

Abstract
Purpose.
The α2β1 integrin plays an important but complex role in angiogenesis and vasculopathies. Published GWAS studies established a correlation between genetic polymorphisms of the α2β1 integrin gene and incidence of diabetic retinopathy. Recent studies indicated that α2-null mice demonstrate superior vascularization in both the wound and diabetic microenvironments. The goal of this study was to determine whether the vasculoprotective effects of α2-integrin deficiency extended to the retina, using the oxygen-induced retinopathy (OIR) model for retinopathy of prematurity (ROP).
Methods.
In the OIR model, wild-type (WT) and α2-null mice were exposed to 75% oxygen for 5 days (postnatal day [P] 7 to P12) and subsequently returned to room air for 6 days (P12–P18). Retinas were collected at postnatal day 7, day 13, and day 18 and examined via hematoxylin and eosin and Lectin staining. Retinas were analyzed for retinal vascular area, neovascularization, VEGF expression, and Müller cell activation. Primary Müller cell cultures from WT and α2-null mice were isolated and analyzed for hypoxia-induced VEGF-A expression.
Results.
In the retina, the α2β1 integrin was minimally expressed in endothelial cells and strongly expressed in activated Müller cells. Isolated α2-null primary Müller cells demonstrated decreased hypoxia-induced VEGF-A expression. In the OIR model, α2-null mice displayed reduced hyperoxia-induced vaso-attenuation, reduced pathological retinal neovascularization, and decreased VEGF expression as compared to WT counterparts.
Conclusions.
Our data suggest that the α2β1 integrin contributes to the pathogenesis of retinopathy. We describe a newly identified role for α2β1 integrin in mediating hypoxia-induced Müller cell VEGF-A production.
Keywords: integrin, angiogenesis, retinopathy, Müller cells
In the oxygen-induced retinopathy model of retinopathy of prematurity, α2β1 integrin contributes to the pathogenesis of retinopathy by mediating hypoxia-induced VEGF production by retinal Müller cells.
Introduction
Angiogenesis, the growth of capillaries from extant blood vessels, is a tightly regulated process and the major mechanism for expansion of the vascular network. Dysregulated angiogenesis is a hallmark of many diseases ranging from cancer to diabetes. In the retina, chronic, pathologic angiogenesis, termed retinal neovascularization (NV), is a central feature of ocular diseases, including diabetic retinopathy, and retinopathy of prematurity (ROP).1
Retinal NV is often preceded by local tissue hypoxia stemming from capillary loss.2–4 Hypoxia initiates stabilization of the hypoxia-inducible factor (HIF) transcription factors, which coordinate cellular responses to hypoxia.5 In Müller cells, the glial cell type that is responsible for maintaining retinal homeostasis and modulating the growth factor microenvironment, retinal hypoxia triggers an HIF-2α–mediated protective program of vasoactive factor secretion.6–8 In pathological conditions, overproduction of growth factors, most prominently VEGF, stimulates retinal NV. Chen et al.9 suggest that α2β1 integrin may have an important role in regulating VEGF production in endothelial cells under conditions of hyperglycemia. In the retina, endothelial cells and many other cell types, including astrocytes, retinal pigment epithelial cells, and ganglion cells, contribute to VEGF overproduction; however, animal models have identified Müller cells as the predominant source of VEGF during retinal NV.10–17
Integrins, a class of obligate α/β heterodimers that serve as cell surface receptors for extracellular matrix ligands, have emerged as important actors in many vascular diseases.18 The α2β1 integrin, a collagen and laminin receptor, has been associated with risk of diabetic retinopathy; multiple genome-wide association studies (GWAS) independently identified an increased risk of diabetic retinopathy among patients with high α2β1 integrin expressing polymorphisms of the ITGA2 gene.19–21 Interesting new work using the diet-induced obesity mouse model indicates that α2-null mice have decreased skeletal muscle capillary rarefaction in the diabetic context.22 As pathological retinal NV is precipitated by capillary loss, the phenotype reported by Kang et al,22 along with the GWAS data, suggested that α2-integrin deficiency may be protective against retinal NV in ocular diseases.23,24
To investigate the hypothesis that α2-integrin deficiency can attenuate retinal NV, we characterized the α2β1 integrin-deficient mouse in the established oxygen-induced retinopathy (OIR) model for ROP. Our findings suggest that α2-integrin deficiency indeed protects the developing retina from oxygen-induced retinopathy; this protective effect appears to be a result of a novel role for α2β1 integrin in mediating hypoxia-induced Müller cell VEGF production.
Materials and Methods
Animals and Animal Models
All animals were housed in pathogen-free conditions at Vanderbilt University Medical Center. All animal experiments were approved by the Vanderbilt University School of Medicine Animal Care and Use Committee, and they were conducted according to the principles expressed in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Generation of α2 integrin-deficient mice on a C57BL6/ was previously described.25 The mice used in these experiments were 99% genetically C57BL/6. All animals were appropriately age-matched.
For OIR, litters were exposed to 75% oxygen for 5 days from postnatal day (P) 7 to P12. and returned to room air for 6 days from (P12 to P18).23,26 At P12 and P18, mice were euthanized and their eyes or retinas were harvested for molecular biology studies, gene expression studies, or morphological analysis. The OIR model, as initially popularized by Smith et al,23 measures NV at P17; recently some groups have observed peak NV at P18 and adopted that time point as their OIR end point.27–29 The central avascular area at P12 and extent of NV at P18 was quantitated using ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). All morphological analyses were conducted in a blinded manner. Additional methodological details are included in the Supplementary Methods.
Quantitative PCR Analysis
For developmental and OIR studies, pairs of wild-type (WT) and α2-null retinas were harvested. Total RNA was recovered following manufacturer's protocol. Quantitative PCR (qPCR) was performed on an Applied Biosystems 7900HT qPCR machine using a 384-plate system, as per manufacture's protocol. Relative fold change in gene expression was defined using the comparative 2−ΔΔCT method (including the amplification efficiency correction factor). Relative RNA quantities were normalized to hypoxanthine-guanine phosphoribosyltransferase (HPRT), ribonuclease P/MRP 30kDa subunit (RPP30), or TATAA-box binding protein based on the gene, determined by NormFinder (Aarhus University Hospital, Aarhus, Denmark), to represent the optimum normalization gene for the set of samples analyzed. Primer pairs for mRNA expression (Supplementary Methods Table S1) analysis were designed using Primer Express 3.0 (Applied Biosystems, Grand Island, NY, USA) and published murine DNA and mRNA sequences (UCSC Genome Bioinformatics, Santa Cruz, CA, USA [http://genome.ucsc.edu/]).
Immunoanalyses
For hematoxylin and eosin (H&E) staining, orbits were removed, fixed in 4% paraformaldehyde (PFA), paraffin-embedded, and sectioned into 6-μm sagittal sections.23 For whole-mount analysis, retinas were dissected and permeabilized.30 For cryosections, orbits were enucleated, fixed in 4% PFA, and embedded in Tissue-Tek optimum cutting temperature compound (Sakura Finetek USA, Inc., Torrance, CA, USA) and sectioned (10 μm).31 Both cryosections and whole mounts were evaluated using immunofluorescence microscopy for identification and quantitation of CD31+ and α2β1 integrin+ endothelial cells, or glial fibrillary acidic protein (GFAP+) and α2β1 integrin+ Müller cells using antibodies listed in Supplementary Methods Table S2.30 Immunofluorescence images were captured with a Nikon Eclipse 80i fluorescence microscope (Melville, NY, USA). Quantitation was performed using ImageJ (National Institutes of Health).
Primary Müller Cell Isolation
Cultures of primary Müller retinal cells were established from retinas of P7 WT and α2-null mice according to previously reported methods.32 Briefly, eyes were enucleated and soaked overnight in medium (Dulbecco's modified Eagle's medium [DMEM] low glucose; HyClone, Logan, UT, USA), supplemented with 1× antibiotic/antimycotic solution (Sigma-Aldrich Corp., St. Louis, MO, USA). The next day, eyes were incubated at 37°C in digestion buffer (the soaking medium plus 0.1% trypsin and 70 U/mL collagenase) for 1 hour before retinas were separated. The retinas were then ground and dissolved in a 10% fetal bovine serum (FBS) containing DMEM growth media. Approximately four to six retinas were used per isolation. Cell growth was monitored for 4 to 5 days until significant cell growth was observed, after which cells were washed repeatedly until a pure population of flat adherent cells remained. The Müller cell content of the cultures was maintained via staining with the marker glutamine synthase and lack of neuronal markers. Cultures were regularly grown at 37°C in a 5% CO2/95% air (20.9% oxygen) atmosphere (normoxia) in a humidified incubator (NuAire, Plymouth, MN, USA) and passages three to six were used for experiments.
Müller Cell VEGF Induction
Wild-type and α2-null mouse Müller cell cultures were grown to 70% subconfluency and then exposed to hypoxia for 0, 4, 8, and 24 hours or hypoxia mimetic CoCl2 for 24 hours. Vascular endothelial growth factor-A protein levels were quantified by ELISA using the Quantikine Immunoassay kit from R&D Systems (MMV00; Minneapolis, MN, USA), according to the manufacturer's protocol. Vascular endothelial growth factor-A mRNA levels were analyzed via qPCR.
Results
In the Retina, α2β1 Integrin Is Most Strongly Expressed in Activated Müller Cells
Earlier reports from our laboratory and our colleagues suggested that the α2β1 integrin was expressed on growth factor–stimulated endothelial cells, but only modestly or not expressed on quiescent adult vessels.7 To temporally define expression during retinal development, the level of α2β1 integrin expression during retinal maturation in WT animals was determined. By quantitative RT-PCR (qRT-PCR) analysis, α2 subunit mRNA was detected at the highest levels at P5 and P7. Only low levels of α2 subunit mRNA were observed in the adult at P18 (Fig. 1A). For comparison, minimal α2 subunit mRNA was detected in α2-null (ITGA2−/−) mice. These data were confirmed via immunofluorescence analysis of α2β1 integrin (red) expression in cross-sections of P5, P7, and P18 retinas (Fig. 1B). In accordance with the mRNA results, α2-integrin staining was broadest at P5 and more limited at P18. Interestingly, at P5 and P7, the α2 integrin subunit (red) was expressed not only at the superficial vascular plexus, where endothelial cells were found, but also in the inner nuclear layer. Interestingly, the α2 integrin subunit (red) was prominently expressed on cells spanning from the inner limiting membrane to the outer limiting membrane in a pattern characteristic of Müller glia (Fig. 1B). At P18, cell type–specific localization of the integrin was evaluated. In the P18 adult retina, α2β1 integrin (red) was only weakly expressed on CD31+ endothelial cells (green) (Fig. 1C). Staining with GFAP (green), a well-established marker of Müller cells, strongly colocalized with α2β1 integrin staining (red) (Fig. 1D).
Figure 1.

α2β1 integrin expression in retinal endothelial and Müller cells. (A) The level of α2 integrin subunit mRNA expression in total retina of WT and ITGA2−/− mice at P5, P7, and P18 was determined by qRT-PCR. Values are normalized to HPRT or RPP30 and displayed relative to WT at P18. Values represent mean ± SEM. (B) Immunofluorescence analysis of α2β1 integrin (red) expression in cross-sections of P5, P7, and P18 WT retinas from unchallenged mice. Integrin expression suggests a pattern indicative of retinal Müller cells. The P5 and P7 images were taken at ×60 magnification. Postnatal day 18 images were taken at ×40 magnification and are representative of five or more retinas taken in three separate trials from separate litters. (C) Immunofluorescence analysis demonstrated limited colocalization of α2β1 integrin expression (red) and CD31 (green) on the vessels in the cross-sections of retinas from P18 WT mice grown in normal air. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Images were taken at ×40 magnification. (D) Analysis of P18 retina cross-section from inset in (B) at ×160 magnification. Staining of α2β1 integrin in red (top) and GFAP in green (middle) are merged to show colocalization in yellow (bottom) of α2β1 integrin and GFAP expression in retinal Müller cells.
α2-Integrin Deletion Impairs Hypoxia-Induced VEGF-A Production in Müller Cells In Vitro
Given the strong expression of α2β1 integrin in retinal Müller cells, we questioned whether α2β1 integrin deletion affected Müller cell function. We evaluated the in vitro response to hypoxia in WT and α2-null primary retinal Müller cells. Wild-type Müller cells secreted significantly higher levels of VEGF-A after 24 hours of incubation in hypoxia than α2-null Müller cells (665.7 vs. 419.6 pg/mL, P = 0.0286 at 24 hours) (Fig. 2A). Similarly, VEGF-A mRNA expression, as measured by qRT-PCR, was higher in WT than α2-null Müller cells after 8 and 24 hours of incubation in hypoxia (5.35- vs. 3.13-fold, P = 0.045 at 8 hours; 9.38- vs. 6.46-fold, P = 0.019 at 24 hours) (Fig. 2B). All fold changes were normalized to VEGF-A expression in WT cells at 0 hours of hypoxia exposure. To determine whether the difference in hypoxia-induced VEGF-A production was HIF transcription factor–dependent, we tested the effect of treatment with 200 μM CoCl2, a hypoxia-mimetic known to specifically activate HIF transcription factors. A 24-hour treatment with 200 μM CoCl2 induced VEGF-A secretion at similar levels in both WT and α2-null Müller cells (551.7 vs. 496.8 pg/mL, P > 0.05), suggesting that the role for α2β1 integrin in the regulation of VEGF-A lies upstream, rather than downstream of HIF transcription factor activity (Fig. 2C). To confirm differential hypoxia-induced HIF-2α activation in WT and α2-null primary retinal Müller cells, we used immunoblot analysis to quantitate HIF-2α levels after 24 hours of culture in normoxic or hypoxic conditions (Fig. 2D). Hypoxia-inducible factor-2α is significantly higher in hypoxia-treated WT compared to α2-null primary retinal Müller cells. Additionally we evaluated the effect of hypoxia on α2β1 integrin expression and found substantially higher levels of the integrin after 24 hours of culture in hypoxic conditions (Fig. 2D). These results were confirmed using In-Cell Western analysis (Supplementary Fig. S1).
Figure 2.
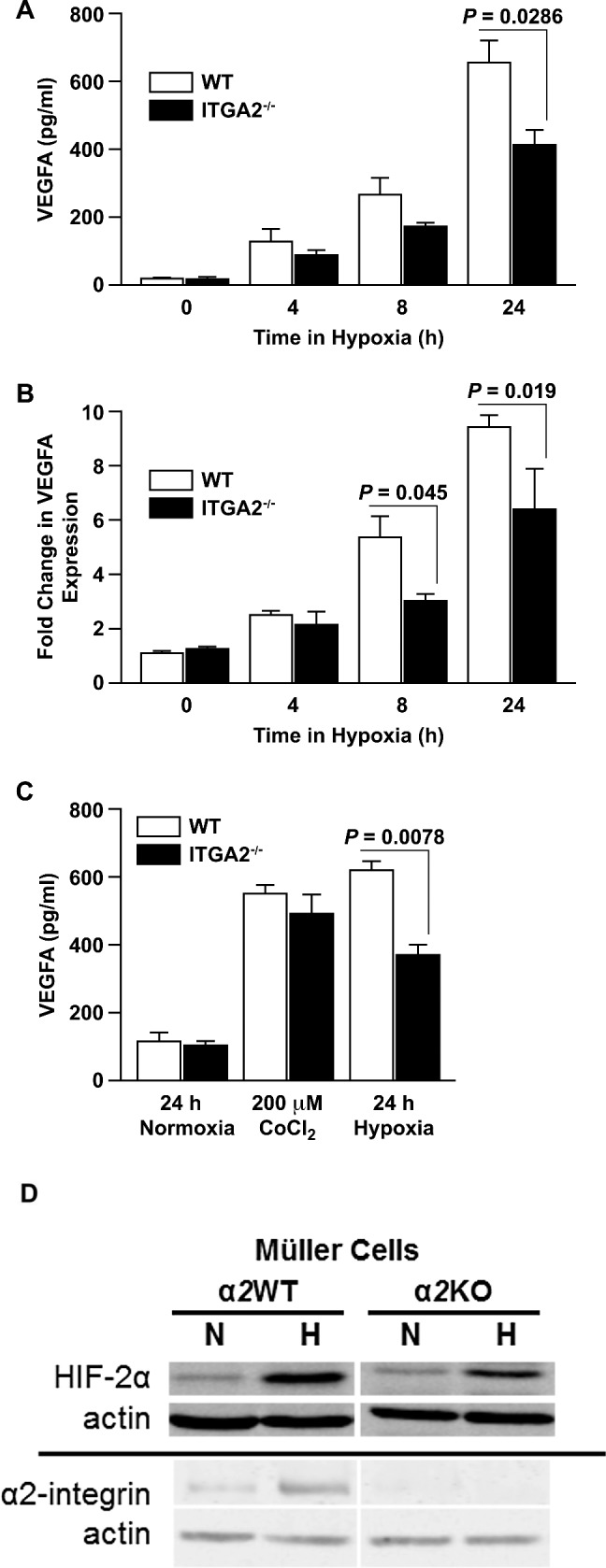
α2β1 deletion impairs hypoxia-dependent VEGF production in Müller cells in vitro. (A) Levels of VEGF-A protein in conditioned media from primary WT and ITGA2−/− Müller cells after incubation in hypoxia for 0, 4, 8, or 24 hours, as determined by ELISA. Data are shown as mean ± SEM for a representative experiment of five separate trials taken with separate isolates of primary Müller cells. (B) Fold changes of VEGF-A mRNA expression from primary WT and ITGA2−/− Müller cells incubated in hypoxic conditions for 0, 4, 8, or 24 hours, as determined by qRT-PCR. Fold changes are relative to WT at 0 hours (mean ± SEM). Data are representative of six separate trials with separate isolates of primary Müller cells. (C) Vascular endothelial growth factor-A protein in conditioned media from primary WT and ITGA2−/− Müller cells after 24 hours in normoxic conditions, treatment with 200 μM CoCl2, or hypoxic challenge. Vascular endothelial growth factor-A concentration was determined by ELISA. Data are shown as a mean ± SEM for a representative of three separate trials with separate isolates of primary Müller cells. (D) Immunoblot analysis of primary WT and ITGA2−/− Müller cells for HIF-2α and α2-integrin expression after 24 hours of culture in normal air and hypoxia.
α2β1 Integrin-Deletion Mitigates Müller Cell Activation and VEGF Production In Vivo
To determine whether the difference in hypoxia-induced Müller cell VEGF production also occurs in vivo, we examined retinas from WT and α2-null mice in the OIR model. Consistent with published literature, Müller cell GFAP expression increased robustly at P18 in response to relative hypoxia in WT mice.33,34 In contrast, GFAP expression had a more modest increase at P18 in α2-null mice after hypoxia (Fig. 3A). To determine whether the hypoxia-induced upregulation of α2-integrin expression that we observed in vitro was also found in vivo, we performed immunofluorescence analysis on retinal cross-sections from WT mice in “relative hypoxia” after the hyperoxia phase of OIR, compared with normal air controls. Specifically, we used P14 mice from the OIR model (P14 is 2 days after hyperoxia, labeled in Fig. 3 as Hypoxia) and from normal air litters. As expected, we observed visibly higher α2β1 integrin in the “hypoxia-treated” mice.
Figure 3.

α2β1 integrin-deletion mitigates Müller cell activation and VEGF production in OIR. (A) Cross-sections of retinas from age-matched P18 WT and ITGA2−/− mice raised in normal air or relative hypoxia (6 days after hyperoxia in OIR) were evaluated by immunofluorescence microscopy using anti-GFAP (Müller cells: red) or anti-CD31 (ECs: green) and DAPI (nuclei: blue). Images were taken at ×40 magnification and are representative of three separate trials. The intensity of GFAP staining was quantified and is shown below (mean ± SEM). Images are at ×40 magnification. (B) Cross-sections of retinas from age-matched WT and ITGA2−/− P14 mice in normal air or relative hypoxia (2 days after hyperoxia in OIR) were evaluated by immunofluorescence microscopy using anti-α2β1 integrin (red) and DAPI (nuclei: blue). Images were taken at ×60 magnification. (C) Lysates from age-matched WT and ITGA2−/− mice after OIR injury at P12, P13, and P18 was quantified by ELISA (mean ± SEM). (D) The level of VEGF-A mRNA expression in age-matched WT and ITGA2−/− retinas at P12, P13, and P18 retinas after OIR was determined by qRT-PCR. Fold changes are relative to WT P12 (mean ± SEM). Results in (B, C) are derived from four similar experiments.
Because Müller cells are the primary source of VEGF in the retina, and our in vitro data suggested decreased VEGF production in α2-null Müller cells, we evaluated VEGF-A levels during the different stages of OIR.17,33,34 At P13, 1 day after hyperoxia, VEGF-A protein and mRNA levels were significantly increased in retinal lysates from WT, compared to α2-null lysates (151.9 vs. 90.7 pg/mL, P = 0.001 at P13; 2.51- vs. 1.64-fold, P = 0.011) (Figs. 3C, 3D). At P18, VEGF-A protein remained elevated in the WT relative to the α2-null animals (113.9 vs. 66.4 pg/mL, P = 0.001) (Fig. 3B). At this later time point, the hypoxia-induced increases in VEGF mRNA levels had diminished in both WT and α2-null animals (Fig. 3D). Vascular endothelial growth factor mRNA fold changes are displayed relative to P12 WT. These results were consistent with our in vitro experiments with primary retinal Müller cells.
α2β1 Integrin-Deletion Protects Against Oxygen-Induced Retinopathy
Reduction in Müller cell VEGF production has been demonstrated to be protective in the OIR model.33,34 To determine whether α2β1-deletion also has a protective phenotype, we examined retinas from WT and α2-null pups exposed to the mouse OIR model. After 5 days of hyperoxic challenge, the central retina was avascular in the WT retina, as expected from published results (Fig. 4A).23,30,33,34 Although α2-null retinas also developed a central avascular area due to capillary loss near the optic nerve, the avascular region was significantly diminished compared to the WT (Fig. 4A), indicating that α2β1 integrin-deletion protected from early vessel attenuation during hyperoxia. At P18, after return to the relatively hypoxic room air environment for 6 days, pathologic NV was significantly increased in WT compared to α2-null retinas (Fig. 4A). This result was corroborated by the observed increase in pathologic neovascular tuft formation in H&E stains (Fig. 4B). In summary, deletion of the α2β1 integrin attenuates retinal vessel loss resulting from hyperoxia and protects from subsequent NV.
Figure 4.

α2β1 integrin deletion protects against OIR. The experimental protocol for OIR is shown as a timeline. (A) Representative immunofluorescence images of retinal flat mounts of age-matched WT and ITGA2−/− mice at P12 (after 75% hyperoxia) and P18 (after return to room air and relative hypoxia). Vessels are identified by GS Lectin (green) staining. Quantitation of central avascular area and neovascular area was determined as described and illustrated (mean ± SEM). Images are ×4 magnification. (B) Representative photograph of H&E-stained retinal cross-section of age-matched WT and ITGA2−/− at P18 following OIR injury. Neovascular nuclei are indicated by arrows. The number of nuclei per retinal area was quantitated (mean ± SEM). Images are ×40 magnification.
α2β1 Integrin Protection Against OIR Is Not a Consequence of Developmental Defects
In addition to their role during vascular stress, Müller cells play a critical role in regulating the growth factors that drive vascularization during retinal development.33 Because α2β1 integrin was important in hypoxia-induced VEGF production in retinal Müller cells, we explored the possibility that a developmental defect in retinal vascularization accounted for the mitigated OIR phenotype in α2-null mice.
Examination of the retinal vasculature at P5 and P7 revealed a modest but statistically significant difference in vascular outgrowth between WT and α2-null retinas (Fig. 5A). Vascular outgrowth was quantitated by comparing the remaining avascular area beyond the vasculature periphery, relative to total retinal area. Deletion of the α2 subunit led to increased avascular area in α2-null compared to WT mice at P5 and P7 (61% vs. 48%, P = 0.0361 at P5 and 27% vs. 18%, P = 0.0011 at P7). By P7, the difference in vascularization between the WT and α2-null retinas was diminished compared to P5. At P18, after scheduled vascular outgrowth was completed, differences in vascularization were assessed by measuring vascular density in P18 retinas and revealed no differences (Fig. 5A). Analysis of vascular density at P9 and P12 also revealed no differences (Supplementary Fig. S2).
Figure 5.

No significant differences in vascularization or VEGF levels were identified during development. (A) Retinal flat mounts of age-matched WT and ITGA2−/− mice at P5, P7, and P18. Vessels were visualized by GS Lectin (green) via immunofluorescence microscopy. For P5 and P7, vascularization was measured by radial vascular outgrowth, as quantitated by pixel percentage of avascular area at retinal periphery relative to total retina area. At P18, radial vascular outgrowth was complete, and so vascularization was measured by vascular density, as quantitated by pixel percentage of vessel area relative to total retina area. Each data point represents the average result from an experiment with a litter of at least four mice (data represent mean ± SEM). The results are representative of three separate trials. Images are ×4 magnification. (B) The level of VEGF-A protein in retinal lysates of P5, P7, and P18 WT and ITGA2−/− animals under normal conditions, as determined by ELISA (mean ± SD). (C) The fold change in mRNA expression of VEGF-A in developing retinas at P5, P7, and P18 was determined by qRT-PCR. Fold changes are relative to WT day 18 and shown as mean ± SEM. All results (B, C) are representative of three similar experiments.
Although interesting, the magnitude of this subtle developmental difference was not considered to be sufficient to influence the OIR phenotype. To ascertain whether the delayed retinal angiogenesis at P5 and P7 was indicative of baseline differences in Müller cell function during development, we examined VEGF levels during normal development. Vascular endothelial growth factor-A protein and mRNA levels in retinal lysates at P5, P7, and P18 were similar in WT and α2-null mice (Figs. 5B, 5C). The absence of VEGF-A differences at these time points indicated that α2β1 integrin deficiency does not affect the growth factor microenvironment, and by extension Müller cell function, under normal conditions.
Discussion
The mouse OIR model mimics pathologic NV observed in ROP and is analogous to proliferative diabetic retinopathy. Epidemiologic studies suggested an association between ITGA2 polymorphisms and diabetic retinopathy in clinical cohorts where preretinal NV is a defining pathologic feature.19–21 These epidemiologic studies suggested that sequence variants associated with high levels of α2β1 integrin expression were an independent risk factor for the development of diabetic retinopathy in individuals with type 2 diabetes, in addition to other cardiovascular diseases in younger patients (younger than 50).19–21,35 Our goal for testing α2-null mice in the OIR model was to evaluate the integrin's effect on retinal NV in a preclinical model of ocular vasculopathy.
In OIR experiments, deletion of the α2β1 integrin provided significant protection against both hyperoxia-induced vascular regression and subsequent neovascular proliferation. The α2-null retina demonstrated less reactive gliosis and substantially decreased VEGF-A secretion.
Under other experimental challenges, including wound healing and tumor implantation, the α2-null mouse demonstrated increased neoangiogenesis.25,36,37 However, in the diabetic microenvironment, α2-null mice exhibited decreased capillary rarefaction. In the OIR model, as in many ocular vasculopathies, pathological retinal NV is preceded by capillary loss and hypoxia. On this basis we originally hypothesized that the α2-null mice would be protected in the OIR model because of superior resistance to hyperoxia-induced capillary loss. However, our survey of α2β1 expression in the retina uncovered very modest levels of CD31-positive, α2β1-positive cells, suggesting low levels of integrin expression in capillaries and other endothelial cells. The expression of α2β1 integrin in a classic Müller glia pattern, and strong colocalization with Müller cell marker GFAP, led us to investigate the effect of α2-integrin deficiency in Müller cell function.
In vitro studies with primary retinal Müller cell cultures demonstrated a decreased ability for α2-null Müller cells to produce VEGF-A in response to hypoxia. Given that NV in OIR is considered to be primarily driven by VEGF-A overproduction, our findings strongly suggest that the integrin's contribution to the pathogenesis of OIR is from the Müller compartment.31,34,38 However, without cell-type–specific α2-null mice, it is not possible to rule out contributing roles in the endothelium or other cell types.
Differences in vascularization and vascular development at P7 can influence response to OIR. Our experiments identified a subtle but statistically significant difference in vascular outgrowth between WT and α2-null retinas. The modest scope of this difference in vascular outgrowth is unlikely to be large enough to influence the OIR phenotype that we observed. Hyperoxia-induced vaso-attenuation of capillaries is most prominent in the central area surrounding the optic nerve, an area in which we observed no significant differences between WT and α2-null vasculature. Further, the absence of differences in VEGF mRNA and protein levels between WT and α2-null retinas suggest normal Müller cell function during retinal development. Previously, Stenzel et al.39 reported that there were no differences in vascular development in α2-null mice. They observed similar vascular sprouting and vascular density at P5 in α2-null mice and heterozygous littermates. Our metric of vascular outgrowth was not reported in their study, and we compared WT mice to α2-null rather than heterozygous α2+/− mice.
These results provide the basis for future investigations into the mechanistic role of the α2β1 integrin in ocular diseases featuring pathologic retinal NV. Our study identified a novel role for the α2β1 integrin in regulating Müller cell function. This newly identified link to glial function may shed new light on studies linking ITGA2 polymorphisms with ischemic stroke.
Full characterization of the molecular mechanism by which the α2β1 integrin is required for hypoxia-induced, Müller cell–mediated VEGF-A induction is outside the scope of this study, but represents another intriguing area for further research. A connection between α2β1 integrin function and VEGF-A induction was first reported by Chen et al.,9 who showed in human umbilical vein endothelial cells that inhibitory antibodies to α2β1 integrin blocked hyperglycemia-induced VEGF-A expression. Our observation of the same phenomena in a distinct cell type (Müller cells as opposed to endothelial cells), in a different species (mouse versus human), with a different stimulus (hypoxia versus hyperglycemia) raises the possibility of a broadly applicable role for the α2β1 integrin in regulating VEGF-A expression. Interestingly, both hyperglycemia and hypoxia are reported to stimulate stabilization and activation of HIF transcription factors.40 To evaluate whether the difference between WT and α2-null Müller function occurred upstream or downstream of HIF transcription factor activation, we tested VEGF-A production after treating Müller cells with CoCl2, an established hypoxia mimetic and chemical inducer of HIF transcription factors. Treatment with CoCl2 caused similar levels of VEGF-A production by WT and α2-null Müller cells, which indicates that the α2-null Müller cells have no defects in VEGF-A expression or secretion, and that the difference must occur upstream of activation of HIF transcription factors.
Recent work indicates that HIF-2α is the HIF family member that is most responsible for Müller cell function during retinopathy.7,8 Culturing in hypoxia for 24 hours caused higher levels of HIF-2α protein in WT compared to α2-null Müller cells. Interestingly, we also observed significant upregulation of α2β1 integrin expression during hypoxia. Induction of α2β1 integrin in response to hypoxia has been reported in other cell types.41 Additionally, Cheli and colleagues42,43 identified PARP-1, a regulator of HIF-2α activity, as a transcriptional coactivator for α2β1 integrin. Based on these reports, it seems likely that HIF-2α may be involved in hypoxia-induced upregulation of α2β1 integrin; however, the potential mechanism by which α2β1 integrin may promote hypoxia-induced HIF-2α activation remains unclear. Recent work characterizing cellular signaling pathways in Müller and other microglial cells offer interesting hypotheses about potential mechanisms for α2β1 regulation of Müller cell–dependent VEGF-A production. The p38, a MAP kinase reported to be activated downstream of α2β1 integrin in mammary epithelial cells, was shown to be phosphorylated in rat Müller cells during hypoxia.44–46 Similarly, phosphorylation of p38 was demonstrated to be critical for VEGF production in mouse embryonic fibroblasts, as well as retinal pigment epithelium.46,47 Interestingly, p38 phosphorylation has been widely implicated in activation of HIF family member, HIF-1α, although the role of p38 in activating HIF-2α is not yet established.47–49 It is tempting to speculate that α2β1 integrin and HIF-2α exist in a feed-forward loop in which hypoxia-induced activation of HIF-2α upregulates α2β1 integrin, potentially in a PARP-1–mediated mechanism. In turn, α2β1 integrin could conceivably promote the stabilization and activation of HIF-2α via P38 phosphorylation or some other downstream signal. Crosstalk between α2β1 integrin and HIF-2α warrants further investigation as a potential mechanism through which the α2β1 integrin mediates hypoxia-induced VEGF-A production in Müller cells.
Acknowledgments
Supported by Grants CA115984, CA098027, and CA70275 from the National Institutes of Health. The authors alone are responsible for the content and writing of the paper.
Disclosure: A. Madamanchi, None; M. Capozzi, None; L. Geng, None; Z. Li, None; R.D. Friedman, None; S.K. Dickeson, None; J.S. Penn, None; M.M. Zutter, None
References
- 1. Barnett JM, McCollum GW, Penn JS. Role of cytosolic phospholipase A(2) in retinal neovascularization. Invest Ophthalmol Vis Sci. 2010; 51: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Steinkuller PG, Du L, Gilbert C, Foster A, Collins ML, Coats DK. Childhood blindness. J AAPOS. 1999; 3: 26–32 [DOI] [PubMed] [Google Scholar]
- 3. Bressler NM, Bressler SB. Preventative ophthalmology. Age-related macular degeneration. Ophthalmology. 1995; 102: 1206–1211 [DOI] [PubMed] [Google Scholar]
- 4. Rahmani B, Tielsch JM, Katz J, et al. The cause-specific prevalence of visual impairment in an urban population. The Baltimore Eye Survey. Ophthalmology. 1996; 103: 1721–1726 [DOI] [PubMed] [Google Scholar]
- 5. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995; 92: 5510–5514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW. Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem. 2004; 89: 1092–1100 [DOI] [PubMed] [Google Scholar]
- 7. Mowat FM, Luhmann UFO, Smith AJ, et al. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS One. 2010; 5: e11103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morita M, Ohneda O, Yamashita T, et al. HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J. 2003; 22: 1134–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S, Chakrabarti R, Keats EC, Chen M, Chakrabarti S, Khan ZA. Regulation of vascular endothelial growth factor expression by extra domain B segment of fibronectin in endothelial cells. Invest Ophthalmol Vis Sci. 2012; 53: 8333–8343 [DOI] [PubMed] [Google Scholar]
- 10. Weidemann A, Krohne TU, Aguilar E, et al. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia. 2010; 58: 1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol. 1995; 113: 1538–1544 [DOI] [PubMed] [Google Scholar]
- 12. Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A. 1995; 92: 10457–10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A. 1995; 92: 905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robbins SG, Rajaratnam VS, Penn JS. Evidence for upregulation and redistribution of vascular endothelial growth factor (VEGF) receptors flt-1 and flk-1 in the oxygen-injured rat retina. Growth Factors Chur Switz. 1998; 16: 1–9 [DOI] [PubMed] [Google Scholar]
- 15. Robbins SG, Conaway JR, Ford BL, Roberto KA, Penn JS. Detection of vascular endothelial growth factor (VEGF) protein in vascular and non-vascular cells of the normal and oxygen-injured rat retina. Growth Factors Chur Switz. 1997; 14: 229–241 [DOI] [PubMed] [Google Scholar]
- 16. Ishida S, Usui T, Yamashiro K, et al. VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med. 2003; 198: 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watkins WM, McCollum GW, Savage SR, Capozzi ME, Penn JS, Morrison DG. Hypoxia-induced expression of VEGF splice variants and protein in four retinal cell types. Exp Eye Res. 2013; 116: 240–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hynes RO. A reevaluation of integrins as regulators of angiogenesis. Nat Med. 2002; 8: 918–921 [DOI] [PubMed] [Google Scholar]
- 19. Petrovic MG, Hawlina M, Peterlin B, Petrovic D. BglII gene polymorphism of the alpha2beta1 integrin gene is a risk factor for diabetic retinopathy in Caucasians with type 2 diabetes. J Hum Genet. 2003; 48: 457–460 [DOI] [PubMed] [Google Scholar]
- 20. Matsubara Y, Murata M, Maruyama T, et al. Association between diabetic retinopathy and genetic variations in alpha2beta1 integrin, a platelet receptor for collagen. Blood. 2000; 95: 1560–1564 [PubMed] [Google Scholar]
- 21. Abhary S, Hewitt AW, Burdon KP, Craig JE. A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes. 2009; 58: 2137–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang L, Ayala JE, Lee-Young RS, et al. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes. 2011; 60: 416–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith LE, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994; 35: 101–111 [PubMed] [Google Scholar]
- 24. Casey R, Li WW. Factors controlling ocular angiogenesis. Am J Ophthalmol. 1997; 124: 521–529 [DOI] [PubMed] [Google Scholar]
- 25. Chen J, Diacovo TG, Grenache DG, Santoro SA, Zutter MM. The alpha(2) integrin subunit-deficient mouse: a multifaceted phenotype including defects of branching morphogenesis and hemostasis. Am J Pathol. 2002; 161: 337–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Connor KM, Krah NM, Dennison RJ, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009; 4: 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mechoulam H, Pierce EA. Expression and activation of STAT3 in ischemia-induced retinopathy. Invest Ophthalmol Vis Sci. 2005; 46: 4409–4416 [DOI] [PubMed] [Google Scholar]
- 28. Recchia FM, Xu L, Penn JS, Boone B, Dexheimer PJ. Identification of genes and pathways involved in retinal neovascularization by microarray analysis of two animal models of retinal angiogenesis. Invest Ophthalmol Vis Sci. 2010; 51: 1098–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukushima Y, Okada M, Kataoka H, et al. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J Clin Invest. 2011; 121: 1974–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Geisen P, Peterson LJ, Martiniuk D, Uppal A, Saito Y, Hartnett ME. Neutralizing antibody to VEGF reduces intravitreous neovascularization and may not interfere with ongoing intraretinal vascularization in a rat model of retinopathy of prematurity. Mol Vis. 2008; 14: 345–357 [PMC free article] [PubMed] [Google Scholar]
- 31. Werdich XQ, McCollum GW, Rajaratnam VS, Penn JS. Variable oxygen and retinal VEGF levels: correlation with incidence and severity of pathology in a rat model of oxygen-induced retinopathy. Exp Eye Res. 2004; 79: 623–630 [DOI] [PubMed] [Google Scholar]
- 32. Hicks D, Courtois Y. The growth and behaviour of rat retinal Müller cells in vitro. 1. An improved method for isolation and culture. Exp Eye Res. 1990; 51: 119–129 [DOI] [PubMed] [Google Scholar]
- 33. Bai Y, Ma J, Guo J, et al. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J Pathol. 2009; 219: 446–454 [DOI] [PubMed] [Google Scholar]
- 34. Yanni SE, McCollum GW, Penn JS. Genetic deletion of COX-2 diminishes VEGF production in mouse retinal Müller cells. Exp Eye Res. 2010; 91: 34–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moshfegh K, Wuillemin WA, Redondo M, et al. Association of two silent polymorphisms of platelet glycoprotein Ia/IIa receptor with risk of myocardial infarction: a case-control study. Lancet. 1999; 353: 351–354 [DOI] [PubMed] [Google Scholar]
- 36. Zweers MC, Davidson JM, Pozzi A, et al. Integrin alpha2beta1 is required for regulation of murine wound angiogenesis but is dispensable for reepithelialization. J Invest Dermatol. 2007; 127: 467–478 [DOI] [PubMed] [Google Scholar]
- 37. Zhang Z, Ramirez NE, Yankeelov TE, et al. alpha2beta1 integrin expression in the tumor microenvironment enhances tumor angiogenesis in a tumor cell-specific manner. Blood. 2008; 111: 1980–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Geisen P, Peterson LJ, Martiniuk D, Uppal A, Saito Y, Hartnett ME. Neutralizing antibody to VEGF reduces intravitreous neovascularization and may not interfere with ongoing intraretinal vascularization in a rat model of retinopathy of prematurity. Mol Vis. 2008; 14: 345–357 [PMC free article] [PubMed] [Google Scholar]
- 39. Stenzel D, Franco CA, Estrach S, et al. Endothelial basement membrane limits tip cell formation by inducing Dll4/Notch signalling in vivo. EMBO Rep. 2011; 12: 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bensellam M, Duvillié B, Rybachuk G, et al. Glucose-induced O2 consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS One. 2012; 7: e29807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keely S, Glover LE, MacManus CF, et al. Selective induction of integrin beta1 by hypoxia-inducible factor: implications for wound healing. FASEB J. 2009; 23: 1338–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheli Y, Williams SA, Ballotti R, Nugent DJ, Kunicki TJ. Enhanced binding of poly(ADP-ribose)polymerase-1 and Ku80/70 to the ITGA2 promoter via an extended cytosine-adenosine repeat. PLoS One. 2010; 5: e8743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gonzalez-Flores A, Aguilar-Quesada R, Siles E, et al. Interaction between PARP-1 and HIF-2α in the hypoxic response. Oncogene. 2014; 33: 891–898 [DOI] [PubMed] [Google Scholar]
- 44. Klekotka PA, Santoro SA. Zutter MM. Alpha 2 integrin subunit cytoplasmic domain-dependent cellular migration requires p38 MAPK. J Biol Chem. 2001; 276: 9503–9511 [DOI] [PubMed] [Google Scholar]
- 45. Klekotka PA, Santoro SA, Wang H, Zutter MM. Specific residues within the alpha 2 integrin subunit cytoplasmic domain regulate migration and cell cycle progression via distinct MAPK pathways. J Biol Chem. 2001; 276: 32353–32361 [DOI] [PubMed] [Google Scholar]
- 46. Klettner A, Westhues D, Lassen J, Bartsch S, Roider J. Regulation of constitutive vascular endothelial growth factor secretion in retinal pigment epithelium/choroid organ cultures: p38, nuclear factor κB, and the vascular endothelial growth factor receptor-2/phosphatidylinositol 3 kinase pathway. Mol Vis. 2013; 19: 281–291 [PMC free article] [PubMed] [Google Scholar]
- 47. Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005; 25: 4853–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mylonis I, Chachami G, Samiotaki M, et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem. 2006; 281: 33095–33106 [DOI] [PubMed] [Google Scholar]
- 49. Hirota K, Semenza GL. Rac1 activity is required for the activation of hypoxia-inducible factor 1. J Biol Chem. 2001; 276: 21166–21172 [DOI] [PubMed] [Google Scholar]