

To the Editor
The treatment for primary B-cell defects with global antibody deficiency is replacement of immunoglobulin.1,2 However, what has not been explored is how to make the decision regarding immunoglobulin replacement for subjects with more modest humoral defects. Suitable immunoglobulin preparations are expensive and, once prescribed, usually result in lifelong therapy. Practical guidelines for initiating immunoglobulin therapy would be of value. Here we describe the development and use of a scoring system based on laboratory and clinical data in a cohort of patients referred for varying degrees of hypogammaglobulinemia.
Laboratory tests included immunoglobulin levels (IgG, IgA, and IgM) and specific antibody responses to both protein and carbohydrate antigens. Serum levels of IgG, IgA, and IgM and responses to tetanus and diphtheria after vaccination were assessed by using the laboratory-assigned protective titers. Post-vaccination pneumococcal responses were considered positive at a minimum of 1.3 μg/mL for any given serotype3 and based on the percentage of positive responses of the total tested.
To include clinical measures, we compiled common features from published resources,1,4–9 including serious bacterial infection (meningitis, sepsis, and osteomyelitis), hospitalizations, pneumonias and upper respiratory tract infections, and antibiotic use. Other features included gastrointestinal symptoms, such as infectious diarrhea, weight loss, failure to thrive, malabsorption, chronic gastroenteritis, inflammatory bowel–like disease, and history of autoimmune disease (commonly immune thrombocytopenia or autoimmune hemolytic anemia). Physical findings, lymphadenopathy, splenomegaly, splenectomy, and evidence of lung disease demonstrated by impaired pulmonary function or evidence of bronchiectasis on chest computed tomography were included.
By using these parameters, a 2-staged scoring system was constructed (Fig 1). A point value between 0 and 5 was assigned according to the degree of severity, with increasing points for lower serum IgG, IgA, and IgM levels and greater loss of antibody. Point values 0 to 5 were assigned for increasing numbers of the clinical events associated with immune deficiency.
FIG 1.

Laboratory and clinical history parameters according to the scoring system for indications for initiation of immunoglobulin therapy in patients with hypogammaglobulinemia. AIHA, Autoimmune hemolytic anemia; FVC, forced vital capacity; ITP, idiopathic thrombocytopenic purpura; TLC, total lung capacity.
The scoring system was then applied retrospectively to the medical records of 65 patients who were referred by their physicians to Mount Sinai Medical Center for evaluation and treatment of varying degrees of hypogammaglobulinemia. Subjects with X-linked agammaglobulinemia, hyper-IgM syndrome, common variable immunodeficiency with very low serum immunoglobulin levels and severe loss of antibody, isolated IgA or IgM deficiency, genetically defined immune deficiency syndromes, and secondary causes of hypogammaglobulinemia were excluded. Of these 65 subjects, 10 had previously been started on intravenous immunoglobulin and were seen in consultation for a second opinion; preimmunoglobulin infusion data were collected. Each subject’s laboratory, clinical, and total cumulative (laboratory plus clinical) scores were tabulated. Subjects were divided into 2 groups for further analysis: those for whom immunoglobulin therapy was recommended and those for whom it was not.
Median values and 25% to 75% interquartile ranges (IQRs) were calculated. A nonparametric significance test, the Mann-Whitney U test with a 2-tailed P value, was applied to determine statistical significance between groups for quantitative immunoglobulins and laboratory, clinical, and cumulative scores. The Fisher exact test was applied to determine statistical significance between numbers of subjects presenting with clinical findings within each group (immunoglobulin recommended or not).
Sixty-five charts of 37 female and 28 male patients were reviewed; these subjects had a median age of 46 years (range, 15–77 years). Median immunoglobulin levels were as follows: IgG, 370 mg/dL (IQR 25% to 75%, 190–565 mg/dL); IgA, 28 mg/dL (IQR 25% to 75%, 12–52 mg/dL); and IgM, 30 mg/dL (IQR 25% to 75%, 12–59 mg/dL; see Table E1 in this article’s Online Repository at www.jacionline.org). Fifty-eight percent (38/65) of subjects had protective antibody titers for either tetanus or diphtheria. The response to pneumococcal serotypes was more varied; overall, sera contained a protective level of antibody to a median of 14.3% of the serotypes tested (range, 0% to 100%). Clinical data are shown in Table E1. Forty patients had a history of 3 or more upper respiratory tract infections per year, 11 had at least 3 or more episodes of pneumonia, and 4 had a significant infection (2 with meningitis, 1 with sepsis, and 1 with empyema); 30 patients had been given 5 or more courses of antibiotics per year or prophylactic antibiotics. Twenty-eight patients had been hospitalized for a condition associated with hypogammaglobulinemia (usually pneumonia) in the 5 years before evaluation. Twelve patients had had either idiopathic thrombocytopenic purpura or autoimmune hemolytic anemia, and 8 had infectious diarrhea, malabsorption, inflammatory bowel–like disease, weight loss, or failure to thrive. Twenty patients had pulmonary function tests performed before evaluation; 1 of these had an FEV1/forced vital capacity ratio or total lung capacity of less than 70% of predicted value, and 6 of 8 subjects who had chest computed tomographic evaluations had evidence of bronchiectasis.
To determine whether the scores from the combined laboratory and clinical data could have been used to help in the decision to treat with replacement immunoglobulin, patients were stratified into 2 groups: those for whom immunoglobulin therapy had been suggested and those for whom it had not. Immunoglobulin had been recommended for 71% (n = 46) of the patients. These patients had a median serum IgG level of 263 mg/dL, IgA level of 17 mg/dL, and IgM level of 21 mg/dL see Table E2 in this article’s Online Repository at www.jacionline.org. Twenty had protective antibody titers for either tetanus or diphtheria but impaired responses to pneumococcal vaccination, with a median of only 7.1% of positive responses to the serotypes tested (range, 0% to 100%). In comparison, the 19 patients for whom immunoglobulin was not recommended had significantly (P <.001) higher serum immunoglobulin levels (median IgG, 575 mg/dL; IgA, 56 mg/dL; and IgM, 79 mg/dL; see Table E2). Of these subjects, 18 had protective antibody titers for either tetanus or diphtheria and positive responses to 35.7% of the pneumococcal serotypes (range, 0% to 100%), demonstrating more preserved humoral immunity (P < .05). Among these 19 patients, 12 had poor/deficient (<50%) responses to polysaccharide antigen, of whom 3 had IgG levels of greater than 600 mg/dL, suggesting a diagnosis of selective antibody deficiency with normal or near-normal immunoglobulin levels. Although patients in the group for whom immunoglobulin was recommended reported more infections, antibiotic use, and hospitalizations, there was no statistical difference in these numbers when compared to those in patients for whom this therapy was not recommended. However, those for whom immunoglobulin was prescribed had a significantly higher incidence of cytopenias (idiopathic thrombocytopenic purpura or autoimmune hemolytic anemia).
Relating the above data to the scoring system, for the 46 subjects for whom immunoglobulin was prescribed, the median laboratory score was 17 and the clinical score was 11, resulting in a cumulative score of 27 (see Fig E1 in this article’s Online Repository at www.jacionline.org). Of this group, 87% (n = 40) had laboratory scores of 10 or greater, and 40 patients had cumulative scores of greater than 16. In contrast, the 19 patients for whom immunoglobulin was not prescribed had median laboratory scores of 7, clinical scores of 10, and cumulative scores of 16. Of these, only 3 patients had laboratory scores of 10 or greater, with 2 of these having slightly low serum IgG levels and poor responses to pneumococcal vaccination. Although the laboratory scores between the groups were highly significant (P <.001), clinical scores taken as a whole were not, mostly because the patients were commonly referred because of frequent respiratory tract infections.
In clinical practice medical conditions common in patients with immune deficiency are also used to supplement laboratory evidence of antibody deficiency, and we have included these in the scoring system. However, frequent infections alone do not necessarily suggest an immune deficit and would not provide a sole basis for prescribing immunoglobulin therapy. Considering the range of laboratory and clinical scores of these patients, we suggest a scoring decision tree to aid in defining the need for immunoglobulin therapy (Fig 2). Beginning with laboratory data, patients with laboratory scores of 10 or more might be candidates for immunoglobulin therapy. In cases in which the laboratory score is less than 10, further considerations, including clinical scores, might be additive. Immunoglobulin therapy might be considered for patients with cumulative scores of greater than 16. For patients with cumulative scores of 10 to 16, the physician’s judgment can prevail based on days missed from school/work, other autoimmune conditions not included here, and the clinician’s experience. However, for patients with laboratory scores of less than 10 points, immunoglobulin replacement might be less likely to be helpful; these patients could be monitored for subsequent immunologic decline, suggestive clinical events, or both.
FIG 2.
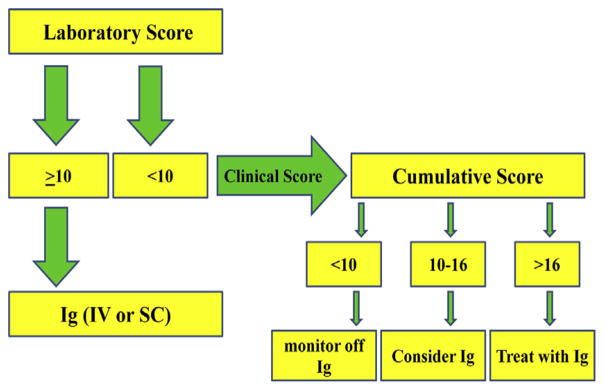
Scoring decision tree. Ig, Immunoglobulin; IV, intravenous; SC, subcutaneous.
Our proposal is meant to be a guide for the initiation of immunoglobulin therapy in adults with modest hypogammaglobulinemia. A patient’s score can be used to monitor his or her clinical progression over time; furthermore, a low score does not rule out the possibility of requiring immunoglobulin therapy in the future.
We acknowledge that this scoring template was designed retrospectively and that the scoring is arbitrary. This proposal requires validation, standardization, and proof of concept in large populations of patients with hypogammaglobulinemia, ideally in different geographic areas and diverse ethnic backgrounds. The majority of our surveyed patients were adults when referred to Mount Sinai, and the proposed scoring parameters might need modification before applying to a pediatric population. Once validated in other populations, our scheme can be used to stratify populations into risk groups and has clinical utility toward current health care policy issues with reimbursement. These recommendations are based on evidence from the current literature and will change with future experience and understanding of the disease process. We encourage and welcome feedback from the immunology community to offer suggestions, recommendations, and personal experience to help develop a consensus.
Supplementary Material
Acknowledgments
This study was conducted under “Dissecting Antibody Deficiencies” GCO#10-1338(0001)(01) ME and funded by Octapharma.
Footnotes
Disclosure of potential conflict of interest: S. Agarwal has no relevant conflicts of interest. C. Cunningham-Rundles has consultancy arrangements with CSL Behring, Baxter, Grifols, and Merck and has received a grant from Octapharma (now completed).
References
- 1.Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–78. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yong PL, Boyle J, Ballow M, Boyle M, Berger M, Bleesing J, et al. Use of intravenous immunoglobulin and adjunctive therapies in the treatment of primary immunodeficiencies: a working group report of and study by the Primary Immunodeficiency Committee of the American Academy of Allergy Asthma and Immunology. Clin Immunol. 2010;135:255–63. doi: 10.1016/j.clim.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Sorensen RU, Moore C. Antibody deficiency syndromes. Pediatr Clin North Am. 2000;47:1225–52. doi: 10.1016/s0031-3955(05)70269-8. [DOI] [PubMed] [Google Scholar]
- 4.Agarwal S, Mayer L. Pathogenesis and treatment of gastrointestinal disease in antibody deficiency syndromes. J Allergy Clin Immunol. 2009;124:658–64. doi: 10.1016/j.jaci.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol. 2011;127:1329–43. e2. doi: 10.1016/j.jaci.2011.02.047. [DOI] [PubMed] [Google Scholar]
- 6.Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125:1354–60. e4. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Stirling RG, Paul E, Hore-Lacy F, Thompson BR, Douglass JA. Longitudinal decline in lung function in patients with primary immunoglobulin deficiencies. J Allergy Clin Immunol. 2011;127:1414–7. doi: 10.1016/j.jaci.2011.03.041. [DOI] [PubMed] [Google Scholar]
- 8.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–7. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Podjasek JC, Abraham RS. Autoimmune cytopenias in common variable immunodeficiency. Front Immunol. 2012;3:189. doi: 10.3389/fimmu.2012.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.