. doi: 10.1016/j.neures.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 91.Dobbs SM, Dobbs RJ, Weller C, Charlett A. Link between Helicobacter pylori infection and idiopathic parkinsonism. Med Hypotheses. 2000;55:93–98. doi: 10.1054/mehy.2000.1110. [DOI] [PubMed] [Google Scholar]
- 92.Woulfe J, Hoogendoorn H, Tarnopolsky M, Muñoz DG. Monoclonal antibodies against Epstein-Barr virus cross-react with alpha-synuclein in human brain. Neurology. 2000;55:1398–1401. doi: 10.1212/wnl.55.9.1398. [DOI] [PubMed] [Google Scholar]
- 93.Jang H, Boltz D, McClaren J, Pani AK, Smeyne M, Korff A, Webster R, Smeyne RJ. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci. 2012;32:1545–1559. doi: 10.1523/JNEUROSCI.5123-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 2009;106:14063–14068. doi: 10.1073/pnas.0900096106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shoji H, Watanabe M, Itoh S, Kuwahara H, Hattori F. Japanese encephalitis and parkinsonism. J Neurol. 1993;240:59–60. doi: 10.1007/BF00838449. [DOI] [PubMed] [Google Scholar]
- 96.Ogata A, Tashiro K, Nukuzuma S, Nagashima K, Hall WW. A rat model of Parkinson's disease induced by Japanese encephalitis virus. J Neurovirol. 1997;3:141–147. doi: 10.3109/13550289709015803. [DOI] [PubMed] [Google Scholar]
- 97.Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, Cron RQ, Shacka JJ, Raman C, Standaert DG. MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci. 2013;33:9592–9600. doi: 10.1523/JNEUROSCI.5610-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cose S, Brammer C, Khanna KM, Masopust D, Lefrancois L. Evidence that a significant number of naive T cells enter non-lymphoid organs as part of a normal migratory pathway. Eur J Immunol. 2006;36:1423–1433. doi: 10.1002/eji.200535539. [DOI] [PubMed] [Google Scholar]
- 99.van der Most RG, Murali-Krishna K, Ahmed R. Prolonged presence of effector-memory CD8 T cells in the central nervous system after dengue virus encephalitis. Int Immunol. 2003;15:119–125. doi: 10.1093/intimm/dxg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 101.Bas J, Calopa M, Mestre M, Mollevi DG, Cutillas B, Ambrosio S, Buendia E. Lymphocyte populations in Parkinson's disease and in rat models of parkinsonism. J Neuroimmunol. 2001;113:146–152. doi: 10.1016/s0165-5728(00)00422-7. [DOI] [PubMed] [Google Scholar]
- 102.Stevens CH, Rowe D, Morel-Kopp MC, Orr C, Russell T, Ranola M, Ward C, Halliday GM. Reduced T helper and B lymphocytes in Parkinson's disease. J Neuroimmunol. 2012;252:95–99. doi: 10.1016/j.jneuroim.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 103.Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord. 2005;11:493–498. doi: 10.1016/j.parkreldis.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 104.Saunders JA, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, Meza JL, Santamaria PM, Bertoni JM, Murman DL, Ali HH, Standaert DG, Mosley RL, Gendelman HE. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson's disease. J Neuroimmune Pharmacol. 2012;7:927–938. doi: 10.1007/s11481-012-9402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Calopa M, Bas J, Callen A, Mestre M. Apoptosis of peripheral blood lymphocytes in Parkinson patients. Neurobiol Dis. 2010;38:1–7. doi: 10.1016/j.nbd.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 106.Niwa F, Kuriyama N, Nakagawa M, Imanishi J. Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson's disease. Geriatr Gerontol Int. 2012;12:102–107. doi: 10.1111/j.1447-0594.2011.00740.x. [DOI] [PubMed] [Google Scholar]
- 107.Fiszer U, Fredrikson S, Mix E, Olsson T, Link H. V region T cell receptor repertoire in Parkinson's disease. Acta Neurol Scand. 1996;93:25–29. doi: 10.1111/j.1600-0404.1996.tb00165.x. [DOI] [PubMed] [Google Scholar]
- 108.Alberio T, Pippione AC, Zibetti M, Olgiati S, Cecconi D, Comi C, Lopiano L, Fasano M. Discovery and verification of panels of T-lymphocyte proteins as biomarkers of Parkinson's disease. Sci Rep. 2012;2:953. doi: 10.1038/srep00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Migliore L, Petrozzi L, Lucetti C, Gambaccini G, Bernardini S, Scarpato R, Trippi F, Barale R, Frenzilli G, Rodilla V, Bonuccelli U. Oxidative damage and cytogenetic analysis in leukocytes of Parkinson's disease patients. Neurology. 2002;58:1809–1815. doi: 10.1212/wnl.58.12.1809. [DOI] [PubMed] [Google Scholar]
- 110.Cornetta T, Palma S, Aprile I, Padua L, Tonali P, Testa A, Cozzi R. Levodopa therapy reduces DNA damage in peripheral blood cells of patients with Parkinson's disease. Cell Biol Toxicol. 2009;25:321–330. doi: 10.1007/s10565-008-9086-6. [DOI] [PubMed] [Google Scholar]
- 111.Blandini F, Mangiagalli A, Cosentino M, Marino F, Samuele A, Rasini E, Fancellu R, Martignoni E, Riboldazzi G, Calandrella D, Frigo GM, Nappi G. Peripheral markers of apoptosis in Parkinson's disease: the effect of dopaminergic drugs. Ann N Y Acad Sci. 2003;1010:675–678. doi: 10.1196/annals.1299.123. [DOI] [PubMed] [Google Scholar]
- 112.Lira A, Kulczycki J, Slack R, Anisman H, Park DS. Involvement of the Fc gamma receptor in a chronic N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of dopaminergic loss. J Biol Chem. 2011;286:28783–28793. doi: 10.1074/jbc.M111.244830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fleisher TA, Oliveira JB. Monogenic defects in lymphocyte apoptosis. Curr Opin Allergy Clin Immunol. 2012;12:609–615. doi: 10.1097/ACI.0b013e3283588da0. [DOI] [PubMed] [Google Scholar]
- 114.Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Research Reviews. 2004;44:65–81. doi: 10.1016/j.brainresrev.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 115.Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Besong-Agbo D, Wolf E, Jessen F, Oechsner M, Hametner E, Poewe W, Reindl M, Oertel WH, Noelker C, Bacher M, Dodel R. Naturally occurring α-synuclein autoantibody levels are lower in patients with Parkinson disease. Neurology. 2013;80:169–175. doi: 10.1212/WNL.0b013e31827b90d1. [DOI] [PubMed] [Google Scholar]
- 117.Han M, Nagele E, DeMarshall C, Acharya N, Nagele R. Diagnosis of Parkinson's disease based on disease-specific autoantibody profiles in human sera. PLoS One. 2012;7:e32383. doi: 10.1371/journal.pone.0032383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Derecki NC, Cardani AN, Yang CH, Quinnies KM, Crihfield A, Lynch KR, Kipnis J. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–1080. doi: 10.1084/jem.20091419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, Burmester GR, Lipsky PE, Dorner T. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. 2002;46:2160–2171. doi: 10.1002/art.10445. [DOI] [PubMed] [Google Scholar]
- 121.Fekete A, Soos L, Szekanecz Z, Szabo Z, Szodoray P, Barath S, Lakos G. Disturbances in B- and T-cell homeostasis in rheumatoid arthritis: suggested relationships with antigen-driven immune responses. J Autoimmun. 2007;29:154–163. doi: 10.1016/j.jaut.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 122.Souto-Carneiro MM, Mahadevan V, Takada K, Fritsch-Stork R, Nanki T, Brown M, Fleisher TA, Wilson M, Goldbach-Mansky R, Lipsky PE. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res Ther. 2009;11:R84. doi: 10.1186/ar2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carvey PM, McRae A, Lint TF, Ptak LR, Lo ES, Goetz CG, Klawans HL. The potential use of a dopamine neuron antibody and a striatal-derived neurotrophic factor as diagnostic markers in Parkinson's disease. Neurology. 1991;41:53–58. doi: 10.1212/wnl.41.5_suppl_2.53. [DOI] [PubMed] [Google Scholar]
- 124.Papachroni KK, Ninkina N, Papapanagiotou A, Hadjigeorgiou GM, Xiromerisiou G, Papadimitriou A, Kalofoutis A, Buchman VL. Autoantibodies to alpha-synuclein in inherited Parkinson's disease. J Neurochem. 2007;101:749–756. doi: 10.1111/j.1471-4159.2006.04365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson's disease. Brain. 2005;128:2665–2674. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- 126.Chen S, Le WD, Xie WJ, Alexianu ME, Engelhardt JI, Siklos L, Appel SH. Experimental destruction of substantia nigra initiated by Parkinson disease immunoglobulins. Arch Neurol. 1998;55:1075–1080. doi: 10.1001/archneur.55.8.1075. [DOI] [PubMed] [Google Scholar]
- 127.He Y, Le WD, Appel SH. Role of Fcγ Receptors in Nigral Cell Injury Induced by Parkinson Disease Immunoglobulin Injection into Mouse Substantia Nigra. Experimental Neurology. 2002;176:322–327. doi: 10.1006/exnr.2002.7946. [DOI] [PubMed] [Google Scholar]
- 128.Cao S, Theodore S, Standaert DG. Fcgamma receptors are required for NF-kappaB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson's disease. Mol Neurodegener. 2010;5:42. doi: 10.1186/1750-1326-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord. 2013;28:14–23. doi: 10.1002/mds.25249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- 132.Houlden H, Singleton AB. The genetics and neuropathology of Parkinson's disease. Acta Neuropathol. 2012;124:325–338. doi: 10.1007/s00401-012-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shin EC, Cho SE, Lee DK, Hur MW, Paik SR, Park JH, Kim J. Expression patterns of alpha-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cells. 2000;10:65–70. doi: 10.1007/s10059-000-0065-x. [DOI] [PubMed] [Google Scholar]
- 134.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, 3rd, Su AI. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Austin SA, Floden AM, Murphy EJ, Combs CK. Alpha-synuclein expression modulates microglial activation phenotype. J Neurosci. 2006;26:10558–10563. doi: 10.1523/JNEUROSCI.1799-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Austin SA, Rojanathammanee L, Golovko MY, Murphy EJ, Combs CK. Lack of alpha-synuclein modulates microglial phenotype in vitro. Neurochem Res. 2011;36:994–1004. doi: 10.1007/s11064-011-0439-9. [DOI] [PubMed] [Google Scholar]
- 138.Tanji K, Mori F, Imaizumi T, Yoshida H, Matsumiya T, Tamo W, Yoshimoto M, Odagiri H, Sasaki M, Takahashi H, Satoh K, Wakabayashi K. Upregulation of alpha-synuclein by lipopolysaccharide and interleukin-1 in human macrophages. Pathol Int. 2002;52:572–577. doi: 10.1046/j.1440-1827.2002.01385.x. [DOI] [PubMed] [Google Scholar]
- 139.Alessandri C, Barbati C, Vacirca D, Piscopo P, Confaloni A, Sanchez M, Maselli A, Colasanti T, Conti F, Truglia S, Perl A, Valesini G, Malorni W, Ortona E, Pierdominici M. T lymphocytes from patients with systemic lupus erythematosus are resistant to induction of autophagy. FASEB J. 2012;26:4722–4732. doi: 10.1096/fj.12-206060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev. 2012;249:205–217. doi: 10.1111/j.1600-065X.2012.01147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 142.Latourelle JC, Sun M, Lew MF, Suchowersky O, Klein C, Golbe LI, Mark MH, Growdon JH, Wooten GF, Watts RL, Guttman M, Racette BA, Perlmutter JS, Ahmed A, Shill HA, Singer C, Goldwurm S, Pezzoli G, Zini M, Saint-Hilaire MH, Hendricks AE, Williamson S, Nagle MW, Wilk JB, Massood T, Huskey KW, Laramie JM, DeStefano AL, Baker KB, Itin I, Litvan I, Nicholson G, Corbett A, Nance M, Drasby E, Isaacson S, Burn DJ, Chinnery PF, Pramstaller PP, Al-hinti J, Moller AT, Ostergaard K, Sherman SJ, Roxburgh R, Snow B, Slevin JT, Cambi F, Gusella JF, Myers RH. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson's disease: the GenePD study. BMC Med. 2008;6:32. doi: 10.1186/1741-7015-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F, Clementi M, Bonifati V, Pezzoli G. Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology. 2007;68:1141–1143. doi: 10.1212/01.wnl.0000254483.19854.ef. [DOI] [PubMed] [Google Scholar]
- 144.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Westerlund M, Belin AC, Anvret A, Bickford P, Olson L, Galter D. Developmental regulation of leucine-rich repeat kinase 1 and 2 expression in the brain and other rodent and human organs: Implications for Parkinson's disease. Neuroscience. 2008;152:429–436. doi: 10.1016/j.neuroscience.2007.10.062. [DOI] [PubMed] [Google Scholar]
- 146.Galter D, Westerlund M, Carmine A, Lindqvist E, Sydow O, Olson L. LRRK2 expression linked to dopamine-innervated areas. Ann Neurol. 2006;59:714–719. doi: 10.1002/ana.20808. [DOI] [PubMed] [Google Scholar]
- 147.Melrose H, Lincoln S, Tyndall G, Dickson D, Farrer M. Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience. 2006;139:791–794. doi: 10.1016/j.neuroscience.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 148.Taymans JM, Van den Haute C, Baekelandt V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem. 2006;98:951–961. doi: 10.1111/j.1471-4159.2006.03919.x. [DOI] [PubMed] [Google Scholar]
- 149.West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 151.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 152.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 153.Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, Moore DJ, Dawson TM, Dawson VL, Chiosis G, Cookson MR, Cai H. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci. 2008;28:3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR, LaVoie MJ, Gris D, Park DS, Angel JB, Shen J, Philpott DJ, Schlossmacher MG. Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. 2011;118:795–808. doi: 10.1007/s00702-011-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Miklossy J, Arai T, Guo JP, Klegeris A, Yu S, McGeer EG, McGeer PL. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol. 2006;65:953–963. doi: 10.1097/01.jnen.0000235121.98052.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Consortium NIG, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Belgian-French IBDC, Wellcome Trust Case Control C. Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, Cui Y, Yan XX, Yang HT, Yang RD, Chu TS, Zhang C, Zhang L, Han JW, Yu GQ, Quan C, Yu YX, Zhang Z, Shi BQ, Zhang LH, Cheng H, Wang CY, Lin Y, Zheng HF, Fu XA, Zuo XB, Wang Q, Long H, Sun YP, Cheng YL, Tian HQ, Zhou FS, Liu HX, Lu WS, He SM, Du WL, Shen M, Jin QY, Wang Y, Low HQ, Erwin T, Yang NH, Li JY, Zhao X, Jiao YL, Mao LG, Yin G, Jiang ZX, Wang XD, Yu JP, Hu ZH, Gong CH, Liu YQ, Liu RY, Wang DM, Wei D, Liu JX, Cao WK, Cao HZ, Li YP, Yan WG, Wei SY, Wang KJ, Hibberd ML, Yang S, Zhang XJ, Liu JJ. Genomewide association study of leprosy. N Engl J Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. [DOI] [PubMed] [Google Scholar]
- 164.Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12:1063–1070. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bonifati V. Autosomal recessive parkinsonism. Parkinsonism & Related Disorders. 2012;18:S4–S6. doi: 10.1016/S1353-8020(11)70004-9. [DOI] [PubMed] [Google Scholar]
- 166.Walden H, Martinez-Torres RJ. Regulation of Parkin E3 ubiquitin ligase activity. Cell Mol Life Sci. 2012;69:3053–3067. doi: 10.1007/s00018-012-0978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 168.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013;110:6400–6405. doi: 10.1073/pnas.1221132110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Alves da Costa C, Checler F. Parkin: Much More than A Simple Ubiquitin Ligase. Neurodgener Dis. 2012;10:49–51. doi: 10.1159/000332803. [DOI] [PubMed] [Google Scholar]
- 171.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 172.Zhu XR, Maskri L, Herold C, Bader V, Stichel CC, Gunturkun O, Lubbert H. Non-motor behavioural impairments in parkin-deficient mice. Eur J Neurosci. 2007;26:1902–1911. doi: 10.1111/j.1460-9568.2007.05812.x. [DOI] [PubMed] [Google Scholar]
- 173.Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Trevino I, O'Brien DE, Casey B, Goldberg MS, Tansey MG. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tran TA, Nguyen AD, Chang J, Goldberg MS, Lee JK, Tansey MG. Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS One. 2011;6:e23660. doi: 10.1371/journal.pone.0023660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mira MT, Alcais A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, Mai CP, Nguyen TH, Nguyen NB, Pham XK, Sarno EN, Alter A, Montpetit A, Moraes ME, Moraes JR, Dore C, Gallant CJ, Lepage P, Verner A, Van De Vosse E, Hudson TJ, Abel L, Schurr E. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature. 2004;427:636–640. doi: 10.1038/nature02326. [DOI] [PubMed] [Google Scholar]
- 176.Ali S, Vollaard AM, Widjaja S, Surjadi C, van de Vosse E, van Dissel JT. PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin Exp Immunol. 2006;144:425–431. doi: 10.1111/j.1365-2249.2006.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Boska MD, Lewis TB, Destache CJ, Benner EJ, Nelson JA, Uberti M, Mosley RL, Gendelman HE. Quantitative 1H magnetic resonance spectroscopic imaging determines therapeutic immunization efficacy in an animal model of Parkinson's disease. J Neurosci. 2005;25:1691–1700. doi: 10.1523/JNEUROSCI.4364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Johnston J, So TY. First-line disease-modifying therapies in paediatric multiple sclerosis: a comprehensive overview. Drugs. 2012;72:1195–1211. doi: 10.2165/11634010-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 180.Benner EJ, Mosley RL, Destache CJ, Lewis TB, Jackson-Lewis V, Gorantla S, Nemachek C, Green SR, Przedborski S, Gendelman HE. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:9435–9440. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gendelman H. Clinical Trials Gov. National Library of Medicine; 2013. [Google Scholar]
- 182.McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson's disease. J Neurosci. 2006;26:9365–9375. doi: 10.1523/JNEUROSCI.1504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McCoy MK, Ruhn KA, Martinez TN, McAlpine FE, Blesch A, Tansey MG. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol Ther. 2008;16:1572–1579. doi: 10.1038/mt.2008.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Harms AS, Barnum CJ, Ruhn KA, Varghese S, Trevino I, Blesch A, Tansey MG. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson's disease. Mol Ther. 2011;19:46–52. doi: 10.1038/mt.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 186.Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 187.Salama M, Ellaithy A, Helmy B, El-Gamal M, Tantawy D, Mohamed M, Sheashaa H, Sobh M, Arias-Carrión O. Colchicine protects dopaminergic neurons in a rat model of Parkinson's disease. CNS Neurol Disord Drug Targets. 2012;11:836–843. doi: 10.2174/1871527311201070836. [DOI] [PubMed] [Google Scholar]
- 188.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov. 2013;12:130–146. doi: 10.1038/nrd3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Larsen CP, Grinyó J, Medina-Pestana J, Vanrenterghem Y, Vincenti F, Breshahan B, Campistol JM, Florman S, Rial Mdel C, Kamar N, Block A, Di Russo G, Lin CS, Garg P, Charpentier B. Belatacept-based regimens versus a cyclosporine A-based regimen in kidney transplant recipients: 2-year results from the BENEFIT and BENEFIT-EXT studies. Transplantation. 2010;90:1528–1535. doi: 10.1097/TP.0b013e3181ff87cd. [DOI] [PubMed] [Google Scholar]
- 190.Rudick R, Polman C, Clifford D, Miller D, Steinman L. Natalizumab: bench to bedside and beyond. JAMA Neurol. 2013;70:172–182. doi: 10.1001/jamaneurol.2013.598. [DOI] [PubMed] [Google Scholar]
- 191.Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol. 2011;29:550–557. doi: 10.1016/j.tibtech.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Furtado J, Isenberg DA. B cell elimination in systemic lupus erythematosus. Clin Immunol. 2013;146:90–103. doi: 10.1016/j.clim.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 193.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]