

Abstract

Using nanofluidic channels in PDMS of cross section 500 nm × 2 μm, we demonstrate the trapping and interrogation of individual, isolated mitochondria. Fluorescence labeling demonstrates the immobilization of mitochondria at discrete locations along the channel. Interrogation of mitochondrial membrane potential with different potential sensitive dyes (JC-1 and TMRM) indicates the trapped mitochondria are vital in the respiration buffer. Fluctuations of the membrane potential can be observed at the single mitochondrial level. A variety of chemical challenges can be delivered to each individual mitochondrion in the nanofluidic system. As sample demonstrations, increases in the membrane potential are seen upon introduction of OXPHOS substrates into the nanofluidic channel. Introduction of Ca2+ into the nanochannels induces mitochondrial membrane permeabilization (MMP), leading to depolarization, observed at the single mitochondrial level. A variety of applications in cancer biology, stem cell biology, apoptosis studies, and high throughput functional metabolomics studies can be envisioned using this technology.
Mitochondria play a multitude of roles in cell life. For example, they generate most of the ATP needed for cellular processes, modulate the concentration of cytoplasmic and mitochondrial matrix calcium, produce and regulate reactive oxygen species, and control intrinsic apoptosis or programmed cell death. Consequently, mitochondrial dysfunctions can significantly affect the body homeostasis. In fact, there is a growing body of evidence that suggest the link between mitochondrial dysfunctions and pathological conditions such as degenerative diseases, diabetes, cancer, and aging.1,2
Advances in live imaging techniques have uncovered large morphological, functional, and behavioral heterogeneity within mitochondrial populations, not just between mitochondria of different cells but also between mitochondria in a single cell.3,4 Studying this heterogeneity can reveal further understanding of mitochondrial functions. However, so far studies on heterogeneity of single mitochondria have not resulted in very conclusive results. Part of this might be attributed to the technical diffculties of imaging individual mitochondria; mitochondria moving out of plane of focus, background fluorescence, not enough control on the chemical environment surrounding each individual mitochondrion, etc. This shows the need for development of innovative, advanced technologies appropriate for single mitochondrion studies.
The advent of microfluidics and nanofluidics can address the shortcomings of traditional approaches in studying isolated mitochondria. The ability to control fluids in small volumes enables assays requiring significantly less sample size, and the precise and controlled delivery of chemicals makes it ideal for testing specific hypotheses involving different processes.
Recently we miniaturized a classic assay of mitochondrial membrane potential requiring only 1000 cells.5,6 However, even though this is a 4 orders of magnitude improvement, it did not get us down to the single mitochondrial or even single cell level. Extensive research this decade has been carried out to assay and analyze individual cells using flow cytometry and microfluidic devices.7 This has given rise to extraordinary technological capabilities, such as the possibility of finding one circulating tumor cell per 109 healthy cells in whole blood for cancer diagnosis.8 In contrast, the field of analysis of individual subcellular organelles is in its infancy. Here we demonstrate a first step in this direction, developing a novel platform to trap single mitochondria inside a nanochannel where they can be subjected to different chemical environments and interrogated via a variety of fluorescent dyes. We demonstrate that single isolated mitochondria remain vital and functional in this trapped state.
EXPERIMENTAL SECTION
Fabrication Process
Silicon wafer with photoresist patterns is used as the mold for soft lithography of PDMS. To fabricate the mold, silicon wafer was cleaned with 120 °C Piranha solution (3:1 mixture of sulfuric acid and hydrogen peroxide) for 1 h, rinsed in DI water, dried with nitrogen, and dehydrated at 200 °C. Wafer was primed with hexamethyldisilazane (HMDS). Channel patterns were fabricated by photolithography of Microposit SC1827 positive photoresist. Photoresist was spin coated on the wafer at 3500 rpm for 30 s, prebaked at 90 °C for 30 min in a convection oven, and exposed to G-Line UV light through a chromium(105 nm)/ glass mask using Karl Suss MA6 mask aligner with soft contact between the mask and the wafer. The dosage of exposure was set at 160 mJ/cm2. Microposit MF-319 was used for developing. The developing takes around 25 s.
We use soft contact for lithography; the contact between the mask and the resist surface is not very close, and there is a slight gap between the two surfaces (compared to vacuum contact). Due to diffraction of light passing through the mask, light that reaches the plane of the wafer does not have a step function intensity distribution; regions of the photoresist near the pattern edges get some light exposure even though they are covered by opaque parts of the mask. This is a well-known phenomenon in positive photoresist lithography that usually leads to trapezoidal cross section of the developed photoresist instead of a rectangular cross section. It can be avoided or reduced by using vacuum contact between the mask and the resist surface, lowering the dosage of exposure and using a photoresist with higher contrast. Here, we are using this phenomenon to get multiple heights with a single lithography step. Due to small width of the channel, the whole width of the channel receives some dosage of exposure; for each channel the edges get more exposure, while the light intensity decreases gradually as it gets closer to the center of the channel. The resist regions that receive a higher exposure dissolve faster in the developer. When the wafer is placed in the developer solution, the regions that are exposed through the transparent parts of the mask have the fastest dissolution rate, while the resist at the trap channel region gets dissolved at a slower rate, with the lowest rate at the center (vertical axis of symmetry); therefore, the trap channels lose some of their height. In case of the larger features (access channels), the edges get some light exposure, but the mask protects the bulk of the feature from light exposure; therefore, larger features keep their original height after the development step. This method is very reproducible, and out of the 10 molds that we fabricated, we could get these multiple height patterns on 7 of them. Using the given lithography parameters the height of the nano-channels ranged from 450 nm to750 nm.
Silicone elastomer and curing agent (Sylgard 184, Dow Corning Co.) were mixed thoroughly at a 10:1 weight ratio. PDMS was degassed for 30 min in a vacuum desiccator and poured over salinized mold to a thickness of 3 mm. The mold was placed in a 70 °C curing oven overnight. After curing, PDMS was easily cut and peeled off from the mold. Inlet and outlet holes were punched with a diameter of 0.63 mm to allow connection to the syringe pump. To seal the channels the chips were exposed to 70 W oxygen plasma treatment at 100 mTorr for 20 s, immediately placed on piranha cleaned glass slides, and left in 70 °C oven for 20 min to complete the bonding process. The oxygen plasma treatment causes the PDMS to become hydrophilic, making it easy to introduce the aqueous solution into the channels after bonding. However, after about a day the sidewalls would become hydrophobic again, and the channels were not reusable.
Fluidic channels were filled with the respiration buffer without mitochondria first, and the buffer containing mitochondria was flown into the channels later.
Mitochondria Isolation and Sample Preparation
Mitochondria were isolated from the human cervical cancer cell line HeLa (ATCC, CCL-2). The adherent cells were cultured and maintained in log growth phase in media consisting of EMEM (ATCC, 30-2003) supplemented with 10% FBS (Invitrogen, 10438-018) and 1% penicillin-streptomycin (ATCC, 30-2300). All other chemicals were obtained from Sigma Aldrich, unless otherwise noted. The mitochondrial isolation protocol was adapted from ref 6. Details of the isolation protocol are discussed in the Supporting Information, p 14.
Imaging
Mitochondria were imaged with Olympus IX71 inverted fluorescence microscope, equipped with a 12 bit monochromatic CCD camera (QIClick-F-M-12), a 60×, 0.7 NA objective, 120 W mercury vapor excitation light source and standard FITC (490 nm-525 nm) and TRITC (557 nm-576 nm) filters. Image analysis was done with ImageJ software. A 3 × 3 median filter was used to remove noise. Images in this paper are false-colored red or green, depending on the filter set used, for clarity. For fluorescence intensity measurements we manually selected the area with the highest intensity at the center of each mitochondrion image and averaged the fluorescence intensity over the selected area. Background fluorescence was removed by choosing 3 neighbor regions with the same area selected for the mitochondria. The fluorescence intensity was averaged over the three regions and subtracted from the mitochondrial intensity. The standard deviation in the background fluorescent intensity causes some small error (less than 10% in most cases) in calculating the fluorescence intensity. This error has been taken into account for the ratio calculations. The resolution of the imaging system and the apparent versus real size of the mitochondria is discussed in the Supporting Information, p 11.
Fluorescence Dyes
Mito Tracker green (MTG), obtained from life technologies, is a mitochondrial selective fluorescent probe that binds to mitochondria proteins regardless of the membrane potential of the mitochondria9 and emits a bright green fluorescence at 519 nm when excited at 490 nm. Mitotracker Green forms a dye-protein complex with free thiol groups inside the mitochondria, yielding significantly higher fluorescence than free dye in aqueous solution. It is reported that this increase could be as much as 40-fold.10 We used MTG to visualize the mitochondria in the channels. To stain the mitochondria, we diluted the dye in dimethyl sulfoxide (DMSO) to a concentration of 100 μM and then diluted it 1000-fold in the respiration buffer to a concentration of 100 nM.
To monitor the membrane potential of the trapped mitochondria, we used 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) obtained from Sigma Aldrich. In energized mitochondria the membrane potential promotes an uptake of JC-1 into the mitochondrial matrix according to the Nernst equation. The high concentration of JC-1 forms aggregates inside the mitochondria. When JC-1 is excited at 488 nm, it emits with peaks at 530 nm (green) and 590 nm (red). The intensity of the red emission strongly depends on the concentration of J-aggregates and therefore on the membrane potential of the mitochondria. Energized mitochondria emit a bright red fluorescence at 590 nm. In mitochondria with low membrane potential the dye does not form aggregates inside the mitochondria and the red fluorescence decreases. It has been shown that there is a significant correlation between the ratio of red to green fluorescence of this dye and the membrane potential of mitochondria.11,12
We also used tetramethylrhodamine methyl ester (TMRM) purchased from Life Technologies in some experiments. TMRM is a lipophilic cationic dye with red-orange fluorescence that is accumulated by mitochondria according to the Nernst equation. The fluorescence intensity of the stained mitochondria can be used to estimate the mitochondrial membrane potential. By monitoring the time-dependence of the fluorescence of the inner mitochondrial membrane, one can obtain qualitative information about the dynamics of the membrane potential. It was observed that TMRM sticks to the PDMS channel and produces a high background fluorescence which makes it difficult to distinguish the mitochondria from background (Figure S-4, Supporting Information).
For JC-1 assays, JC-1 was dissolved in DMSO and then added to the mitochondria sample to a final dye concentration of 300 nM. The solution was mixed and incubated at room temperature for 7 min. Images of JC-1 stained isolated mitochondria on a glass slide are shown in Figure S-1 (Supporting Information). TMRM was added with the same method. We tried various concentrations of TMRM from 100 nM to 2 nM.
RESULTS AND DISCUSSION
Mitochondria Trap Devices
The goal of this project was to design a nanochannel device with dimensions small enough to trap individual mitochondria as a demonstration platform for manipulation and interrogation under controlled conditions at the single mitochondrial level. Mitochondrial morphology is varied, both in vivo and in vitro. A typical morphology is a cigar shape of width from 200 nm to 1 μm.13 For this, standard photolithographically defined PDMS channels have not yet been pushed to the submicrometer scale,14 while nanochannel technology based on etching15 is too small to pass single mitochondria
The mitochondria trapping device is a nanochannel array, with two access channels and two access holes for fluid inlet/ outlet as shown in Figure 1. The nanochannels have a trapezoid cross-section that is 3.5 μm at base and gradually narrows down to around 400 nm. The average width of the channels (the width at middle of the trapezoid) is around 2 μm which is larger than the diameter of mitochondria (0.2−1.2 μm),13 and the height of the nanochannels at its highest point (0.45−0.75 μm) is almost equal to the average diameter of the mitochondria. (The terminology nanochannel has been used to describe channels with dimensions smaller than 1 μm.16 For more discussion please see the Supporting Information, p 17.)
Figure 1.

Geometry of the mitochondria trapping devices. a) Schematic of the device and trapping mechanism. b) Bright field microscope image of the device. c) SEM images of the channels in PDMS.
These dimensions are measured from AFM images (Figure S-8 of the Supporting Information). The nanochannels are 500 μm long and are connected to two 100 μm wide, 2 μm high access channels. The device is fabricated using simple, economic, and high throughput soft lithography of PDMS.
Immobilization of Mitochondria
Mitochondria solution is introduced into the channels with a syringe pump. Individual mitochondria start to get trapped in the nanochannels, and their population gradually goes up as the flow continues. In Figure 2, an example trapping experimental run is shown. Mitochondria stained with MTG are flown into channels with a rate of 10 μL/h. Time-lapse microscopy of the channels is performed at the same time. In all experiments where mitochondria were imaged, their position was fixed after the initial introduction. The chemical environment can be adjusted through additional fluid flow at rates of up to a few μL per minute. This is shown, for example, in Figure 2. Thus, the mitochondria are gently trapped inside the nanochannels, enabling access to fluid for them, but they are not moving.
Figure 2.

Series of time images of MTG labeled mitochondria. In each image, a new mitochondrion appears that has been flown in from the reservoir and trapped inside the nanochannel. Red arrows indicate the addition of new mitochondria. Dashed lines in panel f suggest the outline of the channels. Scale bar is 10 μm.
Trapping Mechanism
The trapping mechanism is not mitochondrial interactions with the channel sidewalls. This was shown by performing a similar experiment with larger channel sizes but otherwise nominally identical conditions. For the larger channels, the mitochondria do not stick to PDMS surfaces (Supporting Information, Figure S-3). It has been shown that oxidized PDMS has negative surface charge.17 Thus, it is likely that an electrostatic repulsion between the mitochondria and the sidewalls prevents adhesion between the negatively charged mitochondrion and negatively charged channel surface.
The mitochondria are physically trapped along the channels (Figure 3). Since the height of the channel at the highest point is similar to the average diameter of mitochondria but the width is around two times larger, the channels act like a filter where fluid can pass through but individual mitochondria are trapped along the channels one by one. Due to variations in size of individual mitochondria and also the trapezoidal cross section of the channels, occasionally we see that one mitochondrion is trapped in some location in the channel, but a smaller mitochondrion can pass along the first mitochondrion and get trapped at a further location. Exact determination of trapping locations requires mathematical simulations, but based on our experimental observations we believe that mitochondria that are already trapped at the channel entrance due to the small height of the channel behave like obstacles that disrupt the flow in the narrow channel and create vertical components in the flow direction (vertical to the channel direction). The flow direction, variations in mitochondria size, and the trapezoidal cross section of the channel result in mitochondria getting trapped at random locations along the channel. We have found experimentally that for a flow rate of 10 μL/h, the concentration of mitochondria in the flow buffer that results in an appropriate number of trapped mitochondria for our imaging setup is around 50 μg/mL of mitochondrial protein. This is discussed in the Supporting Information (Figure S-10). This concentration is over an order of magnitude lower than what is used in most experiments.18,19 The number of trapped mitochondria for each experiment using the described conditions is generally 20−40 in the field of view. It is possible to increase the throughput by increasing the number of nanochannels and using an imaging system with higher resolution and larger field of view.
Figure 3.
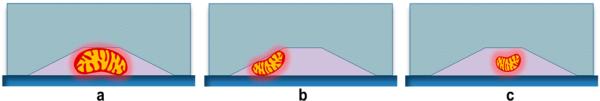
Cross section of the channels and trapping mechanism of mitochondria. a) Larger mitochondria get trapped at the middle of the channels. b) Smaller mitochondria get trapped at the corners, leaving room for other mitochondria to pass. c) Some small mitochondria will pass through the middle of the channel without getting trapped.
Δψm Assays
The mitochondria in our channels are vital and sustain a membrane potential. This can be demonstrated using the fluorescence dye JC-1
In Figure 4a-c, separate green and red fluorescence images of the same mitochondria are shown as well as their superposition over the bright-field image of the nanochannels. JC-1 stained mitochondria are provided with 10 mM sodium succinate and flown into channels with a rate of 10 μL/h. After 2 min the flow is stopped and the channels are imaged. Clearly, the mitochondria are trapped individually in the nanochannels, and a large percentage of them are bright red, indicating the membrane potential is still large. The blue bars in Figure 4d show the histogram of red/green fluorescence intensity for 31 mitochondria that were in the field of imaging. Around 80% of the mitochondria have a ratio of 3 or higher. As a control we performed the same experiment with substrate deprived mitochondria that are expected to have a lower membrane potential. The distribution of red/green fluorescence intensity ratio for mitochondria in this sample is shown by the white bars in Figure 4d. Out of 39 mitochondria, 65% have a ratio of 2 or lower.
Figure 4.

Image of JC-1 stained trapped mitochondria. a) Image taken with TRITC filter. b) Same mitochondria imaged with a FITC filter. c) Overlap of a, b and bright field image of the channels. d) Histogram of ratio of red to green fluorescence for substrate fed mitochondria and substrate deprived mitochondria (Total number of substrate fed mitochondria is 31 and total number of substrate deprived mitochondria is 39).
Time Dependence of Δψm of Individual Mitochondria
To monitor the time dependent membrane potential we performed time-lapse microscopy of stained trapped mitochondria for over ten minutes with continuous illumination.
Mitochondria stained with 30 nM TMRM were introduced into our nanochannels, and an image was captured every 5 s, with an image acquisition time of 2.5 s per image. As can be seen in Figure 5, the fluorescence intensity from TMRM stained mitochondria remains relatively constant indicating that the trapped mitochondria maintain their membrane potential for a long time.
Figure 5.

Normalized TMRM fluorescence intensity from two individual trapped mitochondria imaged every 5 s. Arrows in the inset point to the mitochondria the intensity of which is shown. Dashed lines suggest the outline of the channels.
The substrate response and MMP experiments described next were performed using JC-1. Because of our microscope setup we can only use one filter cube at a time for time-lapse fluorescence microscopy and we cannot measure both red and green simultaneously, so we only monitored the red intensity (which has been reported to be linearly correlated with membrane potential).20 To account for the inaccuracies such as those caused by the apparent size of mitochondria compared to their real size that might be caused by using the red fluorescence only, we are showing the normalized traces for each mitochondrion. Each trace is divided by the fluorescence intensity measured for that mitochondrion at time zero, so that all the traces start at intensity of one. This way we are not comparing different mitochondria, but we are looking at the trend of membrane potential change for one mitochondrion.
Substrate Modulation Δψm of Individual Mitochondria
Without OXPHOS substrates, the electron transport chain is idle, and the membrane potential remains in its basal, resting state. In order to demonstrate the ability to chemically modulate the electron transport chain and the bioenergetic state of the mitochondria in our nanochannels, we performed a series of experiments with and without OXPHOS substrates pyruvate/malate present in the respiration buffer.
In Figure 6, we show two sets of experiments in which the mitochondria are labeled and imaged with JC-1. In (a), substrates are not used. In (b), OXPHOS substrates, 5 mM pyruvate and 5 mM malate, are added to JC-1 stained mitochondria respiration buffer just before flowing the mitochondria in the channels. This activates the electron transport chain and initially increases the mitochondrial membrane potential Δψm. As mitochondria gradually consume the substrates, the substrate concentration decreases; therefore, the membrane potential and fluorescence intensity gradually drop and become identical to the sample without substrates (Supporting Information Figure S-5).
Figure 6.

Fluorescence intensity measurement of JC-1 stained mitochondria. a) Substrates are not used. b) OXPHOS substrates (5 mM pyruvate and 5 mM malate) are added to respiration buffer just before flowing the mitochondria into the nanofluidic channel. This activates the electron transport chain and increases the mitochondrial membrane potential Δψm initially.
Typical normalized red fluorescence intensity of JC-1 stained mitochondria is shown in Figure 6a (each curve shows the fluorescence from a single mitochondrion.) It is observed that for JC-1 stained mitochondria the fluorescence intensity has dropped around 50% during the time span that they were continuously illuminated for photography, and we attribute this to photobleaching of the JC-1 (also reported by other groups)21,22 since experiments with identical buffer but different stain (TMRM) showed no appreciable decay in the membrane potential (Figure 5).
Similarly, in Figure 6b, the fluorescence intensity decays with time but this time with a higher rate, due to gradual consumption of OXPHOS substrates.
Interestingly, for one of the mitochondria (labeled mito4 in Figure 6b), we observed around 20% fluctuation in the fluorescence intensity. Fluctuations (flickering) of membrane potential of individual mitochondria from different cell types have been reported before, although the detailed conditions and causes of this flickering are not fully understood.19,23−25 Our study is the first to observe such flickering in isolated mitochondria from HeLa cell line, and it is surprising that it is observed in substrate fed (rather than basal) mitochondria, which is contradictory to some other studies,18 where removal of substrates resulted in flickering in some mitochondria. Even though JC-1 is a slow response dye,26 it has been shown that the observed flickering when using JC-1 is very similar to TMRM but with a slightly lower frequency.27 It is our belief that the technique demonstrated herein could be useful for future studies to more quantitatively elucidate the flickering of the membrane potential, its causes, and modulators.
Calcium Induced MMP
Calcium in mitochondrial matrix controls the rate of energy production. In case of pathological calcium overload mitochondrial permeability transition pore opens irreversibly causing the mitochondria membrane to become abruptly permeable. This results in mitochondrial depolarization and swelling.
A solution of 10 mM CaCl2 in DI water was prepared and diluted 10 times in respiration buffer to the final calcium concentration of 1 mM. The osmolarity of the final respiration buffer solution with 1 mM CaCl2 is about 310 mOsm, which is well within the physiological osmolarity range for mitochondria.28,29 We used a very high concentration of Ca2+ to make sure that the amount of calcium that reaches the mitochondria is enough to cause the swelling. The solution was pumped into the channels (with a relatively low flow rate of 5 μL h−1 to make sure it will not dislodge the trapped mitochondria) to induce depolarization. In order to avoid the photobleaching of JC-1, we imaged the mitochondria prior to the introduction of calcium solution, started calcium flow, stopped the light exposure, and waited for 4 min. Then we turned on the illumination and imaged the same field again. (The images are shown in Figure S-6 of the Supporting Information.) The red/ green fluorescence intensity of most of the mitochondria had considerably decreased. This clearly indicates the membrane potential has been significantly reduced by the Ca2+.
To quantify this effect, we compared the red fluorescence to green fluorescence intensity ratio prior to and after calcium treatment. The red/green ratio for all 14 mitochondria varied but shifted to a lower value after introduction of the calcium. In Figure 7, the histograms of the ratios (which is indicative of the membrane potential Δψm) for the pre- and post-calcium treatment are plotted. Results for individual mitochondria are shown in Figure S-7 of the Supporting Information. Interestingly, the results indicate the membrane potential has collapsed for all but one mitochondrion, regardless of the initial membrane potential (i.e., red/green ratio). This demonstrates the ability to study heterogeneity and statistical properties of individual mitochondria.
Figure 7.

Histogram showing the distribution of JC-1 fluorescence intensity ratio for the 14 mitochondria involved in the study prior to (red bars) and after calcium treatment (white bars).
Comparison to State of the Art
The vast majority of assays performed on mitochondrial function are on large quantities of mitochondria, typically requiring 107 cells worth of sample.26 Below we compare our work with prior art for the analysis of individual mitochondria, of which there are only a few techniques, none of them suitable for the studies envisioned with our technology.
An alternative technique to immobilize isolated mitochondria involves adhesion to a glass microscope slide.30−32 This technique allows visualization and characterization of up to hundreds of individual mitochondria in a single field of view. A disadvantage of this approach, however, is the large fluorescence background of the fluorophore outside of the mitochondria. If one seeks to quantitatively determine Δψm using potential sensitive dyes, it is necessary to carefully measure the ratio of the dye fluorescence intensity at the inside to the outside of the mitochondria. The measurement is complicated by the large diffuse fluorescence background of the fluorophore outside of the mitochondria. In our nanochannels, the volume of solution outside the mitochondria is minimized, leading to much lower background fluorescence. Another advantage of our method is that mitochondria are physically trapped and cannot move around or move out of the plane of focus, allowing us to do accurate measurements. In glass immobilization method a large portion of the mitochondria do not stably get attached on the glass surface, preventing the accurate measurement of their fluorescence intensity since small movements of mitochondria will result in changes in the measured fluorescence intensity and disrupt the measurement. More importantly, parallel processing of multiple analytes is not possible on a glass slide. In the fluidic channel array, it is possible to provide a large variety of different chemical environments (one in each channel) with suitably designed on-chip microfluidic circuits. For example, each of the 10 channels could be given a linearly varying concentration of Ca2+ by suitable design of a perpendicular microfluidic Ca2+ gradient at the introduction of each channel. This could allow for parallel screening in response to different calcium concentrations at the single mitochondria level, which the glass slide approach cannot easily provide. Generalizations to arbitrary chemical combinatorial conditions are clearly possible.
Several studies have demonstrated the application of flow cytometry to the analysis of individual mitochondria.33,34 Flow cytometry has the advantage of using existing commercially available instruments. Flow cytometry provides a “snap shot” of a single mitochondrial state (e.g., JC-1 fluorescence, forward scatter and side scatter). In this way, statistical analysis of mitochondria under various conditions and states can be obtained. Flow cytometry cannot be used to track the status of individual mitochondria over a long time. In contrast, in our approach, the response of individual mitochondria to a variety of chemical species can be tracked over a long period of time.
Similar to flow cytometry, capillary electrophoresis with laser-induced fluorescence detection allows for analysis of single mitochondria. In this technique, a 50 μm capillary guides individual mitochondria which migrate in response to a high electric field (ca. 200 V cm−1). Using this technique, a variety of mitochondrial properties can be assayed, such as the electro-phoretic mobility, the cardiolipin content, and ROS production.35−40 As in flow cytometry, single mitochondria are analyzed at a snapshot in time as they migrate passed the detection window.
In short, the current approach supplies temporal information, the CE provides chemical information, and the flow cytometry provides statistics on large populations.
CONCLUSION
We present for the first time a nanofluidic chip to trap and study isolated individual mitochondria. This provides a powerful stage for fluorescence imaging of isolated mitochondria in a controlled environment and real time investigation of their behavior under the influence of different chemicals. Multiple applications of the new technology can be envisioned which are discussed in detail in the Supporting Information, p 18. We expect that the application of this and similar technologies to the analysis of subcellular organelles will have a variety of applications in cancer biology, stem cell biology, drug screening, and aging studies, because of the growing consensus of the general importance of functional metabolomics in biology and medicine.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the following grants awarded to D.C.W.: NIH-AG24373, NS21328, AG13154, DK73691, CIRM Comprehensive Grant RC1-00353, and a Doris Duke Clinical Interface Grant 2 005 057. P.B. acknowledges support of this work from NIH National Cancer Institute Grant 1R21CA143351-01 as well as support from the Army Research offce (MURI W911NF-11-1-0024, ARO W911NF-09-1-0319 and DURIP W911NF-11-1-0315). Antonio Davila and Ted Pham are supported by the National Science Foundation Lifechips Integrative Graduate Education and Research Traineeship 0 549 479.
Footnotes
Supporting Information
Figures S-1−S-10, Table S1, and additional text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Wallace DC. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Remedi MS, Nichols CG, Koster JC. Cell Metab. 2006;3:5–7. doi: 10.1016/j.cmet.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Kuznetsov AV, Margreiter R. Int. J. Mol. Sci. 2009;10:1911–29. doi: 10.3390/ijms10041911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anand RK, Chiu DT. Curr. Opin. Chem. Biol. 2012;16:1–9. doi: 10.1016/j.cbpa.2012.05.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim T-S, Dávila A, Wallace DC, Burke P. Lab Chip. 2010;10:1683–88. doi: 10.1039/c001818j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim T-S, Davila A, Jr., Zand K, Wallace DC, Burke PJ. Lab Chip. 2012;12:2719–2725. doi: 10.1039/c2lc40086c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zare RN, Kim S. Annu. Rev. Biomed. Eng. 2010;12:187–201. doi: 10.1146/annurev-bioeng-070909-105238. [DOI] [PubMed] [Google Scholar]
- 8.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Ryan P, Balis UJ, Tompkins RG, Haber DA. Nature. 2011;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pendergrass W, Wolf N, Poot M. Cytometry, Part A. 2004;61:162–9. doi: 10.1002/cyto.a.20033. [DOI] [PubMed] [Google Scholar]
- 10.Presley AD, Fuller KM, Arriaga EAJ. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2003;793:141–150. doi: 10.1016/s1570-0232(03)00371-4. [DOI] [PubMed] [Google Scholar]
- 11.Cossarizza A, Ceccarelli D, Masini A. Exp. Cell Res. 1996;222:84–94. doi: 10.1006/excr.1996.0011. [DOI] [PubMed] [Google Scholar]
- 12.Keil VC, Funke F, Zeug A, Schild D, Müller M. Pflugers Arch. 2011;462:693–708. doi: 10.1007/s00424-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klima J. In: Cytologie: Eine Einf. f. Studierende d. Naturwiss. u. Medizin. Fischer G, editor. Stuttgart; 1967. [Google Scholar]
- 14.Qin D, Xia Y, Whitesides GM. Nat. Protoc. 2010;5:491–502. doi: 10.1038/nprot.2009.234. [DOI] [PubMed] [Google Scholar]
- 15.Liang X, Chou SY. Nano Lett. 2008;8:1472–6. doi: 10.1021/nl080473k. [DOI] [PubMed] [Google Scholar]
- 16.Xia D, Yan J, Hou S. Small. 2012;8:2787–801. doi: 10.1002/smll.201200240. [DOI] [PubMed] [Google Scholar]
- 17.Duffy DC, McDonald JC, Schueller OJ, Whitesides GM. Anal. Chem. 1998;70:4974–84. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 18.Vergun O, Votyakova TV, Reynolds IJ. Biophys. J. 2003;85:3358–3366. doi: 10.1016/S0006-3495(03)74755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hattori T, Watanabe K, Uechi Y, Yoshioka H, Ohta Y. Biophys. J. 2005;88:2340–2349. doi: 10.1529/biophysj.104.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cossarizza A, Ceccarelli D, Masini A. Exp. Cell Res. 1996;222:84–94. doi: 10.1006/excr.1996.0011. [DOI] [PubMed] [Google Scholar]
- 21.Lecoeur H, Chauvier D, Langonné A, Rebouillat D, Brugg B, Mariani J, Edelman L, Jacotot E. Apoptosis. 2004;9:157–69. doi: 10.1023/B:APPT.0000018798.03705.69. [DOI] [PubMed] [Google Scholar]
- 22.McGill A, Frank A, Emmett N, Turnbull DM, Birch-Machin MA, Reynolds NJ. FASEB J. 2005;19:1012–4. doi: 10.1096/fj.04-2664fje. [DOI] [PubMed] [Google Scholar]
- 23.Duchen MR, Leyssens A, Crompton MJ. Cell Biol. 1998;142:975–988. doi: 10.1083/jcb.142.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huser J, Blatter LA. Biochem. J. 1999;343:311–317. [PMC free article] [PubMed] [Google Scholar]
- 25.Kurz FT, Aon MA, O'Rourke B, Armoundas AA. Proc. Natl. Acad. Sci. U. S. A. 2010;107:14315–20. doi: 10.1073/pnas.1007562107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholls DG. Methods Mol. Biol. (N. Y., NY, U. S.) 2012;810:119–33. doi: 10.1007/978-1-61779-382-0_8. [DOI] [PubMed] [Google Scholar]
- 27.Buckman JF, Reynolds IJ. J. Neurosci. 2001;21:5054–5065. doi: 10.1523/JNEUROSCI.21-14-05054.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halestrap A. Biochim. Biophys. Acta. 1989;973:355–82. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- 29.Trounce IA, Kim YL, Jun AS, Wallace DC. Methods Enzymol. 1996;42:484–508. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 30.Huser J, Rechenmacher CE, Blatter LA. Biophys. J. 1998;74:2129–2137. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uechi Y, Yoshioka H, Morikawa D, Ohta Y. Biochem. Biophys. Res. Commun. 2006;344:1094–1101. doi: 10.1016/j.bbrc.2006.03.233. [DOI] [PubMed] [Google Scholar]
- 32.Higuchi Y, Miura T, Kajimoto T, Ohta Y. FEBS Lett. 2005;579:3009–13. doi: 10.1016/j.febslet.2005.04.054. [DOI] [PubMed] [Google Scholar]
- 33.Medina JM, López-Mediavilla C, Orfao A. FEBS Lett. 2002;510:127–132. doi: 10.1016/s0014-5793(01)03260-4. [DOI] [PubMed] [Google Scholar]
- 34.Lecoeur H, Langonné A, Baux L, Rebouillat D, Rustin P, Prévost M-C, Brenner C, Edelman L, Jacotot E. Exp. Cell Res. 2004;294:106–17. doi: 10.1016/j.yexcr.2003.10.030. [DOI] [PubMed] [Google Scholar]
- 35.Strack A, Duffy CF, Malvey M, Arriaga EA. Anal. Biochem. 2001;294:141–147. doi: 10.1006/abio.2001.5148. [DOI] [PubMed] [Google Scholar]
- 36.Fuller KM, Arriaga EA. Curr. Opin. Biotechnol. 2003;14:35–41. doi: 10.1016/s0958-1669(02)00008-3. [DOI] [PubMed] [Google Scholar]
- 37.Duffy CF, MacCraith B, Diamond D, O'Kennedy R, Arriaga EA. Lab Chip. 2006;6:1007–1011. doi: 10.1039/b601896c. [DOI] [PubMed] [Google Scholar]
- 38.Johnson RD, Navratil M, Poe BG, Xiong G, Olson KJ, Ahmadzadeh H, Andreyev D, Duffy CF, Arriaga EA. Anal. Bioanal. Chem. 2007;387:107–118. doi: 10.1007/s00216-006-0689-6. [DOI] [PubMed] [Google Scholar]
- 39.Kostal V, Arriaga EA. Electrophoresis. 2008;29:2578–2586. doi: 10.1002/elps.200700917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kostal V, Katzenmeyer J, Arriaga EA. Anal. Chem. 2008;80:4533–4550. doi: 10.1021/ac8007384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.