

Abstract
Atopic dermatitis (AD) is an inflammatory skin disease characterized by increased Th2 cytokine expression. AD skin lesions are often exacerbated by Staphylococcus aureus mediated secretion of the lytic virulence factor, alpha toxin. In the current study, we report that alpha toxin induced cell death is greater in the skin from patients with AD compared to controls. Furthermore, we find that keratinocyte differentiation and Th2 cytokine exposure influence sensitivity to S. aureus alpha toxin induced cell death. Differentiated keratinocytes are protected from cell death, while cells treated with Th2 cytokines have increased sensitivity to alpha toxin induced lethality. Our data demonstrates that downstream effects mediated by Th2 cytokines are dependent upon host expression of STAT6. We determine that Th2 cytokines induce biochemical changes that decrease levels of acid sphingomyelinase, an enzyme that cleaves sphingomyelin, an alpha toxin receptor. Furthermore, Th2 cytokines inhibit production of lamellar bodies, organelles critical for epidermal barrier formation. Finally, we determine that sphingomyelinase and its enzymatic product, phosphocholine, prevent Th2 mediated increases in alpha toxin induced cell death. Therefore, our studies may help explain the increased propensity for Th2 cytokines to exacerbate S. aureus induced skin disease, and provide a potential therapeutic target for treatment of AD.
INTRODUCTION
Atopic dermatitis (AD) is a chronic inflammatory skin disease associated with significant morbidity (Bieber, 2008). Colonization and recurrent infection with the bacterial pathogen, Staphylococcus aureus, are characteristic findings in AD (Boguniewicz and Leung, 2011); however, the mechanisms underlying this increased susceptibility remain poorly understood. Studies on the pathophysiology of AD reveal reduced epidermal barrier function attributable to defects in the keratinocyte differentiation program (Elias and Wakefield, 2011; Kuo et al., 2013; McAleer and Irvine, 2013). Furthermore, AD skin is characterized by over-expression of the Th2 cytokines, IL-4 and IL-13 (Hamid et al., 1994). These cytokines have been shown to inhibit keratinocyte differentiation causing decreased expression of the protein, filaggrin (FLG) (Howell et al., 2007). Recently, Th2 cytokines have also been shown to inhibit generation of ceramides (Sawada et al., 2012), hydrophobic lipids also critical for epidermal function.
Although Th2 cytokines are highly expressed in acute AD skin lesions (Hamid et al., 1994), the effect of these cytokines on cell death induced by S. aureus has not yet been explored. Cell death induced by S. aureus is primarily mediated by secretion of alpha toxin (Bubeck Wardenburg et al., 2007; Taubler et al., 1963), a destructive pore-forming toxin that is often found in the lesions of patients with severe AD (Travers et al., 2001; Wichmann et al., 2009). Alpha toxin induced cell lysis requires that the host express the lipid, sphingomyelin, on its cell surface (Schwiering et al., 2013; Watanabe et al., 1987). It has been reported that alpha toxin specifically recognizes the phosphocholine head group of sphingomyelin (Valeva et al., 2006). Following binding, alpha toxin heptamerizes and becomes irreversibly inserted into the cell membrane (Valeva et al., 1997).
Different cell types display varying sensitivities to staphylococcal alpha toxin (Valeva et al., 1997). Recent studies have shown that keratinocyte death induced by alpha toxin can be modulated through the process of cell differentiation (Brauweiler et al., 2013). It is not currently known, however, whether Th2 cytokines also modulate alpha toxin induced cell death. The current study was therefore carried out to compare the effects of alpha toxin in Th2 cytokine treated and control human keratinocytes, and to determine processes that may contribute to host defense.
RESULTS
Staphylococcal alpha toxin induces cell death in AD skin biopsies
Control and acute AD skin biopsies were harvested and treated for 24 hours with media (Fig. 1A top panels) or S. aureus alpha toxin (lower panels). Compared to normal biopsies, skin from AD patients shows an increase in pyknotic nuclei and decreased cytoplasmic staining, which are indicative of cell damage. (Yellow arrows in the lower right panel show pyknosis, condensed chromatin and decreased cytoplasm in alpha toxin treated AD skin). In AD skin, alpha toxin induced cell damage was observed throughout the skin keratinocyte layers. Measurement of cell death in skin biopsies was quantitated by lactate dehydrogenase (LDH) release and is shown in Fig. 1B. While small increases in spontaneous cell death were observed in AD skin, a prominent 5-fold increase in alpha toxin induced cell death was observed in AD skin (mean: 25.9%) compared to normal skin (mean: 3.59%). This difference in alpha toxin induced cell death was statistically significant (p < 0.001).
Fig 1. Increased staphylococcal alpha toxin induced cell death in atopic dermatitis skin.

(A) Panels show skin biopsies from normal and AD patients treated with media or alpha toxin, and stained by H&E. Increased cell damage (indicated by pyknotic cells with reduced cytoplasmic staining) is observed in AD biopsies compared to controls. Scale bar = 50 μm. (B) Quantification of cell death induced by alpha toxin treatment (12.5ng/ml 24 h) in skin biopsies from normal (n = 5) and AD patients (n = 5) was determined by LDH release. The LDH release induced by alpha toxin was calculated by subtracting out the baseline induced by media. Mean percent alpha toxin induced cell death ± SEM in control skin is 3.59% ± 1.20 (* p < .05 as compared to media cultured biopsies): mean alpha toxin induced cell death in AD is 25.9% ± 3.31, and was significantly different from control skin (*** p < 0.001 as compared to media cultured biopsies).
Th2 cytokines increase staphylococcal alpha toxin induced keratinocyte death
Atopic dermatitis is strongly associated with increased levels of the inflammatory Th2 cytokines, IL-4 and IL-13 (Hamid et al., 1994). We therefore examined the direct effect of these cytokines, as well as the Th1 cytokine, interferon gamma (IFN-γ), on keratinocyte sensitivity to staphylococcal alpha toxin. Modulation of alpha toxin induced cell death was determined by LDH release. Fig 2A shows that undifferentiated primary human epidermal keratinocytes were highly sensitive to alpha toxin, and that IL-4/IL-13 modestly increased both basal and alpha toxin induced cell death (p< 0.05). A striking effect, however, was induced by IFN-γ, which caused significant protection from alpha toxin in undifferentiated cells (p< 0.001). To evaluate whether keratinocyte differentiation influenced sensitivity to alpha toxin, keratinocytes were differentiated in vitro by raising the calcium concentration. Differentiation was monitored through induction of filaggrin (FLG) expression (Suppl. Fig. 1). A comparison of Figs. 2A and 2B shows that keratinocytes differentiated with calcium were resistant to alpha toxin induced cell death. Fig. 2B shows that although differentiated cells were protected from alpha toxin, keratinocytes differentiated in the presence of IL-4/IL-13 had significantly increased alpha toxin induced cell death (p < 0.001). Therefore, IL-4/IL-13 exerted effects that blocked the differentiation induced protection from alpha toxin.
Fig 2. Th2 cytokines increase staphylococcal alpha toxin induced keratinocyte death.
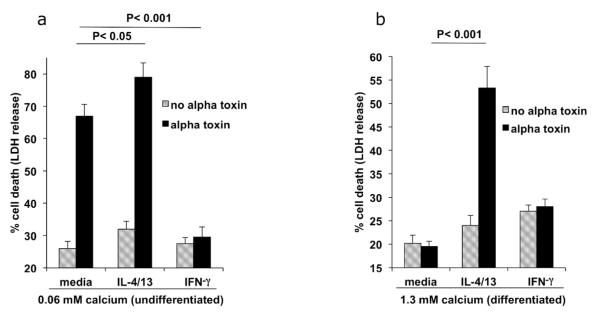
(A) Undifferentiated primary human keratinocytes were cultured in the presence of media, IL-4/IL-13, or IFN-γ. Cell death induced by 100 ng/ml alpha toxin was measured by LDH release (n = 4). IFN-γ caused significant protection from alpha toxin induced cell death (p < 0.001). Th2 cytokines caused a modest increase in cell death in undifferentiated cells (p < 0.05). (B) Primary keratinocytes treated with media, IL-4/13, or IFN-γ, were differentiated with calcium for four days. Cell death induced by 100 ng/ml alpha toxin induced was measured by LDH release (n = 4). Th2 cytokines caused a significant increase in alpha toxin induced death in differentiated cells, p < 0.001.
STAT6 mediates the increased cell death induced by Th2 cytokines
A potent increase in alpha toxin induced cell death was observed upon treatment with Th2 cytokines. In the remainder of our studies, we focus on determining the molecular events induced by Th2 cytokines that influence alpha toxin induced cytotoxicity. It has been documented that STAT6 mediates signaling through IL-4 and IL-13 (Albanesi et al., 2007; Kim et al., 2008; Travagli et al., 2004). We therefore used siRNA directed against STAT6 to show the specificity of the Th2 cytokines, IL-4 and IL-13, in potentiating alpha toxin induced cell death. Knockdown of STAT6 in differentiated cells is shown in Figs. 3A, B, and C. Fig. 3D demonstrates up-regulation of the differentiation marker, FLG, in differentiated cells, as well as FLG inhibition by IL-4/IL-13. Notably, following siRNA knockdown of STAT6, keratinocytes expressed FLG even in the presence of IL-4/IL-13. In Fig. 3E, we show that differentiated keratinocytes treated with IL-4/IL-13 are sensitive to alpha toxin induced cell death. However, keratinocytes treated with STAT6 siRNA were protected from the effects of these cytokines. The 4-fold reduction in cell death induced by alpha toxin in STAT6 knockdown cells was significant (p < 0.001). Therefore, the increased alpha toxin induced cell death mediated by IL-4/13 requires expression of STAT6.
Fig 3. Cell death induced by Th2 cytokines is mediated by STAT6.

Primary keratinocytes were transfected with control (non-targeting) or STAT6 siRNA. Transfected cells were then differentiated with calcium in the presence or absence of IL-4/13. (A) Following siRNA transfection, 80% percent knockdown of STAT6 was demonstrated by RTPCR. (B) Western blot analysis of STAT6 protein levels after siRNA knockdown. (C) Quantitation of reduction in STAT6 protein. Bars represent ratios of band density of STAT6 protein to that of actin loading control and are means of 3 experiments. (D) FLG expression in cytokine treated cells determined by RT-PCR (n = 3). (E) Control or STAT6 siRNA transfected cultures were treated with 100 ng/ml alpha toxin and cell death was measured by LDH release (n = 4). STAT6 siRNA protected IL-4/13 treated cells from alpha toxin induced cell death (p < 0.001).
Sphingomyelinase and phosphocholine protect Th2 cytokine treated keratinocytes from alpha toxin binding and induced death
Alpha toxin recognition of the membrane lipid, sphingomyelin, is critical for binding to the host cell (Valeva et al., 2006). Levels of sphingomyelin on the host cell surface can be reduced by sphingomyelinase (SMase), which cleaves the phosphocholine head group from sphingomyelin to produce ceramide (Suppl. Fig. 2). Importantly, although alpha toxin readily binds to membranes composed of sphingomyelin, it cannot bind to ceramide (Valeva et al., 2006). Treatment of cells with exogenously applied SMase, has been shown to convert 95% of surface sphingomyelin into ceramide without adversely affecting cell viability (Uchida et al., 2002). We therefore tested whether the addition of exogenous SMase could directly protect Th2 cytokine treated cells from the lethal effects of alpha toxin. Fig. 4A shows that Th2 cytokine exposed cells are sensitive to alpha toxin induced cell death. In contrast, keratinocytes treated exogenously with SMase were protected from alpha toxin induced lethality. These results indicated that SMase activity was sufficient to protect keratinocytes from alpha toxin, and suggest that SMase causes reduced expression of an alpha toxin receptor.
Fig 4. SMase and phosphocholine protect Th2 cytokine treated cells from alpha toxin induced death and alpha toxin binding.

(A) Primary keratinocytes treated with Th2 cytokines were incubated in the presence or absence of exogenous SMase (0.5 units/ml) for 1hour. Keratinocytes were then treated with the indicated concentrations of alpha toxin, and cell death was measured by LDH release (n = 4). (B) keratinocytes treated with Th2 cytokines were incubated in the presence or absence of SMase for one hour. Cells were then incubated with alpha toxin for one additional hour. Alpha toxin heptamer binding was determined by immunoblotting the cell lysates. (C) Quantitation of alpha toxin heptamer binding. Bars represent means of 3 experiments. (D) 10 mM phosphocholine or media control was pre-incubated with alpha toxin for 1 hour and then the mixture was added to primary keratinocytes treated with Th2 cytokines. Keratinocytes cell death was measured by LDH release. Statistically significant differences in cell death were observed between the two groups at alpha concentrations over 100 ng/ml (*** p < 0.001, n = 4).
The number of alpha toxin receptors is reflected by the amount of alpha toxin heptamer that irreversibly attaches to the host cell surface. Therefore, we next analyzed the effect of exogenous SMase treatment on alpha toxin binding to host cells (Figs. 4B, C). In comparison to untreated cells, cells pretreated for an hour with SMase bound significantly less alpha toxin heptamer (compare lanes 2,3 with lanes 5,6). The reduced binding demonstrates that SMase can mediate a protective effect by preventing alpha toxin attachment to the cell surface.
SMase treatment releases free phosphocholine. Because alpha toxin interacts primarily with the phosphocholine head group on sphingomyelin, we next tested whether free phosphocholine could act as a decoy to prevent alpha toxin binding to the cell membrane. Fig. 4D shows that cells treated with millimolar concentrations of phosphocholine are protected from alpha toxin mediated lysis, particularly at low alpha toxin concentrations.
Th2 cytokines reduce acid SMase expression, lamellar body formation and ceramide levels
We next tested whether Th2 cytokines directly inhibit SMase expression. We examined cellular acid SMase (ASMase) expression, as this is a predominant SMase that regulates sphingomyelin conversion into ceramide in keratinocytes. Keratinocytes differentiated in the presence or absence of cytokines were stained with anti-ASMase antibody and expression was determined by immunofluorescence. Fig. 5A shows that cells differentiated in the absence of cytokines have prominent staining of ASMase. In contrast, ASMase staining in Th2 cytokine treated cells is significantly reduced. Quantification (Fig. 5B) reveals a 3.5 fold reduction in the number of ASMase positive cells (p < 0.001). In normal differentiated cells, ASMase localizes to discrete granules, known as lamellar bodies (Grayson et al., 1985; Pillai et al., 1988). Our data shows that ASMase staining in differentiated cells localizes in punctate granules, consistent with association into lamellar bodies. Staining with Oil Red O, a lipid/lamellar body indicator, is prominent in differentiated cells, but is markedly reduced by Th2 cytokine treatment (Fig. 5C), thus indicating a reduction in lamellar body formation. Upon Th2 cytokine treatment a six-fold reduction in the number of Oil Red O positive cells was observed (p < 0.001) (Fig. 5D).
Fig 5. Th2 cytokines reduce ASMase levels, lamellar body formation, and ceramides.

Keratinocytes were differentiated in the presence or absence of Th2 cytokines. (A) ASMase staining (red), scale bar = 15 μm. (B) Quantification of the percent ASMase positive cells. (C) Lamellar granule formation (Oil Red O staining, Hematoxylin counterstain), scale bar = 40 μm. (D) Quantification of Oil Red O positive cells. (E) Keratinocytes were left undifferentiated (U), or differentiated for five (D5), seven (D7), or nine (D9) days in the absence (-) or presence (+) of Th2 cytokines. Lipids were extracted and analyzed by high-performance TLC and detected by fluorescence with 0.2% 8-anilino-1-naphthalenesulfonic acid (ANSA). Ceramide standard (Stnd) is indicated with an arrow. Ceramides induced upon differentiation were decreased by Th2 cytokines at all time points tested (compare lanes 2, 4, and 6 with 3, 5, and 7). (F) Keratinocytes were differentiated for 5 days in the presence or absence of Th2 cytokines and levels of ceramides were quantitated (n = 3).
An additional marker of keratinocyte differentiation, the up-regulation of ceramide levels, was also inhibited by Th2 cytokines as determined by analysis of cellular lipid content by thin layer chromatography (TLC) (compare lanes 2, 4, and 6 with 3, 5, and 7 in Fig. 5E). Quantification of ceramide levels in keratinocytes differentiated for five days in the presence and absence of Th2 cytokines is shown in Fig. 5F.
Quantification of levels of A Disintegrin And Metalloproteinase domain-containing protein 10 (ADAM10), an additional receptor for alpha toxin (Wilke and Bubeck Wardenburg, 2010), was also performed. Our results indicate that ADAM10 expression was not significantly modulated by Th2 cytokines (Suppl. Figs. 3A and B).
STAT6 is required for Th2 cytokine mediated inhibition of lipid processing events
Our data demonstrates that Th2 cytokines interfere with lipid processing by blocking production of ASMase, lamellar bodies, and ceramides. To determine whether STAT6 plays a role in these processes, we transfected cells with siRNA directed against STAT6, and then differentiated the keratinocytes in the presence or absence of Th2 cytokines. Our data indicates that STAT6 siRNA reverses the inhibition of lipid processing mediated by Th2 cytokines, leading to increased levels of ASMase (Fig.6A), lamellar bodies (Fig. 6B) and ceramides (Fig. 6C).
Fig 6. Changes in lipid metabolism induced by Th2 cytokines are mediated by STAT6.

Primary keratinocytes were transfected with control (non-targeting) or STAT6 siRNA. Transfected keratinocytes were differentiated in the presence or absence of Th2 cytokines. (A) Cells were stained with antibodies directed against ASMase and the percent positive cells were quantitated. (B) Cells were stained with Oil Red O to show lamellar bodies and the percent positive cells were quantitated. (C) Total lipids were extracted and ceramide levels were quantitated as described in Figure 5 and the change in ceramide content compared to media control was determined (n = 3).
DISCUSSION
S. aureus colonization and infection of the skin are recurrent complications in the pathogenesis of AD (Boguniewicz and Leung, 2011). Although inflammatory Th2 cytokines are highly expressed in acute AD skin lesions (Gittler et al., 2012; Suarez-Farinas et al., 2013), the effect of Th2 cytokines on cell death induced by S. aureus has not yet been explored. Here, we report that Th2 cytokines increase S. aureus alpha toxin induced cytotoxicity. We also show that AD skin is more sensitive to alpha toxin induced cell death. Therefore, the increased cell death in AD skin correlates with increased exposure to Th2 cytokines.
Previous studies have shown that Th2 cytokines induce signaling events through STAT6 (Albanesi et al., 2007; Kim et al., 2008; Travagli et al., 2004). We show here that STAT6 siRNA blocks the decrease in FLG expression induced by Th2 cytokines. Furthermore, STAT6 siRNA prevents the Th2 cytokine mediated reduction in ceramide levels, ASMase expression, and lamellar body formation. Cumulatively, STAT6 siRNA reverses the inhibition of differentiation induced by Th2 cytokines and protects keratinocytes from alpha toxin induced cell death.
We propose that Th2 cytokines induce changes at the molecular level that modulate keratinocyte sensitivity to alpha toxin. Our previous studies have shown that Th2 cytokines inhibit differentiation signals leading to decreased expression of filaggrin (Howell et al., 2007; Kim et al., 2013). We show here that keratinocytes exposed to Th2 cytokines also have reduced levels of ASMase, as well as reduced levels of ceramide. Since ASMase functions to degrade sphingomyelin, one could hypothesize that increased levels of sphingomyelin may remain on Th2 cytokine treated cells, rendering cells more sensitive to alpha toxin. We were unable, however, to determine levels of sphingomyelin on the cell surface (data not shown) as measurements were complicated by significant pools of sphingomyelin stored intracellularly inside lamellar bodies (Grayson et al., 1985; Wertz et al., 1984). We also found that lamellar body production, which normally occurs during keratinocyte differentiation (Pillai et al., 1988), was inhibited by Th2 cytokines. Since lamellar bodies are also involved in the transport of ASMase (Grayson et al., 1985), the reduction in lamellar bodies may result in reduced surface ASMase in Th2 cytokine treated cells.
It is interesting to note that the altered development caused by Th2 cytokines correlates with defects often associated with AD. Indeed, AD skin reportedly has reduced ASMase activity (Jensen et al., 2004) and lamellar body secretion (Pilgram et al., 2001), as well as reduced levels of ceramides (Di Nardo et al., 1998; Hatano et al., 2005; Janssens et al., 2011). Since, Th2 cytokines also reduce levels of tight junction proteins (De Benedetto et al., 2011), and filaggrin expression, these cumulative changes may result in aberrant barrier function, allowing greater alpha toxin penetration.
In addition, we observe increased basal cell death in Th2 cytokine treated primary keratinocytes. Therefore, these cytokines may contribute to elevated basal LDH release observed in AD skin. In addition, although none of our patients were overtly infected, AD skin can also be colonized with S. aureus, and contain measurable levels of alpha toxin (Travers et al., 2001), which may also contribute to increased basal LDH release.
We find that the increased cell death induced by Th2 cytokines can be blocked by exogenous SMase, a treatment that also prevents alpha toxin binding to the cell surface. Th2 cytokine treated keratinocytes can be protected from alpha toxin by treatment with phosphocholine. This protection may be due to fact that alpha toxin specifically interacts with the phosphocholine head group on sphingomyelin (Valeva et al., 2006). Since alpha toxin released by S. aureus plays a critical role in exacerbation of the keratinocytic lesions associated with AD (Wichmann et al., 2009), it remains possible that phosphocholine could be developed into therapy for treatment of S. aureus mediated skin disease
In contrast to Th2 cytokines, we find that exposure to the Th1 cytokine, IFN-γ, inhibits alpha toxin cytotoxicity. Intriguingly, IFN-γ treatment in leukemia cells has been shown to activate SMase, and generate ceramide (Kim et al., 1991). These data support a hypothesis in which protection from alpha toxin may be obtained by activation of cellular SMase.
S. aureus causes approximately 500,000 infections and 20,000 deaths each year in the United States alone (Klevens et al., 2007). Since infections are becoming increasingly antibiotic resistant, a better understanding of the factors that modulate alpha toxin induced lethality may help to reduce disease. Our current study demonstrates that Th2 cytokine exposed keratinocytes, as well as AD skin, have increased susceptibility to S. aureus alpha toxin induced cell death. We find that the molecular signaling events induced by Th2 cytokines are mediated through host expression of STAT6. Finally, we observe that phosphocholine can prevent the increased alpha toxin induced cell death induced by Th2 cytokines. Therefore, topical application of phosphocholine may be a useful therapeutic approach for prevention of S. aureus mediated exacerbation of skin lesions associated with AD.
MATERIALS AND METHODS
Patients and Human Skin Explant Cultures
Subjects included patients with acute lesional AD (erythematous lesions, less than 3 days in onset) and healthy individuals. All patients gave written informed consent prior to participation. The studies were approved by the Institutional Review Board at National Jewish Medical and Research Center in Denver, and were conducted according to the Declaration of Helsinki Principles. Four punch biopsies (2 mm) were obtained from skin of each donor and were placed in a 96-well plate and grown in EpiLife (Cascade Biologics; Portland, OR) with serum free media. All biopsies were initially cultured for 2 hours in the presence of media alone to allow for spontaneously occurring LDH release. The media was then removed and two biopsies from each donor were then cultured in fresh media containing no alpha toxin, and two biopsies were cultured in media containing 12.5ng/ml alpha toxin. Total incubation time was 20 hours. After the treatment period, media was removed and analyzed for LDH release. One of two of the skin biopsies from each group was used to determine total LDH release (Cyto-Tox One Kit, Promega; Madison, WI) and the other was submerged in 10% buffered formalin for sectioning and H&E staining.
Keratinocyte cell culture
Primary human keratinocytes (Cascade Biologics) were grown in serum-free keratinocyte growth medium (EpiLife; Cascade Biologics), with 1% human keratinocyte growth supplement (Cascade Biologics), 0.06 mM CaCl2, and antibiotics. When indicated, cells were treated with alpha toxin (Sigma) for 24 hours. IL-4 and IL-13 cytokines (50 ng/ml each) and IFN–γ (25ng/ml) were from R&D systems (Minneapolis, MN). For LDH assays, keratinocytes were plated at 20,000 per well in a 96 well plate and were allowed to adhere overnight before treatment. Keratinocytes were treated with cytokines or media for 24 hours and then left undifferentiated or differentiated with media containing 1.3 mM calcium for 4 days. LDH release was determined using the Cyto-Tox One Kit from Promega according to the manufacturer’s instructions.
Quantitative real-time PCR (RT-PCR)
Total RNA was isolated by RNeasy Mini Kits (Qiagen, Inc.; Valencia, CA) according to the manufacturer’s protocol. One microgram of RNA was reverse-transcribed using the Qiagen Quantiscript kit according to manufacturers protocol. RT-PCR was performed and analyzed by the dual-labeled fluorogenic probe method by using an ABI Prism 7300 sequence detector (Applied Biosystems; Foster City, CA). Primers and probes for human FLG, actin, STAT6, and ADAM10 were purchased from Applied Biosystems. Amplification reactions were performed in MicroAmp optical plates (Applied Biosystems) in a 25-μL volume as previously described (Howell et al., 2007). All reactions were normalized to beta actin.
siRNA transfection
Third-passage keratinocytes of 50-60% confluence were transfected according to the manufacturer’s instructions using Lipofectamine 2000 (Invitrogen) with 20 μM non-targeting or STAT6 Smartpool siRNA (Dharmacon) in antibiotic free media.
Immunoblot analysis
For analysis of alpha toxin binding and heptamerization, keratinocytes were treated with alpha toxin for one hour, washed with PBS, and harvested (without trypsin) in hypotonic lysis buffer with 1% Triton X-100, containing protease inhibitor (Complete, Roche). Cellular debris was pelleted by centrifugation and clarified lysates were resuspended in Laemmli buffer and proteins resolved on a 5-15% gradient gel (Biorad). (The alpha-toxin heptamer is stable during electrophoreses.) Transferred proteins were blotted with anti-alpha toxin antibody (Sigma), and detected by enhanced chemiluminescence (Amersham). Scanned images were quantitated with Image J software. For immunoblotting ADAM10 (antibody from Millipore), cells were harvested, lysed and probed as described above, but without alpha toxin treatment.
Cell staining and microscopy
Immunofluorescence staining was performed using keratinocytes grown on coverslips. Cells were left untreated or treated with Th2 cytokines for 24 hours prior to addition of 1.3 mM calcium (to induce differentiation). Cells differentiated for 3 days were fixed with paraformaldehyde and permeabilized briefly with .5% Triton X-100. After blocking with BSA, sections were incubated with primary ASMase antibody (Abcam; Cambridge, MA) for 2 hours at room temperature. Secondary antibody (Jackson labs; West Grove, PA) was added for 1 hour. Images were taken with a Leica Microscope at 40× magnification using SlideBook software. Oil Red O (Sigma; Saint Louis, MO) staining was performed on cells grown on coverslips and fixed with paraformaldehyde. Cells were counterstained with Hematoxylin (Sigma) and images obtained with a light microscope at 20× magnification. Quantification was performed in a blinded fashion from printed images by counting 6 fields with 100 cells per field.
Measurement of ceramide levels
Cells were left untreated or treated with Th2 cytokines for 24 hours prior to differentiation with 1.3 mM calcium for 5-9 days. Total cell lipids were harvested in chloroform/methanol/PBS (1:2:0.8) as described (Sawada et al., 2012). After centrifugation the bottom organic layer was collected, evaporated, re-dissolved in chloroform/methanol (2/1) solvent and fractionated by High Performance Thin Layer Chromatography (TLC) on silica gel 60 plates (EMD-Millipore; Billerica, MA) using a solvent system of chloroform/methanol/water (80:10:1). Ceramide bands were determined based on co-migration with control standards (Matreya; Pleasant Gap, PA). Lipids were detected with 0.2% 8-anilino-1-naphthalenesulfonic acid (Sigma) and illuminated by fluorescence with UV light on a BioRad Molecular Imager. Quantitation of ceramides was performed on three replicates of samples differentiated in the presence or absence of Th2 cytokines for 5 days using Quantity One software (BioRad Laboratories).
Sphingomyelinase and phosphocholine treatment
Keratinocytes were treated with Th2 cytokines for 24 hours. SMase (0.5 units/ml) from B. cereus (Sigma) was incubated with keratinocytes for one hour prior to the addition of alpha toxin. After 24-hour incubation, LDH analysis was performed. 10 mM phosphocholine (Bachem; Bubendorf, Switzerland) was pre-incubated with alpha toxin for 1 hour at room temperature. The alpha toxin/phosphocholine mixture was then added to Th2 treated keratinocytes for 24 hours and LDH analysis was performed.
Statistical analyses
All statistical analysis was conducted using Graph Pad Prism. Comparisons of expression levels were performed using analysis of variance (ANOVA) techniques and Student’s t tests as appropriate.
Supplementary Material
Supplemental Fig. 1 Filaggrin levels are reduced in Th2 cytokine treated cells. Primary kera4nocytes were treated with media, IL-4/13, or IFN-γ for 24 hours. Cells were then harvested (undifferen4ated) or treated with 1.3 mM calcium (differen4ated) for 5 addi4onal days. Filaggrin (FLG) mRNA was measured by RT-PCR and normalized to ac4n.
Supplemental Fig. 2 Space filling model. Sphingomyelin (which binds alpha toxin) can be cleaved by sphingomyelinase to generate ceramide (which cannot bind alpha toxin) and free phosphocholine.
Supplemental 3 ADAM10 expression is unchanged by Th2 cytokines (A) Kera4nocytes were differen4ated for 5 days in the presence or absence of IL-4/13 (n = 3). ADAM10 mRNA was measured by RT-PCR and normalized to ac4n. (B) Protein levels were measured by Western blot.
ACKNOWLEDGEMENTS
The authors thank Cliff Hall and Joanne Streib for excellent technical support. We would also like to thank Dr. Dennis Voelker for help in development of the lipid analysis assays. We thank Shih-Yun Lyman for her assistance in the preparation of this manuscript and the authors wish to acknowledge The Edelstein Family Foundation for their generous support of this work. This research was supported by NIH grants R01 AR41256 and The Atopic Dermatitis Research Network (NIH/NIAID contract NIH/NIAID HHSN272201000020C). This research was also supported in part by Colorado Clinical and Translational Sciences Institute (CCTSI), and in part by Colorado Grant UL1RR025780 from NCRR/NIH and UL1 TR000154 from NIH/NCATS.
Abbreviations
- AD
atopic dermatitis
- ADAM10
A Disintegrin And Metalloproteinase domain-containing protein 10
- ASMase
Acid Sphingomyelinase
- FLG
Filaggrin
- IFN-γ
Interferon gamma
- IL
interleukin
- LDH
Lactate dehydrogenase
- SMase
Sphingomyelinase
- STAT6
Signal Transducer and Activator of Transcription 6
- Th1
T Helper type 1
- Th2
T Helper type 2
- TLC
Thin layer chromatography
Footnotes
CONFLICT OF INTEREST The authors state no conflict of interest.
REFERENCES
- Albanesi C, Fairchild HR, Madonna S, et al. IL-4 and IL-13 negatively regulate TNF-alpha- and IFN-gamma-induced beta-defensin expression through STAT-6, suppressor of cytokine signaling (SOCS)-1, and SOCS-3. J Immunol. 2007;179:984–92. doi: 10.4049/jimmunol.179.2.984. [DOI] [PubMed] [Google Scholar]
- Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–94. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242:233–46. doi: 10.1111/j.1600-065X.2011.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauweiler AM, Bin L, Kim BE, et al. Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal alpha-toxin-induced keratinocyte death. J Allergy Clin Immunol. 2013;131:421–7. e1–2. doi: 10.1016/j.jaci.2012.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck Wardenburg J, Bae T, Otto M, et al. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–6. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- De Benedetto A, Rafaels NM, McGirt LY, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:773–86. e1–7. doi: 10.1016/j.jaci.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A, Wertz P, Giannetti A, et al. Ceramide and cholesterol composition of the skin of patients with atopic dermatitis. Acta Derm Venereol. 1998;78:27–30. doi: 10.1080/00015559850135788. [DOI] [PubMed] [Google Scholar]
- Elias PM, Wakefield JS. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Clin Rev Allergy Immunol. 2011;41:282–95. doi: 10.1007/s12016-010-8231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittler JK, Shemer A, Suarez-Farinas M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012;130:1344–54. doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson S, Johnson-Winegar AG, Wintroub BU, et al. Lamellar body-enriched fractions from neonatal mice: preparative techniques and partial characterization. J Invest Dermatol. 1985;85:289–94. doi: 10.1111/1523-1747.ep12276826. [DOI] [PubMed] [Google Scholar]
- Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest. 1994;94:870–6. doi: 10.1172/JCI117408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano Y, Terashi H, Arakawa S, et al. Interleukin-4 suppresses the enhancement of ceramide synthesis and cutaneous permeability barrier functions induced by tumor necrosis factor-alpha and interferon-gamma in human epidermis. J Invest Dermatol. 2005;124:786–92. doi: 10.1111/j.0022-202X.2005.23651.x. [DOI] [PubMed] [Google Scholar]
- Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007;120:150–5. doi: 10.1016/j.jaci.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens M, van Smeden J, Gooris GS, et al. Lamellar lipid organization and ceramide composition in the stratum corneum of patients with atopic eczema. J Invest Dermatol. 2011;131:2136–8. doi: 10.1038/jid.2011.175. [DOI] [PubMed] [Google Scholar]
- Jensen JM, Folster-Holst R, Baranowsky A, et al. Impaired sphingomyelinase activity and epidermal differentiation in atopic dermatitis. J Invest Dermatol. 2004;122:1423–31. doi: 10.1111/j.0022-202X.2004.22621.x. [DOI] [PubMed] [Google Scholar]
- Kim BE, Bin L, Ye YM, et al. IL-25 Enhances HSV-1 Replication by Inhibiting Filaggrin Expression, and Acts Synergistically with Th2 Cytokines to Enhance HSV-1 Replication. J Invest Dermatol. 2013;133:2678–85. doi: 10.1038/jid.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BE, Leung DY, Boguniewicz M, et al. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126:332–7. doi: 10.1016/j.clim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Linardic C, Obeid L, et al. Identification of sphingomyelin turnover as an effector mechanism for the action of tumor necrosis factor alpha and gamma-interferon. Specific role in cell differentiation. J Biol Chem. 1991;266:484–9. [PubMed] [Google Scholar]
- Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama. 2007;298:1763–71. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- Kuo IH, Yoshida T, De Benedetto A, et al. The cutaneous innate immune response in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131:266–78. doi: 10.1016/j.jaci.2012.12.1563. [DOI] [PubMed] [Google Scholar]
- McAleer MA, Irvine AD. The multifunctional role of filaggrin in allergic skin disease. J Allergy Clin Immunol. 2013;131:280–91. doi: 10.1016/j.jaci.2012.12.668. [DOI] [PubMed] [Google Scholar]
- Pilgram GS, Vissers DC, van der Meulen H, et al. Aberrant lipid organization in stratum corneum of patients with atopic dermatitis and lamellar ichthyosis. J Invest Dermatol. 2001;117:710–7. doi: 10.1046/j.0022-202x.2001.01455.x. [DOI] [PubMed] [Google Scholar]
- Pillai S, Bikle DD, Hincenbergs M, et al. Biochemical and morphological characterization of growth and differentiation of normal human neonatal keratinocytes in a serum-free medium. J Cell Physiol. 1988;134:229–37. doi: 10.1002/jcp.1041340208. [DOI] [PubMed] [Google Scholar]
- Sawada E, Yoshida N, Sugiura A, et al. Th1 cytokines accentuate but Th2 cytokines attenuate ceramide production in the stratum corneum of human epidermal equivalents: an implication for the disrupted barrier mechanism in atopic dermatitis. J Dermatol Sci. 2012;68:25–35. doi: 10.1016/j.jdermsci.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Schwiering M, Brack A, Stork R, et al. Lipid and phase specificity of alpha-toxin from S. aureus. Biochim Biophys Acta. 2013;1828:1962–72. doi: 10.1016/j.bbamem.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Suarez-Farinas M, Dhingra N, Gittler J, et al. Intrinsic atopic dermatitis shows similar TH2 and higher T17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013 doi: 10.1016/j.jaci.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubler JH, Kapral FA, Mudd S. Role of Alpha-Toxin in Lesion Formation by Staphylococcus Aureus on Sutures Subcutaneously Implanted in Mice. J Bacteriol. 1963;86:51–7. doi: 10.1128/jb.86.1.51-57.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travagli J, Letourneur M, Bertoglio J, et al. STAT6 and Ets-1 form a stable complex that modulates Socs-1 expression by interleukin-4 in keratinocytes. J Biol Chem. 2004;279:35183–92. doi: 10.1074/jbc.M403223200. [DOI] [PubMed] [Google Scholar]
- Travers JB, Norris DA, Leung DY. The keratinocyte as a target for staphylococcal bacterial toxins. J Investig Dermatol Symp Proc. 2001;6:225–30. doi: 10.1046/j.0022-202x.2001.00045.x. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Murata S, Schmuth M, et al. Glucosylceramide synthesis and synthase expression protect against ceramide-induced stress. J Lipid Res. 2002;43:1293–302. [PubMed] [Google Scholar]
- Valeva A, Hellmann N, Walev I, et al. Evidence that clustered phosphocholine head groups serve as sites for binding and assembly of an oligomeric protein pore. J Biol Chem. 2006;281:26014–21. doi: 10.1074/jbc.M601960200. [DOI] [PubMed] [Google Scholar]
- Valeva A, Walev I, Pinkernell M, et al. Transmembrane beta-barrel of staphylococcal alpha-toxin forms in sensitive but not in resistant cells. Proc Natl Acad Sci U S A. 1997;94:11607–11. doi: 10.1073/pnas.94.21.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Tomita T, Yasuda T. Membrane-damaging action of staphylococcal alpha-toxin on phospholipid-cholesterol liposomes. Biochim Biophys Acta. 1987;898:257–65. doi: 10.1016/0005-2736(87)90065-4. [DOI] [PubMed] [Google Scholar]
- Wertz PW, Downing DT, Freinkel RK, et al. Sphingolipids of the stratum corneum and lamellar granules of fetal rat epidermis. J Invest Dermatol. 1984;83:193–5. doi: 10.1111/1523-1747.ep12263553. [DOI] [PubMed] [Google Scholar]
- Wichmann K, Uter W, Weiss J, et al. Isolation of alpha-toxin-producing Staphylococcus aureus from the skin of highly sensitized adult patients with severe atopic dermatitis. Br J Dermatol. 2009;161:300–5. doi: 10.1111/j.1365-2133.2009.09229.x. [DOI] [PubMed] [Google Scholar]
- Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A. 2010;107:13473–8. doi: 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1 Filaggrin levels are reduced in Th2 cytokine treated cells. Primary kera4nocytes were treated with media, IL-4/13, or IFN-γ for 24 hours. Cells were then harvested (undifferen4ated) or treated with 1.3 mM calcium (differen4ated) for 5 addi4onal days. Filaggrin (FLG) mRNA was measured by RT-PCR and normalized to ac4n.
Supplemental Fig. 2 Space filling model. Sphingomyelin (which binds alpha toxin) can be cleaved by sphingomyelinase to generate ceramide (which cannot bind alpha toxin) and free phosphocholine.
Supplemental 3 ADAM10 expression is unchanged by Th2 cytokines (A) Kera4nocytes were differen4ated for 5 days in the presence or absence of IL-4/13 (n = 3). ADAM10 mRNA was measured by RT-PCR and normalized to ac4n. (B) Protein levels were measured by Western blot.