

Abstract
Ultra violet (UV) irradiation, in particular UVB, is the single most important carcinogen for skin tumor formation. UVB induces genetic mutations and immune suppression, which lead to abnormal cell proliferation and eventually tumor formation. Previously studies from our group and others demonstrated that both global and epidermal specific VDR knock out mice are predisposed to either chemical (DMBA)-or long-term UVB-induced skin tumor formation, paralleled by an increase in β-catenin signaling. Using primary cultured human keratinocytes, we further demonstrated that 1,25(OH)2-dihydroxyvitamin D3 (1,25(OH)2D3) suppresses cyclin D1 and Gli1 which are regulated by β-catenin/TCF signaling and have a critical role in epidermal carcinogenesis. Blockage of VDR by siRNA resulted in hyperproliferation of keratinocytes, and increased expression of cyclin D1 and Gli1. In addition, we also showed that 1,25(OH)2D3/VDR directly regulates transcriptional activity of β-catenin/TCF signaling using the –catenin reporter TopGlow. Using K14 driven tamoxifen-induced cre recombinase to delete both VDR and β-catenin in keratinocytes of mice following the first hair follicle cycle, we found that ablation of epidermal specific β-catenin cannot rescue VDR null mice from UVB-induced skin tumor formation. Further study using VDR or β-catenin single null mice is necessary to compare with the data from double null mice.
Keywords: UVB, Wnt/β-catenin, Skin tumor
1. Introduction
Accumulating evidence suggests that chronic sun exposure is the single most important etiological factor for the pathogenesis of non-melanoma skin cancer (NMSC), including squamous cell carcinoma (SCC) and basal cell carcinoma (BCC) [1–4]. Epidemiological data also show an association between the development of malignant melanoma (MM) and short-term intense UV-exposure, particularly when sunburns occur in youth [5,6]. The solar UV-spectrum can be divided into several bands with different physical and biological properties: UVC (wavelength < 280 nm), UVB (280–310 nm) and UVC (315–400 nm). While the predominant part of the short-wave, high-energy and destructive UV-spectrum (UVC and part of UVB) can be absorbed by the ozone layer, UVA radiation penetrates deeper into the dermis and deposits 30–50% of its energy in the dermal papillare (resulting in skin aging and solar elastosis). On the other hand, the majority of UVB can be absorbed by the epidermis (resulting in skin cancer development) [7]. Both UVA and UVB radiation induce DNA damage, resulting in mutagenic photoproducts including cyclobutane pyrimidine dimer (CpD) and 6-4-photoproducts. In fact, gene mutation has been implicated for the pathogenesis of skin cancer including p53 mutations (actinic keratosis, SCC) [7], and mutations in the patched (PTCH)/sonic hedgehog pathway (BCC) [8–10]. Together, sun exposure-induced premature skin aging, sunburns, immunesuppression and activation of latent viruses, which alone or in concert with each other could contribute to photocarcinogenesis. However, the mechanism of UVB-induced skin cancer at the molecular level remains largely unknown.
In addition to the DNA damage, sunlight induces production of vitamin D whose metabolite 1α,25(OH)2D3 has significant protective effect against the development of various types of cancer [11]. Recently, studies from our group and others demonstrated that global vitamin D receptor (VDR) mice are predisposed to either chemical (DMBA) or UVB-induced skin tumor formation [12–14], indicating a role of 1α,25(OH)2D3/VDR as tumor suppressor in skin. However, the mechanism of VDR protection against chemical or UVB-induced skin tumor is not clear.
1α,25(OH)2D3 acts via binding to its corresponding receptor VDR. VDR belongs to the subfamily of nuclear hormone receptors which requires heterodimerization with RXR (retinoid X receptor) for effective DNA interaction [15]. VDR is encoded by a relatively large gene encompassing 2 promoter regions, 7 protein-coding exons and 6 un-translated exons [16,17]. It has an extensive promoter region capable of generating multiple tissue-specific transcripts [18] In addition, VDR extends its signaling by directly or indirectly interacting with many other proteins; one such important protein is β-catenin, in which VDR and β-catenin are cross regulated via interaction between the activator function-2 (AF-2) domain of the VDR and C-terminus of β-catenin [19].
β-Catenin is a crucial component in the Wnt/β-catenin signaling pathway controlling the expression of specific target genes that regulate cell proliferation, cell fate and differentiation [20]. Furthermore, the interaction of β-catenin with VDR has been shown to contribute to at least some types of skin cancer [21]. Over-expression of β-catenin in which exon 3 is deleted or mutated leads to hyperproliferation of hair follicles eventually causing hair follicle tumors (pilomatricomas, trichofolliculomas) [22,23]. Very interestingly, deletion of VDR results in increased β-catenin activity. Since β-catenin is an oncogene for several types of cancer, we hypothesized that the predisposition of UVB-induced skin cancer is due to increased β-catenin signaling, a hypothesis that we tested by deleting both VDR and β-catenin in the skin of mice exposed to UVB.
2. Materials and methods
2.1. Animals
All animal experimentation in this study has been approved by the San Francisco VA Medical Center Animal Review Committee.
Mice homozygous for floxed VDR (kindly provided by Dr. Shigeaki Kato, Molecular and Cellular Biosciences, University of Yokyo, Japan, bred into the C57BL/6 background) were bred with mice expressing K14ERtam cre recombinase (Jackson Lab), which enabled us to selectively knockout the VDR in skin using parenteral application of tamoxifen (TM). These epiVDRKO were then crossed with mice expressing a floxed β-catenin (exon 2–6 deletion) (kindly provided by Dr. Matthias Hebrok, Diabetes Center, Department of Medicine, UCSF), to produce epidermal-specific VDR and β-catenin double knock out (DKO) mice. Genotyping was performed by PCR with different primers designed to amplify the mutant VDR or β-catenin, or wild type (WT) DNA (Table 1).
Table 1.
Primers used in this study.
| Gene | Primer sequence | Purpose | |
|---|---|---|---|
| Floxed VDR | F* R |
TCT GAC TCC CAC AAG TGT ACC ACG G ATG GAC AGG AAC ACA CAG CAT CA |
Genotype |
| Floxed β-catenin | F R |
AAG GTA GAG TGA TGA AAG TTG TT CAC CAT GTC CTC TGT CTA TTC |
Genotype |
| VDR | F R |
ACCCTGGTGACTTTGACCG GGCAATCTCCATTGAAGGGG |
qPCR |
| β-Catenin | F R |
CCCAGTCCTTCACGCAAGAG CATCTAGCGTCTCAGGGAACA |
qPCR |
| L19 | F R |
TCACCCTCAGGAACACGATTG GGATCTCCTGGATTCGAGGATTAT |
qPCR |
F: forward; R: reverse.
2.2. UVB irradiation and tumor monitoring
Dorsal skin was shaved with electric clippers 24 h before UVB exposure. Mice were TM injected (ip.) after weaning, and irradiated 3 times per week, with 1–2 days between treatments, and re-shaved as needed. The dorsal skin was exposed to UV irradiation from a band of eight FS-40 fluorescent lamps (Daavlin, Bryan, OH) as reported previously [12]. Briefly, mice were irradiated initially at the dose of 120 mJ/cm2. The dose was then increased to 25% per week for 5 weeks, up to 400 mJ/cm2 for 9 weeks, followed by 200 mJ/cm2 until week 40 [14]. After UVB treatment for 33 weeks, mice were examined weekly for tumor development by visual inspection and palpation. Mice bearing a skin growth 1 mm or larger, persisting for more than 7 days, were scored positive.
2.3. Epidermal preparation and RT-qPCR
Mice epidermal preparation, RNA extraction and RT-qPCR procedures were described in detail previously [24].
3. Results and discussion
3.1. Generation of epidermal-specific VDR and β-catenin DKO mice
Using a Cre-lox P system, we first generated conditional mice that specifically delete both VDR and β-catenin expression in epidermis. Floxed mice in which lox P sites were inserted into the introns upstream and down stream of either exon 2 of the VDR gene (Fig. 1A), or exons 2–6 of the β-catenin gene (Fig. 1B), were bred with transgenic mice expressing Cre recombinase under the control of the keratin 14 promoter. The resulting mice homozygous floxed for both VDR and β-catenin and expressing the Cre transgene were designated as double knock out (DKO). These were compared with the control littermates that have both floxed alleles but no Cre (WT). Upon TM injection, Cre recombinase will be activated and the floxed VDR and β-catenin genes will be deleted. This was confirmed by genotyping (data not shown) and the reduction of the epidermal VDR and β-catenin mRNAs (Fig. 1, C–D). One week after TM injection, both DKO and WT mice began UVB irradiation for 40 weeks (Fig. 1E).
Fig. 1.

Generation of conditional VDR and β-catenin null mice. (A and B) The gene-targeting strategy to delete VDR or β-catenin from keratinocytes by using. Cre-loxP system. (C and D). Decreased expression of VDR or β-catenin mRNA was shown by RT-qPCR. (E) Method of UVB-induced skin tumor formation in K14 driven, TM-induced VDR and β-catenin null mice.
3.2. Increased UVB-induced skin tumor formation in DKO mice
Previously we and other groups have reported that global VDRKO mice are predisposed for chemical or UVB-induced skin tumor formation [12–14]. To examine whether the increased skin tumor formation in VDRKO mice are attributable to the increased Wnt/β-catenin signaling, we generated the conditional DKO mice lacking both VDR and β-catenin. These mice are viable and fertile; however, with time some DKO (30–40%) mice showed increase pigmentation (Fig. 2A) and developed skin lesions on nose/face even without UVB exposure (data not shown). At the end of 40 week UVB irradiation, we observed an increase in skin tumor formation in DKO mice compared with their WT littermates (Fig. 2B–D and Table 2). Of 15 WT mice, only 2 mice (13%) developed a small tumor (<2 mm), while among 8 DKO mice, 4 (50%) were found to bear at least 1 tumor, and 2 mice were found to have 2–3 tumors for each mouse (Fig. 2E). In addition, the tumors found in DKO are larger in size (2.5–4.5 mm). Histologic analysis is currently underway to examine the type of tumor.
Fig. 2.
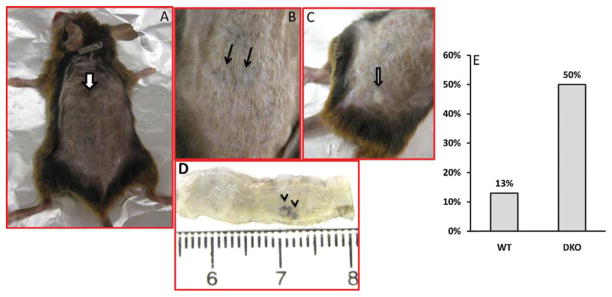
Conditional deletion of VDR and β-catenin in epidermis promotes UVB-induced skin tumors. (A) The whole body of DKO mouse shows pigmentation (arrow) and hair, two solid dark-brown tumors (B and D) and one white, exophytic tumor (C). (E) The graph shows the increased rate (%) of skin tumor formation in DKO vs. WT control.
Table 2.
| Genotype | No. of mice | % with tumors | Tumor burden (Average) | Response latency (week) |
|---|---|---|---|---|
| WT | 15 | 13 (2/15) 1 | 1 | 39–40 |
| DKO | 8 | 50 (4/8) | 1.75 | 36–37 |
Our results of increased UVB-induced skin tumor formation in DKO mice are surprising. Although a wealth of evidence indicates a role of β-catenin as an oncogene in hair follicle tumor formation because of its gain-of-function [23,25–27], this is the first example that lack of endogenous β-catenin can also result in tumor formation. Since global VDRKO mice are predisposed to UVB-induced skin tumor formation, and β-catenin is the co-activator of VDR signaling, it is possible that ablation of both proteins synergistically leads to photocarcinogenesis by some unknown mechanism. In this regard, the inhibitory effect of 1,25(OH)2-D3/VDR on Wnt/β-catenin signaling seems not to play a major role in preventing photocarcinogenesis. A thorough comparison of the outcome of long-term UVB irradiation on epiVDR and epiβ-catenin single KO mice will provide important comparisons.
Acknowledgments
The authors appreciate the tech help from Alicia Menendez. This study is supported by the NIH grant R01 AR055924, VA Merit Review, and DOD grant CA110338.
Abbreviations
- BCC
basal cell carcinoma
- CpD
cyclobutane pyrimidine dimer
- DKO
VDR and β-catenin double knock out
- MM
malignant melanoma
- NMSC
non-melanoma skin cancer
- SCC
squamous cell carcinoma
- 1,25(OH)2D3
1,25(OH)2-dihydroxyvitamin D3
- VDR
vitamin D receptor
- TM
tamoxifen
- UVB
ultra violet band B
- WT
wild type
References
- 1.Green A, Battistutta D. Incidence and determinants of skin cancer in a high-risk Australian population. International Journal of Cancer. 1990;46(3):356–361. doi: 10.1002/ijc.2910460303. [DOI] [PubMed] [Google Scholar]
- 2.Kricker A, Armstrong BK, English DR. Sun exposure and non-melanocytic skin cancer. Cancer Causes and Control. 1994;5(4):367–392. doi: 10.1007/BF01804988. [DOI] [PubMed] [Google Scholar]
- 3.Neale RE, et al. Basal cell carcinoma on the trunk is associated with excessive sun exposure. Journal of the American Academy of Dermatology. 2007;56(3):380–386. doi: 10.1016/j.jaad.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 4.Tremezaygues L, et al. Cutaneous photosynthesis of vitamin D: an evolutionary highly-conserved endocrine system that protects against environmental hazards including UV-radiation and microbial infections. Anticancer Research. 2006;26(4A):2743–2748. [PubMed] [Google Scholar]
- 5.Gandini S, et al. Meta-analysis of risk factors for cutaneous melanoma: III family history, actinic damage and phenotypic factors. European Journal of Cancer. 2005;41(14):2040–2059. doi: 10.1016/j.ejca.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Osterlind A, Engholm G, Jensen OM. Trends in cutaneous malignant melanoma in Denmark 1943–1982 by anatomic site. APMIS. 1988;96(11):953–963. doi: 10.1111/j.1699-0463.1988.tb00968.x. [DOI] [PubMed] [Google Scholar]
- 7.Tremezaygues L, Reichrath J. From the bench to emerging new clinical concepts: our present understanding of the importance of the vitamin D endocrine system (VDES) for skin cancer. Dermato-Endocrinology. 2011;3(1):11–17. doi: 10.4161/derm.3.1.14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie J, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 9.Johnson RJ, et al. Peripheral blood stem cell transplantation in myeloma using CD34 selected cells. Bone Marrow Transplantation. 1996;17(5):723–727. [PubMed] [Google Scholar]
- 10.Gailani MR, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nature Genetics. 1996;14(1):78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 11.Vuolo L, et al. Vitamin D and cancer. Frontiers in Endocrinology (Lausanne) 2012;3:58. doi: 10.3389/fendo.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teichert AE, et al. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor-null mice. Journal of Investigative Dermatology. 2011;131(11):2289–2297. doi: 10.1038/jid.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23(12):2103–2109. doi: 10.1093/carcin/23.12.2103. [DOI] [PubMed] [Google Scholar]
- 14.Ellison TI, et al. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. Journal of Investigative Dermatology. 2008;128(10):2508–2517. doi: 10.1038/jid.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kliewer SA, et al. Retinoid X receptor interacts with nuclear receptors in retinoic acid: thyroid hormone and vitamin D3 signalling. Nature. 1992;355(6359):446–449. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker AR, et al. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(10):3294–3298. doi: 10.1073/pnas.85.10.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyamoto K, et al. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Molecular Endocrinology. 1997;11(8):1165–1179. doi: 10.1210/mend.11.8.9951. [DOI] [PubMed] [Google Scholar]
- 18.Crofts LA, et al. Multiple promoters direct the tissue-specific expression of novel N-terminal variant human vitamin D receptor gene transcripts. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10529–10534. doi: 10.1073/pnas.95.18.10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah S, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Molecular Cell. 2006;21(6):799–809. doi: 10.1016/j.molcel.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 20.Grigoryan T, et al. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes and Development. 2008;22(17):2308–2341. doi: 10.1101/gad.1686208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmer HG, et al. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS ONE. 2008;3(1):e1483. doi: 10.1371/journal.pone.0001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gat U, et al. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 1998;95(5):605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- 23.Chan EF, et al. A common human skin tumour is caused by activating mutations in beta-catenin. Nature Genetics. 1999;21(4):410–413. doi: 10.1038/7747. [DOI] [PubMed] [Google Scholar]
- 24.Jiang YJ, et al. IL-1alpha accelerates stratum corneum formation and improves permeability barrier homeostasis during murine fetal development. Journal of Dermatological Science. 2009;54(2):88–98. doi: 10.1016/j.jdermsci.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Huelsken J, Birchmeier W. New aspects of Wnt signaling pathways in higher vertebrates. Current Opinion in Genetics and Development. 2001;11(5):547–553. doi: 10.1016/s0959-437x(00)00231-8. [DOI] [PubMed] [Google Scholar]
- 26.Ito M, et al. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature. 2007;447(7142):316–320. doi: 10.1038/nature05766. [DOI] [PubMed] [Google Scholar]
- 27.Malanchi I, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452(7187):650–653. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]