

Abstract
The multifunctional cytokine tumor necrosis factor-α (TNF-α) is known to play an important role in inflammatory and immunological responses in human skin. Although it has been documented that reactive oxygen species (ROS) are involved in TNF-α-induced signaling pathways associated with certain inflammatory diseases, their role in TNF-α signaling cascades has not been examined in primary human keratinocytes used as a model of inflammatory skin disease and psoriasis. Employing a series of in vitro and in cellulo approaches, we have demonstrated that in primary human keratinocytes (i) TNF-α rapidly induces ROS generation, IκB degradation, NF-κB p65 nuclear translocation, and ultimately production of inflammatory cytokines; (ii) TNF-α-induced cytokine production is mediated both by the mammalian target of rapamycin signaling pathway via NF-κB activation and by ROS; (iii) TNF-α-dependent NF-κB activation (that is, IκB degradation and NF-κB p65 nuclear translocation) is not mediated by ROS; and (iv) a cell-penetrating derivative of the antioxidant enzyme, catalase, as well as taurine and N-acetyl-cysteine attenuate the TNF-α-induced production of cytokines. These latter results suggest that catalase and perhaps other antioxidants should be considered as part of a more specific and effective therapy for the treatment of inflammatory skin diseases, including psoriasis.
INTRODUCTION
Skin, the outermost layer of the body, is the largest human organ. It plays an important protective role by providing an interactive boundary between the body and the environment. Skin is repeatedly exposed to ionizing and UV radiation and systemically introduced to potentially toxic dietary and drug metabolites, all of which may influence its health and appearance (Sander et al., 2004). Many of these environmental agents and endogenous metabolites are natural oxidants. They can directly or indirectly drive the production of a variety of reactive oxygen species (ROS) through a number of physiological processes (Kohen, 1999; Trouba et al., 2002). The surface of the skin is especially sensitive to ROS. Many studies have documented their role in skin aging, the development of inflammatory skin disorders, and carcinogenesis (Briganti and Picardo, 2003; Sander et al., 2004; Okayama, 2005).
One particular skin pathology, psoriasis, is an incurable disorder that has been defined as a chronic inflammatory disease largely mediated by immune cells (Nickoloff and Nestle, 2004). Although it affects 2–4% of the general population worldwide, its etiology remains largely unknown. However, it can be triggered by stress or trauma, and by bacterial, fungal, or viral infection of the skin (Krueger, 2002; Lebwohl, 2003). Psoriasis is generally characterized by hyperproliferation, abnormal differentiation, and altered maturation of the epidermis, as well as inflammation in the epidermis and dermis (Banno et al., 2004; Chamian and Krueger, 2004). The pathogenesis of psoriasis has been reported to be driven by activated T cells or antigen-presenting cells, which release a number of cytokines and chemokines. These then bind to surface receptors and signal keratinocytes to hyperproliferate and abnormally differentiate. This, in turn, causes the accumulation of additional cytokines and inflammatory mediators in the lesional skin (Barker, 1991; Nickoloff, 2001; Gudjonsson et al., 2004).
Tumor necrosis factor-α (TNF-α) is a major mediator in the pathogenesis of psoriasis. It is a multifunctional cytokine that is believed to play an important role in mediating inflammation, immune responses, and apoptosis (Tracey and Cerami, 1994; Aggarwal and Natarajan, 1996; Locksley et al., 2001). It has been reported that there are much higher levels of TNF-α found in the skin of patients with psoriatic lesions as compared to that of normal subjects (Mease, 2004; De Rycke et al., 2005; Veale et al., 2005). In addition to TNF-α, other inflammatory cytokines and a transcription factor, NF-κB, are found to be highly expressed in psoriatic patients (Bachelez, 2005). One of the molecular mechanisms proposed is that TNF-α produced locally within psoriatic lesions causes NF-κB activation, subsequently leading to increased expression of other cytokines as well as of TNF-α itself. Thus, the view is that a TNF-α→NF-κB activation→increased TNF-α/inflammatory cytokine expression positive feedback loop exists that maintains and may enhance the inflammatory process within psoriatic plaques (Barnes and Karin, 1997; Bachelez, 2005; Punzi et al., 2007). A number of studies support the proposed importance of TNF-α-positive feedback loops in the pathophysiology of psoriasis (Mease, 2005; Nash and Florin, 2005; Saripalli and Gaspari, 2005). However, there is also the speculation that increased ROS production and deficient function of cellular antioxidant systems play a critical role in the etiology of the disease (Antille et al., 2002; Briganti and Picardo, 2003). Importantly, these points have yet to be examined in a cell model of inflammatory skin disease, specifically primary human keratinocytes treated with TNF-α.
Although the extent to which keratinocytes themselves contribute to the TNF-α load seen in psoriatic skin remains a matter of some debate, these cells can clearly produce the cytokine and other inflammatory mediators (for example, see Johansen et al., 2006), and therefore a molecular analysis of the underlying mechanisms of such production is clearly warranted. The overall aim of this study was to investigate, in primary human keratinocytes, the role of ROS in psoriasis-associated TNF-α signaling cascades. We show that treatment with TNF-α (i) stimulates ROS generation; (ii) activates NF-κB signaling through IκB degradation and NF-κB p65 nuclear translocation; and (iii) increases mRNA expression of the cytokines, IL-8, IL-6, and TNF-α itself. We implicate the serine/threonine protein kinase, mammalian target of rapamycin (mTOR), in these processes and also show that quenching hydrogen peroxide and downstream ROS with a cell-penetrating catalase derivative or other antioxidants is an effective means of reducing inflammatory cytokine gene expression. Taken together, these results shed light on the biochemistry and pathophysiology of inflammatory skin diseases, including psoriasis, at the level of the keratinocyte, and may provide previously unknown targets for therapeutic intervention.
RESULTS
TNF-α treatment induces hydrogen peroxide/ROS generation
To evaluate whether or not cellular hydrogen peroxide production was involved in TNF-α-mediated signaling, we measured levels of the ROS produced in primary human keratinocytes. First, we found that treatment with TNF-α caused a time-dependent increase in hydrogen peroxide production; this occurred within 5 minutes of TNF-α stimulation and reached a maximum at approximately 20 minutes (Figure 1). Having identified a “threshold time” (~20 minutes), we addressed ROS production in a dose–response analysis (Figure 2). We found that TNF-α increases ROS production in a dose-dependent (0–100 ng ml−1) manner. These findings correlate well with results showing TNF-α-induced ROS generation in other human cell types (Schulze-Osthoff et al., 1992; Goossens et al., 1995) as well as in canine keratinocytes (Kohler et al., 2001). However, to the best of our knowledge, we are the first group to demonstrate these phenomena in primary human keratinocytes.
Figure 1. Time course of hydrogen peroxide production induced by TNF-α in primary human keratinocytes.

Cells (2 × 105 ml−1) were treated with 10 ng ml−1 TNF-α for the indicated times. The amount of hydrogen peroxide produced in cells was determined using oxidation of the fluorogenic indicator Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine). Fluorescence units data presented reflect subtraction of untreated control cell values and are the means±SEM of triplicate samples. Similar results were seen in two additional experiments.
Figure 2. Dose response of hydrogen peroxide production induced by TNF-α in primary human keratinocytes.

Cells (2 × 105 ml−1) were treated with varying amounts of TNF-α (from 0 to1,000 ng ml−1) for 20 minutes, and the amount of hydrogen peroxide produced in cells was determined as in Figure 1. Results presented are the means±SEM of triplicate samples. Similar results were seen in two additional experiments.
TNF-α stimulates NF-κB activation in human primary keratinocytes
The NF-κB is a ubiquitous transcription factor for genes encoding proinflammatory and inflammatory cytokines, chemokines, and growth factors (Liou and Baltimore, 1993; Baldwin, 1996; Ghosh and Karin, 2002; Takao et al., 2003). In resting cells, IκB forms a complex with NF-κB that masks the nuclear localization sequences of NF-κB and thus prevents it from entering the nucleus. Upon cell stimulation, for example after TNF-α treatment, IκB proteins are rapidly phosphorylated and degraded by the proteasome pathway, and the newly freed NF-κB molecule translocates into the nucleus to activate expression of the multiple target genes noted above. The phosphorylation and degradation of IκB are thus the important steps in the pathway leading to NF-κB activation.
To assess activation of NF-κB following TNF-α exposure, we examined IκB degradation and NF-κB nuclear translocation as the relevant parameters. IκB degradation was evaluated by western blot analysis using an anti-IκBα antibody. Our results show that degradation of IκBα induced by TNF-α occurs very rapidly, with some 70% of the molecule being lost in the first 15 minutes (Figure 3). Next, we studied the effect of TNF-α on nuclear translocation of the NF-κB p65 subunit. To accomplish this, we performed laser scanning indirect immunofluorescence confocal microscopy using an antibody to the p65 molecule. As shown in Figure 4, the immunofluorescence staining pattern showed quite clearly the translocation of p65 into the nucleus of human keratinocytes after treatment with TNF-α. As expected, this molecule remained in the cytoplasm of untreated control cells.
Figure 3. Degradation of IκB(α) induced by TNF-α in primary human keratinocytes.

Cells (2 × 106 ml−1) were treated with 10 ng ml−1 TNF-α for the indicated times, and cell lysates were prepared as described in Materials and Methods. (a) Western blot analysis of IκBα or tubulin using appropriate polyclonal antibodies. (b) Densitometric analysis of the immunoreactive bands obtained after western blotting. Results are presented as IκBα/tubulin normalized ratios. Similar results were seen in duplicate experiments.
Figure 4. Nuclear translocation of the NF-κB p65 subunit induced by TNF-α in primary human keratinocytes.
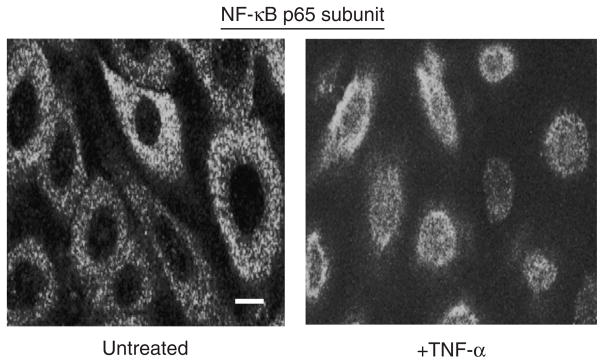
Cells were stimulated with TNF-α (10 ng ml−1) for 1 hour. Nuclear translocation of the NF-κB p65 subunit was assessed by indirect immunofluorescence confocal microscopy using anti-p65 subunit antibodies and appropriate fluorescently tagged secondary antibodies. A representative image of several obtained is included here. Bar=10 μm. Similar results were seen in duplicate experiments.
Induction of TNF-α, IL-8, and IL-6 mRNA expression following TNF-α stimulation
Although it has been well documented that high concentrations of TNF-α are present in skin lesions of patients with psoriasis (Mease, 2004; De Rycke et al., 2005; Veale et al., 2005), to date the effect of TNF-α on cytokine expression in primary human keratinocytes has not been determined. To address this point, we examined whether or not TNF-α is able to increase the (mRNA) expression of itself or other potent inflammatory cytokines in primary human (cultured) keratinocytes. We performed real-time reverse transcriptase-PCR (RT-PCR) (Figure 5) and found that in response to TNF-α, the message levels for TNF-α, IL-8, and IL-6 were all significantly increased. For IL-6, this induction was nearly 40-fold as compared to untreated controls (Figure 5).
Figure 5. Effects of TNF-α and rapamycin on expression of TNF-α, IL-8, and IL-6.

Primary human keratinocytes were treated with TNF-α (10 ng ml−1) for 6 hours. Total RNA was then isolated and mRNA analysis performed by real-time RT-PCR. Where indicated, cells were pretreated with rapamycin (20 nM) for 1 hour and then challenged with TNF-α. Data are expressed as fold induction of relevant mRNAs as compared to untreated controls. Representative results (means±SEM) from one of three independent experiments are shown.
Role of the mTOR pathway in TNF-α-induced cytokine upregulation
mTOR is, as mentioned above, a conserved serine/threonine protein kinase and a member of the cellular phosphatidylinositol 3-kinase/AKT pathway. We investigated its role in NF-κB activation and ROS production induced by TNF-α by using rapamycin (sirolimus), a well-characterized immuno-suppressant and potent inhibitor of the signaling cascade. As described above, the phosphorylation and subsequent degradation of IκB play a critical role in regulating NF-κB translocation to the nucleus and subsequent transcriptional regulation. In Figure 6, we asked if rapamycin has any effect on TNF-α-induced degradation of IκBα; clearly, it did. IκBα is largely stabilized by the pretreatment.
Figure 6. Effects of rapamycin on TNF-α-induced IκB degradation in primary human keratinocytes.

Cells (2 × 106 ml−1) were pretreated or not with rapamycin (20 nM) for 1 hour and then challenged with 10 ng ml−1 TNF-α for 30 minutes. (a) Western blot analysis, and (b) densitometric measurements and normalization of IκBα and tubulin levels were performed as outlined in Figure 3. Similar results were seen in duplicate experiments.
Furthermore, we showed that rapamycin inhibits mRNA expression of TNF-α, IL-8, and IL-6 (Figure 5). These results support the notion that there is a connection between the mTOR signaling cascade, NF-κB activation, and inflammatory cytokine expression. Importantly, such a link between mTOR and NF-κB has already been put forth (Ghosh et al., 2006), and is supported by our observations.
Finally, our analysis of the mTOR pathway showed that rapamycin has only modest effects on TNF-α-induced ROS production in these cells (Figure 7). One interpretation of these results is that ROS production lies upstream of the mTOR to NF-κB signaling cascades in TNF-α-activated primary human keratinocytes.
Figure 7. Effects of rapamycin and antioxidants on hydrogen peroxide production induced by TNF-α in primary human keratinocytes.

Cells (2 × 105 ml−1) were pretreated or not with rapamycin (Rap) (20 nM), catalase (Cat*) (1 μM), taurine (Taur) (40mM), or N-acetyl-cysteine (NAC) (5mM) for 1 hour and then challenged with TNF-α (10 ng ml−1) for 30 minutes. Hydrogen peroxide production was measured using DCF-DA as outlined in Materials and Methods. Fluorescence unit data presented reflect subtraction of untreated control cells values and are the means of triplicate samples. Similar results were seen in two additional experiments.
Involvement of ROS in TNF-α signaling in primary human keratinocytes
The results presented thus far have demonstrated that in primary human keratinocytes, treatment with TNF-α rapidly induces ROS generation, activation of the mTOR and NF-κB signaling and transcription pathways, and mRNA expression of the proinflammatory cytokines TNF-α, IL-8, and IL-6. We next asked what the effects of TNF-α would be on cells pretreated with a targeted antioxidant. To analyze this point, we employed a transducible form of catalase as a specific ROS scavenger that efficiently decomposes hydrogen peroxide. This genetically engineered catalase molecule (hereafter called Cat*) is modified with a cell-penetrating peptide (specifically, an arginine nonamer) at its amino terminus and a high efficiency peroxisomal targeting signal 1 (specifically, serine–lysine–leucine) at its carboxy terminus. This enzyme rapidly enters cells and efficiently traffics to the peroxisome to metabolize hydrogen peroxide. The strategy of restoring or supplementing peroxisomal catalase in cells has been shown to re-establish oxidative balance, to renew mitochondrial integrity (by repolarizing the organelle), and to delay senescence (Wood et al., 2006; Koepke et al., 2007; Terlecky and Koepke, 2007). Our results indicate that Cat* prophylaxis significantly reduces the amount of hydrogen peroxide and related ROS present in TNF-α-treated keratinocytes (Figure 7). Furthermore, these cells are unable to induce expression of TNF-α mRNA to the level seen in non-antioxidant pretreated cells (Figure 8).
Figure 8. Effect of hydrogen peroxide on TNF-α-induced gene expression in primary human keratinocytes.

Cells were pretreated or not with 3-aminotriazole (3-AT) (1 μM), hydrogen peroxide (2.5 μM), catalase (Cat*) (1 μM), taurine (Taur) (40mM), or N-acetyl-cysteine (NAC) (5mM) for 1 hour and then challenged with TNF-α (10 ng ml−1) for 6 hours. Total RNA was then isolated and TNF-α mRNA analysis performed by real-time RT-PCR. Data are expressed as fold induction of relevant mRNAs as compared to untreated controls. Representative results (means±SEM) from one of three independent experiments are shown.
Importantly, similar results were seen with taurine, a sulfur-containing β-amino acid, and N-acetyl cysteine, the acetylated precursor of both L-cysteine and reduced glutathione (Figures 7 and 8). Both molecules possess antioxidant properties and elicit a variety of effects on cells (Petty et al., 1990; Huxtable, 1992; Atmaca, 1998). Taurine’s demonstrated ability to control release of proinflammatory cytokines in a number of organisms is consistent with the effects seen in Figure 8.
The obvious suggestion is that hydrogen peroxide and related ROS are stimulating expression of inflammatory cytokines in this model system. To further test this, cells were pretreated with hydrogen peroxide directly or with 3- aminotriazole, a specific inhibitor of catalase. In both cases, the resultant oxidative load present in cells enhanced TNF-α’s ability to induce cytokine expression (Figure 8).
We next asked if activation of NF-κB in response to TNF-α was mediated by ROS. This does not appear to be the case, as cells pretreated with Cat*, taurine, or N-acetyl-cysteine still degrade IκB(α) in response to TNF-α (Figure 9). Therefore, even though ROS certainly plays a role in the expression of inflammatory cytokines induced by TNF-α, it apparently is not through IκB degradation and accompanying NF-κB activation/nuclear translocation. Rather, it appears that there exists a redox-independent IκB degradation/NF-κB activation step involved in TNF-α-activated primary human keratinocytes.
Figure 9. Effects of antioxidants on TNF-α-induced IκB degradation in primary human keratinocytes.

Cells (2 × 106 ml−1) were pretreated or not with catalase (Cat*) (1 μM), taurine (Taur) (40mM), N-acetyl-cysteine (NAC) (5mM) for 1 hour and subsequently challenged with TNF-α (10 ng ml−1) for 30 minutes. (a) Western blot analysis, and (b) densitometric measurements and normalization of IκBα and tubulin levels were performed as outlined in Figure 3. Similar results were seen in duplicate experiments.
DISCUSSION
The pathogenesis of psoriasis is still not completely understood. It is certainly a disease of immune cells with some, as yet to be clearly defined, contribution by skin cells including keratinocytes. Collectively, these cells conspire to potentiate the development of chronic skin inflammation through the production of cytokines (Bhalerao and Bowcock, 1998; Krueger et al., 2001; Krueger, 2002; Lebwohl, 2003; Nickoloff and Nestle, 2004; Albanesi et al., 2005). Among these is TNF-α, the major proinflammatory cytokine of psoriasis, which activates cells in a positive feedback loop producing other inflammatory mediators, including ROS and various other cytokines (Gottlieb et al., 2005; Lowes et al., 2005; Pelle et al., 2005). It is also thought that an insufficient antioxidant system, together with increased levels of ROS, contributes to the pathogenesis (Antille et al., 2002; Briganti and Picardo, 2003; Baz et al., 2003; Bickers and Athar, 2006).
One aim of this work was to gain a better understanding of the interactions between TNF-α, ROS, and antioxidants in the initiation and progression of the disease. Our studies employing primary human keratinocytes clearly support a role for ROS in TNF-α-induced cytokine expression. We demonstrate rapid TNF-α-dependent ROS generation (Figures 1 and 2). In addition, we show that quenching the resultant oxidative stress with either a cell-penetrating, peroxisomally targeted, catalase derivative or the antioxidants taurine or N-acetyl-cysteine significantly reduces the accompanying elevated cytokine expression (Figure 8). The suggestion is that disturbances in the oxidant–antioxidant system in human skin may play an important role in the pathogenesis of psoriasis. As current systemic medications for psoriasis have been reported to have significant side effects, we propose that targeted antioxidant prophylaxis, akin to the strategy employed here with a transducible catalase derivative, is worth considering as a potential treatment modality.
Our results also implicate the critical cellular kinase, mTOR, in response to TNF-α. This enzyme participates in, and often regulates, a variety of cellular functions, including protein synthesis and autophagy, as well as (cell) growth, survival, and proliferation (Hay and Sonenberg, 2004; Inoki et al., 2005; Ghosh et al., 2006). The molecule and associated complex partners integrate a variety of upstream signals and modulate a number of downstream effectors. Perhaps best underscoring the importance of mTOR and interaction complex members is the now well-established role of these molecules, when dysregulated, in cancer (Tokunaga et al., 2004; Tsang et al., 2007). In primary human keratinocytes treated with TNF-α, mTOR appears to regulate NF-κB activation (Figure 6). Interestingly, (inhibiting) mTOR has little effect on TNF-α’s ability to elicit ROS formation (Figure 7), suggesting that distinct mechanisms are at work. Therefore, TNF-α’s stimulation of inflammatory cytokine expression employs both mTOR-dependent and mTOR-independent reactions.
Considering NF-κB activation further, it was unexpected that quenching TNF-α-induced ROS did not inhibit the process (Figure 9). Indeed, IκBα was degraded (and thus NF-κB activated) as well or better in the presence of the antioxidant. At first glance, this seemed inconsistent with a number of observations in other cellular contexts, suggesting a role for ROS in NF-κB activation (Schreck et al., 1991; Bubici et al., 2006; Gloire et al., 2006; Nakano et al., 2006). Although the answer is not completely clear, we speculate that perhaps NF-κB-induced transcription of relevant genes is redox sensitive. Therefore, the ROS-requiring step lies downstream of NF-κB activation. Further experimentation will be required to confirm this conjecture.
In summary, our findings suggest a rather straightforward series of biochemical relationships. In primary human keratinocytes, TNF-α induces ROS, stimulates mTOR, and activates NF-κB, resulting in expression of inflammatory cytokines. What should not be lost is that the pathways may interact or may function independently. The intricacy presumably reflects the complexity of psoriasis and related inflammatory skin diseases. Having identified some of the relevant pathways in the overall response of human primary keratinocytes to TNF-α, a more careful dissection of specific components and underlying molecular mechanisms may now begin.
MATERIALS AND METHODS
Reagents and antibodies
TNF-α was obtained from R&D Systems (Minneapolis, MN). DMEM/F12, fetal bovine serum, trypsin–EDTA, phosphate-buffered saline (PBS), and other products for cell culture were supplied by Invitrogen (Carlsbad, CA). Serum-free culture EpiLife Medium and EpiLife Defined Growth Supplement for human primary keratinocytes culture were purchased from Cascade Biologics Inc. (Portland, OR). The Amplex Red reagent kit for measuring hydrogen peroxide was purchased from Invitrogen (Carlsbad, CA). N-acetyl-cysteine, rapamycin, taurine, and 2′7′-dichlorofluorescein diacetate (DCF-DA) were purchased from Sigma-Aldrich (St Louis, MO). Antibodies to IκBα and NF-κB p65 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Anti-catalase antibodies were generated under contract by our laboratory. Anti-tubulin antibodies were kindly provided by Dr Raymond Mattingly (Department of Pharmacology, Wayne State University School of Medicine).
Transducible catalase-SKL: plasmid preparation and enzyme purification
A transducible form of the human antioxidant enzyme, catalase, was created by adding a cell-penetrating peptide, specifically nine arginine residues, to the protein’s amino terminus. To accomplish this, a double-stranded oligonucleotide encoding nine arginine residues was produced by annealing a pair of synthesized complementary oligonucleotides (Operon Biotechnologies Inc., Huntsville, AL) flanked by restriction enzyme sites. The oligonucleotides were (top strand) 5′-tatgCGCCGTCGCCGTCGCCGTCGCCG TCGCctgca-3′ and (bottom strand) 5′-gGCGACGGCGACGGCGAC GGCGACGGCGca-3′. An NdeI restriction site (lowercase letters) is encoded at the upstream end, whereas a PstI site is at the downstream end of the double-stranded oligonucleotides. The double-stranded oligonucleotide was directly ligated into NdeI–PstI- digested pQE-2 (Qiagen, Chatsworth, CA).
In addition, the enzyme’s carboxy-terminal peroxisomal targeting signal 1 was altered from the naturally occurring alanine–asparagine– leucine to the more efficient serine–lysine–leucine (SKL). To generate this plasmid, the human catalase gene was PCR amplified from a full-length cDNA clone (Invitrogen, Paisley, UK). The forward primer, 5′-AAActgcagATGGCTGACAGCCGGGATCCCGCC-3′, complemented the amino-terminal sequence of human catalase along with a PstI restriction site (lowercase letters). The reverse primer was designed to produce the SKL amino-acid sequence (5′-GGGCGCaagcttTCACAGTTTCGATTTCTCCCTTGCCGCCAAGT-3′). A HindIII restriction site (lowercase letters) was also incorporated downstream of the stop codon.
The catalase-SKL gene amplified by PCR was digested appropriately, and ligated into the pQE-2 vector containing the sequence encoding the nine arginine residues. Ligation products were transformed into the E. coli strain DH5α, and the recovered plasmids were confirmed to be correct by restriction analysis and DNA sequencing. The sequence-verified (His)6-tagged cell-penetrating catalase-SKL construct was then expressed in the E. coli strain Rosetta 2 (DE3)pLysS (Novagen, Madison, WI), according to the manufacturer’s instructions.
Recombinant catalase protein was purified using a modified denaturing protocol. To prepare denatured fusion proteins, the induced cells were harvested and lysed by freeze/thawing in a binding buffer (5mM imidazole, 500mM NaCl, and 20mM Tris-HCl, pH 7.9) containing 2 M urea. After the removal of the cell debris by centrifugation, the clarified cell extracts were loaded onto a Ni-NTA column. The column was washed first with the binding buffer containing 2 M urea and then with a wash buffer (30mM imidazole, 500mM NaCl, and 20mM Tris-HCl, pH 7.9). The proteins were released by an elution buffer (500mM imidazole, 500mM NaCl, and 20mM Tris-HCl, pH 7.9), followed by desalting with a PD10 column. The purified transducible recombinant catalase fusion protein (in PBS containing 20% glycerol) was then aliquoted and stored at −80 °C.
Primary human keratinocyte isolation from neonatal foreskin
Neonatal foreskins from healthy males were the source of primary human keratinocytes. They were obtained through an Internal Review Board protocol approved by the Human Investigative Committee at the Wayne State University School of Medicine. Skin samples were obtained and pooled together from three or more surgically circumcised foreskins. Isolation of keratinocytes was performed essentially as described by Liu and Karasek (1978). Briefly, skin samples were washed, cut, scraped, and treated with a dispase solution to prepare a largely epidermal layer. The tissue was then trypsinized, filtered, centrifuged, and the resultant keratinocytes cultured and seeded as required. The cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 in a serum-free low-Ca2+ (0.06mM) EpiLife keratinocyte growth medium (Cascade Biologics Inc.) supplemented with bovine hydrocortisone, insulin, pituitary extract, and transferrin, as well as recombinant human epidermal growth factor (Cascade Biologics Inc.). The medium was changed every other day. For all experiments, cells were seeded at passage numbers 3–5 into 60mm dishes (2 × 106 cells per well), 24-well plates (5 × 105 cells per well), or 96-well plates (1 × 105 cells per well), and were treated upon reaching 80–90% confluence. In experiments designed to examine effects of TNF-α, cells were grown in standard medium without growth factors for 24 hours to avoid the effects of supplements in the growth medium and to obtain quiescent cells with low levels of activated NF-κB.
Measurement of intracellular hydrogen peroxide/ROS
Production of intracellular hydrogen peroxide was determined using either DCF-DA (Bass et al., 1983; LeBel et al., 1992; Ohba et al., 1994) or the Amplex Red reagent (Molecular Probes/Invitrogen, Carlsbad, CA). The method employed in a given experiment is indicated in the accompanying figure legend.
With the DCF-DA assay, the medium was removed from cells, which were then washed and incubated with 50 μM DCF-DA in keratinocyte growth medium at 37 °C for 30 minutes. The DCF-DA was then removed and the cells were washed once again. A fluorescence microplate reader was used to take a time zero (Ft0) point at an emission wavelength of 535nm and an excitation wavelength of 490 nm. Immediately thereafter, the cells were subjected to TNF-α treatment with or without pretreatment with rapamycin, catalase, taurine, or N-acetyl-cysteine in keratinocyte growth medium, and a time course was initiated. At each time point, the fluorescence intensity was again determined. For data analysis, the percentage increase in fluorescence per well was calculated by the formula [(Ftx–Ft0)/Ft0100], where Ftx is fluorescence at time x minutes and Ft0 is fluorescence at time 0 minute.
Measurement of intracellular hydrogen peroxide was also directly determined using Amplex Red hydrogen peroxide/peroxidase assay kit. The principle behind this assay is that Amplex Red is a derivative of 10-acetyl-3,7-dihydroxyphenoxazine, which becomes highly fluorescent upon oxidation by hydrogen peroxide in the presence of horseradish peroxidase. Experimentally, after a given treatment, equal numbers of cells were washed twice with PBS (pH 7.4) and harvested into reaction buffer containing the Amplex Red reagent for 30 minutes, according to the manufacturer’s protocol. Fluorescence was recorded in a microplate reader (Molecular Devices, Sunnyvale, CA) set at 530nm excitation and 590nm emission wavelengths.
Cytokine mRNA expression determined by real-time quantitative RT-PCR
At the end of a particular treatment, human primary keratinocyte samples were harvested, treated with lysis buffer, and total RNA was extracted according to the RNeasy Mini Kit manual (Qiagen). RNA concentrations and 260/280 ratios were assessed using a DU530 spectrophotometer (Beckman, Fullerton, CA). Reverse transcription of cDNA was performed using random hexamers as primers according to the SuperScript II Reverse Transcriptase System Reagents kit manual (Invitrogen, Carlsbad, CA).
To assess the relative transcript levels of specific genes, the cDNA products were subjected to real-time SYBR-green-based quantitative PCR as described by Lee et al. (2006). The primers used for these studies were generated from GenBank (http://www.ncbi.nlm.nih.gov/Genbank; provided in the public domain by the National Center for Biotechnology Information, Bethesda, MD) with Gene Runner software (Hastings Software, Hastings, NY). Primer sequences used were as indicated in Table 1. We confirmed that primers amplified the appropriate products by testing under real-time PCR conditions. Each assay typically was repeated twice, with each sample run in triplicate. Signals from each sample were normalized to values obtained for the 18S rRNA gene, which was run as a “housekeeping gene” simultaneously with the experimental samples. Cycle threshold numbers (Ct) were recorded from the exponential phase of PCR amplification. The relative expression of cytokine mRNA was calculated using the comparative 2-ΔΔCt method, as previously described (Livak and Schmittgen, 2001). The results from each run for each sample were averaged and expressed as the relative level of the mRNA transcript.
Table 1.
Primer sequences for real-time PCR
| Gene | Primer | Sequence | Product size (bp) |
|---|---|---|---|
| TNF-α | Sense | 5′-CTACTCCCAGGTTCTCTTCAA-3′ | 137 |
| TNF-α | Antisense | 5′-GCAGAGAGGAGGTTGACTTTC-3′ | |
| IL-6 | Sense | 5′-ATGAACTCCTTCTCCACAAGC-3′ | 156 |
| IL-6 | Antisense | 5′-CTACATTTGCCGAAGAGCCCCTG-3′ | |
| IL-8 | Sense | 5′-ATCAAGGCGCATGTGAACTC-3′ | 140 |
| IL-8 | Antisense | 5′-AGAGCCCCAGATCCGATTTT-3′ | |
| 18S rRNA | Sense | 5′-CGGCTACCACATCCAAGGAA-3′ | 178 |
| 18S rRNA | Antisense | 5′-GCTGGAATTACCGCGGCT-3′ |
TNF-α, tumor necrosis factor-α.
Detection of NF-κB activation by western blot analysis
After human primary keratinocytes were incubated under the appropriate experimental conditions, cells were harvested in 2× SDS-PAGE sample buffer and subjected to western blot analysis. Briefly, equal amounts of whole-cell extracts were resolved by 10% SDS-PAGE and transferred onto a nitrocellulose membrane. After nonspecific binding sites were quenched using blocking buffer (5% non-fat dry milk, 0.5% Tween-20 in 20mM Tris-buffered saline, pH 7.6), the membrane was incubated overnight with primary antibodies specific for IκBα or tubulin. The anti-IκBα antibodies were used at a 1:500 dilution, and the anti-tubulin antibodies were used at a 1:200 dilution. The blot was washed and incubated again with horseradish peroxidase-conjugated anti-goat or anti-rabbit IgG secondary antibodies. After washing once again, protein levels were determined by chemiluminescence using an ECL detection system. The intensities of the bands were measured using a Fujifilm LAS1000plus luminescent image analyzer.
NF-κB p65 subunit translocation detected by indirect immunofluorescence confocal microscopy
Human keratinocytes grown on glass coverslips were fixed with 4% (w/v) formaldehyde for 10 minutes and permeabilized in 1% (v/v) Triton X-100 for 5 minutes. The cells were blocked with 4% (w/v) BSA for 1 hour and then incubated with primary antibodies for 1 hour and secondary antibodies for 45 minutes. All reactions were conducted in PBS; rabbit anti-p65 antibodies were used at a 1:500 dilution and CY3-conjugated goat anti-rabbit secondary antibodies were used at a 1:250 dilution. Coverslips were mounted in PBS containing anti-fade mounting media and examined using a confocal fluorescence microscope (Zeiss LSM510) at the Core Facility, Department of Pharmacology, Wayne State University School of Medicine.
Acknowledgments
We acknowledge the Center for Molecular and Cellular Toxicology with Human Applications in Michigan (funded by NIEHS Grant P30 ES06639).
Abbreviations
- DCF-DA
2′7′-dichlorofluorescein diacetate
- mTOR
mammalian target of rapamycin
- ROS
reactive oxygen species
- RT
reverse transcriptase
- TNF-α
tumor necrosis factor-α
Footnotes
CONFLICT OF INTEREST
SRT is a cofounder of EXT Life Sciences Inc., a Michigan-based biotechnology company owned, in part, by the Wayne State University. SRT retains an equity interest in the company. Transducible catalase-SKL, one of the antioxidants examined in this report, is covered by a United States patent pending, which is owned by the Wayne State University. Other authors state no conflict of interest.
References
- Aggarwal BB, Natarajan K. Tumor necrosis factors: developments during the last decade. Eur Cytokine Netw. 1996;7:93–124. [PubMed] [Google Scholar]
- Albanesi C, Scarponi C, Giustizieri ML, Girolomoni G. Keratinocytes in inflammatory skin diseases. Curr Drug Targets Inflamm Allergy. 2005;4:329–34. doi: 10.2174/1568010054022033. [DOI] [PubMed] [Google Scholar]
- Antille C, Sorg O, Lübbe J, Saurat JH. Decreased oxidative state in non-lesional skin of atopic dermatitis. Dermatology. 2002;204:69–71. doi: 10.1159/000051814. [DOI] [PubMed] [Google Scholar]
- Atmaca G. Antioxidant effect of sulfur-containing amino acids. Yonsei Med J. 1998;45:776–8. doi: 10.3349/ymj.2004.45.5.776. [DOI] [PubMed] [Google Scholar]
- Bachelez H. Immunopathogenesis of psoriasis: recent insights on the role of adaptive and innate immunity. J Autoimmun. 2005;25(Suppl):69–73. doi: 10.1016/j.jaut.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J Biol Chem. 2004;279:32633–42. doi: 10.1074/jbc.M400642200. [DOI] [PubMed] [Google Scholar]
- Barker JN. The pathophysiology of psoriasis. Lancet. 1991;338:227–30. doi: 10.1016/0140-6736(91)90357-u. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol. 1983;130:1910–7. [PubMed] [Google Scholar]
- Baz K, Cimen MY, Kokturk A, Yazici AC, Eskandari G, Ikizoglu G, et al. Oxidant/antioxidant status in patients with psoriasis. Yonsei Med J. 2003;44:987–90. doi: 10.3349/ymj.2003.44.6.987. [DOI] [PubMed] [Google Scholar]
- Bickers DR, Athar M. Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol. 2006;126:2565–75. doi: 10.1038/sj.jid.5700340. [DOI] [PubMed] [Google Scholar]
- Bhalerao J, Bowcock AM. The genetics of psoriasis: a complex disorder of the skin and immune system. Hum Mol Genet. 1998;7:1537–45. doi: 10.1093/hmg/7.10.1537. [DOI] [PubMed] [Google Scholar]
- Briganti S, Picardo M. Antioxidant activity, lipid peroxidation and skin diseases. What’s new. J Eur Acad Dermatol Venerol. 2003;17:663–9. doi: 10.1046/j.1468-3083.2003.00751.x. [DOI] [PubMed] [Google Scholar]
- Bubici C, Papa S, Dean K, Franzoso G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene. 2006;25:6731–48. doi: 10.1038/sj.onc.1209936. [DOI] [PubMed] [Google Scholar]
- Chamian F, Krueger JG. Psoriasis vulgaris: an interplay of T lymphocytes, dendritic cells, and inflammatory cytokines in pathogenesis. Curr Opin Rheumatol. 2004;16:331–7. doi: 10.1097/01.bor.0000129715.35024.50. [DOI] [PubMed] [Google Scholar]
- De Rycke L, Vandooren B, Kruithof E, DeKeyser F, Veys EM, Baeten D. Tumor necrosis factor alpha blockade treatment down-modulates the increased systemic and local expression of Toll-like receptor 2 and Toll-like receptor 4 in spondylarthropathy. Arthritis Rheum. 2005;52:2146–58. doi: 10.1002/art.21155. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109:S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Tergaonkar V, Rothlin CV, Correa RG, Bottero V, Bist P, et al. Essential role of tuberous sclerosis genes TSC1 and TSC2 in NF-kappaB activation and cell survival. Cancer Cell. 2006;10:215–26. doi: 10.1016/j.ccr.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Gloire G, Legrand-Poels S, Piette J. NF-kB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci USA. 1995;92:8115–9. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb AB, Chamian F, Masud S, Cardinale I, Abello MV, Lowes MA, et al. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J Immunol. 2005;175:2721–9. doi: 10.4049/jimmunol.175.4.2721. [DOI] [PubMed] [Google Scholar]
- Gudjonsson JE, Johnston A, Sigmundsdottir H, Valdimarsson H. Immunopathogenic mechanisms in psoriasis. Clin Exp Immunol. 2004;135:1–8. doi: 10.1111/j.1365-2249.2004.02310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Huxtable RJ. Physiological actions of taurine. Physiol Rev. 1992;72:101–63. doi: 10.1152/physrev.1992.72.1.101. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Li Y, Guan KL. Signaling by target of rapamycin proteins in cell growth control. Microbiol Mol Biol Rev. 2005;69:79–100. doi: 10.1128/MMBR.69.1.79-100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen C, Funding AT, Otkjaer K, Kragballe K, Jensen UB, Madsen M, et al. Protein expression of TNF-α in psoriatic skin is regulated at a posttranscriptional level by MAPK-activated protein kinase 2. J Immunol. 2006;176:1431–8. doi: 10.4049/jimmunol.176.3.1431. [DOI] [PubMed] [Google Scholar]
- Koepke JI, Nakrieko KA, Wood CS, Boucher KK, Terlecky LJ, Walton PA, et al. Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic. 2007;8:1590–600. doi: 10.1111/j.1600-0854.2007.00633.x. [DOI] [PubMed] [Google Scholar]
- Kohen R. Skin antioxidants: their role in aging and in oxidative stress—new approaches for their evaluation. Biomed Pharmacother. 1999;53:181–92. doi: 10.1016/S0753-3322(99)80087-0. [DOI] [PubMed] [Google Scholar]
- Kohler HBK, Huchzermeyer B, Martin M, De Bruin A, Meier B, Nolte I. TNF-α dependent NF-κB activation in culture canine keratinocytes is partly mediated by reactive oxygen species. Vet Dermatol. 2001;12:129–37. doi: 10.1046/j.1365-3164.2001.00237.x. [DOI] [PubMed] [Google Scholar]
- Krueger G, Koo J, Lebwohl M, Menter A, Stern RS, Rolstad T. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280–4. [PubMed] [Google Scholar]
- Krueger JG. The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol. 2002;46:1–23. doi: 10.1067/mjd.2002.120568. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescein as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–31. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Lebwohl M. Psoriasis. Lancet. 2003;361:1197–204. doi: 10.1016/S0140-6736(03)12954-6. [DOI] [PubMed] [Google Scholar]
- Lee CL, Ow DS, Oh SK. Quantitative real-time polymerase chain reaction for determination of plasmid copy number in bacteria. J Microbiol Methods. 2006;65:258–67. doi: 10.1016/j.mimet.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Liou HC, Baltimore D. Regulation of the NF-kappa B/rel transcription factor and Ikappa B inhibitor system. Curr Opin Cell Biol. 1993;5:477–87. doi: 10.1016/0955-0674(93)90014-h. [DOI] [PubMed] [Google Scholar]
- Liu SC, Karasek M. Isolation and growth of adult human epidermal keratinocytes in cell culture. J Invest Dermatol. 1978;71:157–62. doi: 10.1111/1523-1747.ep12546943. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, et al. Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a) Proc Natl Acad Sci USA. 2005;102:19057–62. doi: 10.1073/pnas.0509736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease P. TNF-alpha therapy in psoriatic arthritis and psoriasis. Ann Rheum Dis. 2004;63:755–8. doi: 10.1136/ard.2004.020719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease PJ. Psoriatic arthritis therapy advances. Curr Opin Rheumatol. 2005;17:426–32. doi: 10.1097/01.bor.0000166382.96024.85. [DOI] [PubMed] [Google Scholar]
- Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13:730–7. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- Nash PT, Florin TH. Tumour necrosis factor inhibitors. Med J Aust. 2005;183:205–8. doi: 10.5694/j.1326-5377.2005.tb06998.x. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ. Creation of psoriatic plaques: the ultimate tumor suppressor pathway. A new model for an ancient T-cell-mediated skin disease. Viewpoint. J Cutan Pathol. 2001;28:57–64. doi: 10.1034/j.1600-0560.2001.280201.x. [DOI] [PubMed] [Google Scholar]
- Nickoloff BJ, Nestle FO. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest. 2004;113:1664–75. doi: 10.1172/JCI22147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba M, Shibanuma M, Kuroki T, Nose K. Production of hydrogen peroxide by transforming growth factor-β1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J Cell Biol. 1994;126:1079–88. doi: 10.1083/jcb.126.4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayama Y. Oxidative stress in allergic and inflammatory skin diseases. Curr Drug Targets Inflamm Allergy. 2005;4:517–9. doi: 10.2174/1568010054526386. [DOI] [PubMed] [Google Scholar]
- Pelle E, Mammone T, Maes D, Frenkel KV. Keratinocytes act as a source of reactive oxygen species by transferring hydrogen peroxide to melanocytes. J Invest Dermatol. 2005;124:793–7. doi: 10.1111/j.0022-202X.2005.23661.x. [DOI] [PubMed] [Google Scholar]
- Petty MA, Kintz J, DiFrancesco GF. The effects of taurine on atherosclerosis development in cholesterol-fed rabbits. Eur J Pharmacol. 1990;180:119–27. doi: 10.1016/0014-2999(90)90599-2. [DOI] [PubMed] [Google Scholar]
- Punzi L, Podswiadek M, Sfriso P, Oliviero F, Fiocco U, Todesco S. Pathogenetic and clinical rationale for TNF-blocking therapy in psoriatic arthritis. Autoimmun Rev. 2007;6:524–8. doi: 10.1016/j.autrev.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int J Dermatol. 2004;43:326–35. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- Saripalli YV, Gaspari AA. Focus on: biologics that affect therapeutic agents in dermatology. J Drugs Dermatol. 2005;4:233–45. [PubMed] [Google Scholar]
- Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–58. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267:5317–23. [PubMed] [Google Scholar]
- Takao J, Yudate T, Das A, Shikano S, Bunkobara M, Ariizumi K, et al. Expression of NF-kappaB in epidermis and the relationship between NF-kappaB activation and inhibition of keratinocyte growth. Br J Dermatol. 2003;148:680–8. doi: 10.1046/j.1365-2133.2003.05285.x. [DOI] [PubMed] [Google Scholar]
- Terlecky SR, Koepke JI. Drug delivery to peroxisomes: employing unique trafficking mechanisms to target protein therapeutics. Adv Drug Deliv Rev. 2007;59:739–47. doi: 10.1016/j.addr.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Tokunaga C, Yoshino K, Yonezawa K. mTOR integrates amino acid-and energy-sensing pathways. Biochem Biophys Res Commun. 2004;313:443–6. doi: 10.1016/j.bbrc.2003.07.019. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Ann Rev Med. 1994;45:491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- Trouba KJ, Hamadeh HK, Amin RP, Germolec DR. Oxidative stress and its role in skin disease. Antioxid Redox Signal. 2002;4:665–73. doi: 10.1089/15230860260220175. [DOI] [PubMed] [Google Scholar]
- Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–24. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Veale DJ, Ritchlin C, FitzGerald O. Immunopathology of psoriasis and psoriatic arthritis. Ann Rheum Dis. 2005;64(Suppl 2):ii26–9. doi: 10.1136/ard.2004.031740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CS, Koepke JI, Teng H, Boucher KK, Katz S, Chang P, et al. Hypocatalasemic fibroblasts accumulate hydrogen peroxide and display age-associated pathologies. Traffic. 2006;7:97–107. doi: 10.1111/j.1600-0854.2005.00358.x. [DOI] [PubMed] [Google Scholar]