

Introduction
The advent of the polymerase chain reaction (PCR) radically transformed biological science from the time it was first discovered (Mullis, 1990). For the first time, it allowed for specific detection and production of large amounts of DNA. PCR-based strategies have propelled huge scientific endeavors such as the Human Genome Project. The technique is currently widely used by clinicians and researchers to diagnose diseases, clone and sequence genes, and carry out sophisticated quantitative and genomic studies in a rapid and very sensitive manner. One of the most important medical applications of the classical PCR method is the detection of pathogens. In addition, the PCR assay is used in forensic medicine to identify criminals. Because of its widespread use, it is important to understand the basic principles of PCR and how its use can be modified to provide for sophisticated analysis of genes and the genome.
The PCR Process
PCR is a simple, yet elegant, enzymatic assay, which allows for the amplification of a specific DNA fragment from a complex pool of DNA. Dr. Kary Mullis, who discovered the PCR assay, stated it “lets you pick the piece of DNA you’re interested in and have as much of it as you want” (Mullis, 1990). PCR can be performed using source DNA from a variety of tissues and organisms, including peripheral blood, skin, hair, saliva, and microbes. Only trace amounts of DNA are needed for PCR to generate enough copies to be analyzed using conventional laboratory methods. For this reason, PCR is a sensitive assay.
Each PCR assay requires the presence of template DNA, primers, nucleotides, and DNA polymerase. The DNA polymerase is the key enzyme that links individual nucleotides together to form the PCR product. The nucleotides include the four bases – adenine, thymine, cytosine, and guanine (A, T, C, G) – that are found in DNA. These act as the building blocks that are used by the DNA polymerase to create the resultant PCR product. The primers in the reaction specify the exact DNA product to be amplified. The primers are short DNA fragments with a defined sequence complementary to the target DNA that is to be detected and amplified. These serve as an extension point for the DNA polymerase to build on.
The above mentioned components are mixed in a test tube or 96-well plate and then placed in a machine that allows repeated cycles of DNA amplification to occur in three basic steps. The machine is essentially a thermal cycler. It has a thermal block with holes, into which the test tubes or plates holding the PCR reaction mixture are inserted. The machine raises and lowers the temperature of the block in discrete, precise and pre-programmed steps (Weier & Gray, 1988). The reaction solution is first heated above the melting point of the two complementary DNA strands of the target DNA, which allows the strands to separate, a process called denaturation. The temperature is then lowered to allow the specific primers to bind to the target DNA segments, a process known as hybridization or annealing. Annealing between primers and the target DNA occurs only if they are complementary in sequence (e.g. A binding to G). The temperature is raised again, at which time the DNA polymerase is able to extend the primers by adding nucleotides to the developing DNA strand (Figure 1). With each repetition of these three steps, the number of copied DNA molecules doubles.
Figure 1.
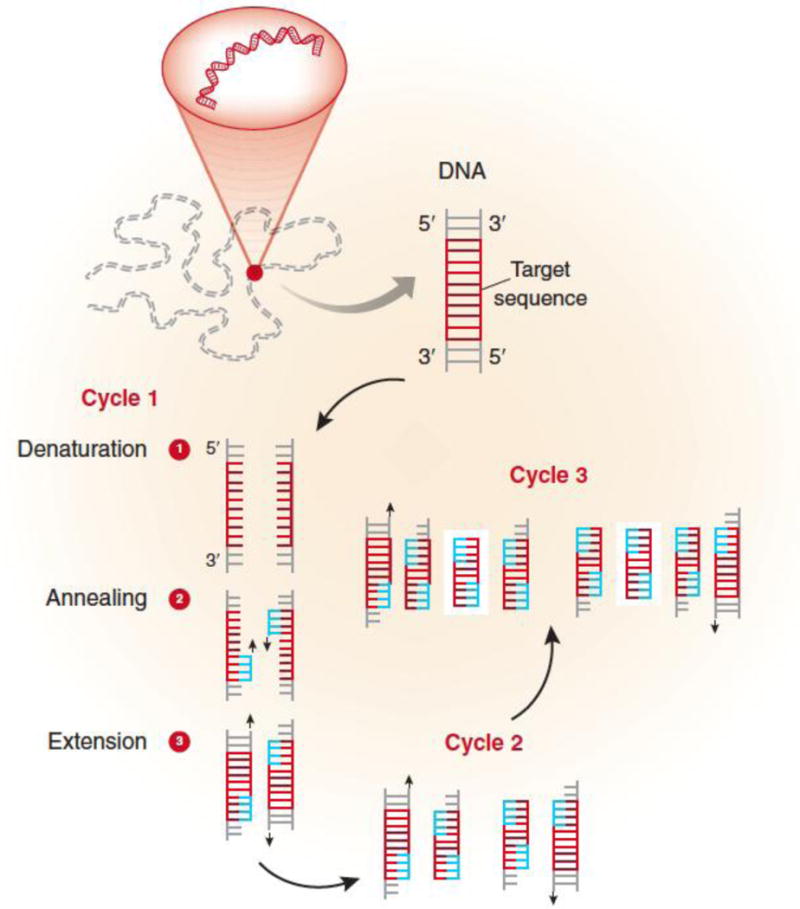
Schematic presentation of the PCR principle
Analysis of the PCR product
There are two main methods of visualizing the PCR products: (1) staining of the amplified DNA product with a chemical dye such as ethidium bromide, which intercalates between the two strands of the duplex or (2) labeling the PCR primers or nucleotides with fluorescent dyes (fluorophores) prior to PCR amplification. The latter method allows the labels to be directly incorporated in the PCR product. The most widely used method for analyzing the PCR product is the use of agarose gel electrophoresis, which separates DNA products on the basis of size and charge. Agarose gel electrophoresis is the easiest method of visualizing and analyzing the PCR product. It allows for the determination of the presence and the size of the PCR product (Figure 2). A predetermined set of DNA products with known sizes are run simultaneously on the gel as standardized molecular markers to help determine the size of the product.
Figure 2. Visualization of the PCR products on an agarose gel.
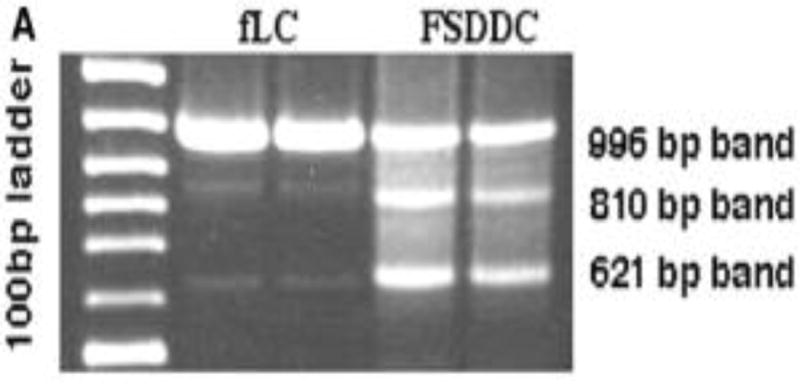
From Riedl et al, 2004: Identification of an alternatively spliced mouse Langerin transcript. (a) Ethidium bromide-stained agarose gel showing PCR products from full-length mouse Langerin obtained from C57BL/6 fresh LC (fLC) and from fetal skin-derived dendritic cells (FSDDC).
Typically, when PCR is used to detect the presence or absence of a specific DNA product, it is called qualitative PCR. Qualitative PCR is a good technique to use when PCR is performed for cloning purposes or to identify a pathogen. For example, in the report by Dworkin et al, qualitative PCR was used to detect the presence of Merkel cell polyomavirus in cutaneous squamous cell carcinoma (SCC) in immunocompetent individuals (2009). Using genomic DNA isolated from SCCs excised from immunocompetent individuals and primers specific to virus genes, the investigators were able to demonstrate the presence of a 351base pair (bp) viral gene in 6 out of 16 samples tested, by the presence of a PCR-product band about 351 bp long, as seen on a 2% agarose gel with ethidium bromide (Figure 3). The experiment also included template DNA from a polyomavirus containing plasmid as a positive control (P) and a negative water control (W). The first lane marked by (M) is the molecular marker, which is used to identify the size of the detected PCR product. The presence of a viral specific gene detected by PCR is marked by (+); absence of viral gene is marked by (−).
Figure 3. Qualitative PCR detects the presence of Merkel cell polyomavirus in cutaneous squamous cell carcinoma (SCC) in immunocompetent individuals.
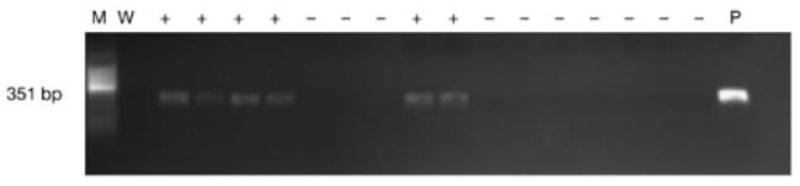
From Drowkin et al, 2009: MCPyV detection. (a) The presence of MCPyV in SCCs, genomic normal DNA, and adjacent skin DNA was determined by PCR using VP1 primers. A representational result is shown with 6 of 16 samples tested showing a PCR product at 351 bp. All experiments included DNA from an MCPyV plasmid as a positive control (P) and a negative water control (W). M, molecular weight marker; +, positive for virus; −, negative for virus.
Quantitative PCR
Quantitative real-time or qRT-PCR provides information beyond mere detection of DNA. It indicates how much of a specific DNA or gene is present in the sample. qRT-PCR allows for both detection and quantification of the PCR product in real-time, while it is being synthesized (Van Guilder et al, 2008). The two common methods used to detect and quantify the product include (1) fluorescent dyes that non-specifically intercalate with double-stranded DNA and (2) sequence-specific DNA probes consisting of fluorescently labeled reports. These permit detection only after hybridization of the probe with its complementary DNA target. Real-time PCR can be combined with reverse transcription, which allows messenger RNA to be converted into cDNA (i.e., reverse transcription), after which quantification of the cDNA is performed with qPCR (Valasek & Ripa, 2005). Quantification of the desired gene during the exponential amplification avoids problems that are associated with end-point PCR, which is analyzation after completion of the final PCR cycle.
Analysis of tumors is an ideal example of the use of PCR. It can be used to isolate and amplify DNA of tumor suppressor genes or proto-oncogenes. In turn, quantitative PCR can be utilized to quantify the amount of the particular gene isolated. On the other hand, quantitative PCR can be used to analyze single cells and quantify any combination of DNA, mRNAs, and proteins. (Stahlberg et al, 2012)
Advantages and limitations of PCR
There are multiple advantages to PCR. First, it is a simple technique to understand and to use, and it produces results rapidly (Bolognia et al, 2008). It is a highly sensitive technique with the potential to produce millions to billions of copies of a specific product for sequencing, cloning, and analysis. This is true of qRT-PCR as well, but qRT-PCR has the advantage of quantification of the synthesized product. Thus, it can be used to analyze alterations of gene expression levels in tumors, microbes, or other disease states.
Although PCR is a valuable technique, it does have limitations. Because PCR is a highly sensitive technique, any form of contamination of the sample by even trace amounts of DNA can produce misleading results (Bolognia et al, 2008; Smith & Osborn, 2009). In addition, in order to design primers for PCR, some prior sequence data is needed. Therefore, PCR can only be used to identify the presence or absence of a known pathogen or gene. Another limitation is that the primers used for PCR can anneal non-specifically to sequences that are similar, but not completely identical to target DNA. In addition, incorrect nucleotides can be incorporated into the PCR sequence by the DNA polymerase, albeit at a very low rate.
SUMMARY AND FUTURE DIRECTION
PCR is a simple and widely used process in which minute amounts of DNA can be amplified into multiple copies. In addition to the rapidity with which this assay works, it is able to quantitatively demonstrate how much of a particular sequence is present. As with all methods, the validity of the results should be compared with the specificity associated with the method. The future of PCR is promising, combining various assays and approaches to produce greater insight into various gene combinations (Botes et al, 2011). For example, in a study to link distinct taxa within the microbial community to specific metabolic processes, stable isotope probing was combined with qPCR (Postoller et al, 2011; Smith & Osborn, 2009). Microarray experiments can be validated by qPCR approaches due to its rapidity.
WHAT PCR DOES.
PCR is a very sensitive technique that allows rapid amplification of a specific segment of DNA.
PCR makes billions of copies of a specific DNA fragment or gene, which allows detection and identification of gene sequences using visual techniques based on size and charge.
Modified versions of PCR have allowed quantitative measurements of gene expression with techniques called real-time PCR
LIMITATIONS
The DNA polymerase used in the PCR reaction is prone to errors and can lead to mutations in the fragment that is generated
The specificity of the generated PCR product may be altered by nonspecific binding of the primers to other similar sequences on the template DNA
In order to design primers to generate a PCR product, some prior sequence information is usually necessary.
References
- 1.Mullis KB. The unusual origin of the polymerase chain reaction. Scientific American. 1990;262(4):56–61. 64–5. doi: 10.1038/scientificamerican0490-56. [DOI] [PubMed] [Google Scholar]
- 2.Weier HU, Gray JW. A programmable system to perform the polymerase chain reaction. DNA. 1988;7(6):441–7. doi: 10.1089/dna.1.1988.7.441. [DOI] [PubMed] [Google Scholar]
- 3.Dworkin AM, et al. Merkel cell polyomavirus in cutaneous squamous cell carcinoma of immunocompetent individuals. J Invest Derm. 2009;129(12):2868–74. doi: 10.1038/jid.2009.183. [DOI] [PubMed] [Google Scholar]
- 4.VanGuilder HD, Vrana KE, Freeman WM. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques. 2008;44(5):619–26. doi: 10.2144/000112776. [DOI] [PubMed] [Google Scholar]
- 5.Valasek MA, Repa JJ. The power of real-time PCR. Advances in physiology education. 2005;29(3):151–9. doi: 10.1152/advan.00019.2005. [DOI] [PubMed] [Google Scholar]
- 6.Stahlberg A, Thomsen C, Ruff D, et al. Quantitative PCR analysis of DNA, RNAs, and Proteins in the same single cell. Clin Chem. 2012;58:1682–1691. doi: 10.1373/clinchem.2012.191445. [DOI] [PubMed] [Google Scholar]
- 7.Botes M, de Kwaadsteniet M, Cloete TE. Application of quantitative PCR for the detection of microorganisms in water. Food Microbiol. 2011 Aug;28(5):848–61. doi: 10.1007/s00216-012-6399-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.AUTHOR/S. TITLE OF CHAPTER. In: Bolognia J, Jorizzo J, Rapini R, editors. Dermatology. Vol. 2008. Elsevier; Spain: 2008. PAGE RANGE. [Google Scholar]
- 9.Postollec F, Falentin H, Pavan D, et al. Recent advances in quantitative PCR (qPCR) applications in food microbiology. Food Microbiol. 2011 Aug;28(5):848–61. doi: 10.1016/j.fm.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 10.Smith C, Osborn M. Advantages and limitations of quantitative PCR (qPCR)-based approaches in microbial ecology. FEMS Microbiol Ecol. 2009 Jan;67(1):6–20. doi: 10.1111/j.1574-6941.2008.00629.x. [DOI] [PubMed] [Google Scholar]