

Abstract
Eradicating cancer stem-like cells (CSC) may be essential to fully eradicate cancer. Metabolic changes in CSC could hold a key to their targeting. Here we report that the dietary micronutrient selenium can trigger apoptosis of CSC derived from chronic or acute myelogenous leukemias when administered at supraphysiological but non-toxic doses. In leukemia CSC, selenium treatment activated ATM-p53-dependent apoptosis accompanied by increased intracellular levels of reactive oxygen species. Importantly, the same treatment did not trigger apoptosis in hematopoietic stem cells. Serial transplantation studies with BCR-ABL-expressing CSC revealed that the selenium status in mice was a key determinant of CSC survival. Selenium action relied upon the endogenous production of the cyclooxygenase-derived prostaglandins Δ12-PGJ2 and 15d-PGJ2. Accordingly, non-steroidal anti-inflammatory drugs and NADPH oxidase inhibitors abrogated the ability of selenium to trigger apoptosis in leukemia CSC. Our results reveal how selenium-dependent modulation of arachidonic acid metabolism can be directed to trigger apoptosis of primary human and murine CSC in leukemia.
Keywords: Leukemia stem cells, NSAID, selenoproteins, eicosanoids, metabolism
Introduction
Leukemia is a hierarchical disease where leukemia stem cells (LSCs) occupy the apex and give rise to bulk leukemia cells that are responsible for the pathology of the disease. However, unlike LSCs, bulk leukemia cells are unable to initiate leukemia when transplanted into secondary recipients. Because of this property, LSCs represent a challenge to current therapy in that they can actively resist chemotherapy regimens and cause relapse of the disease (1, 2). In CML, the BCR-ABL1 fusion protein generated from the 9:22 translocation is capable of generating LSCs when expressed in hematopoietic stem cells (HSCs) (3). Although current treatment using tyrosine kinase inhibitors (TKIs) can block the production of bulk leukemia cells and lead to remission of CML, these agents do not affect LSCs (1). Therefore, new therapies are needed that target LSCs to prevent relapse following traditional TKI therapy.
Cyclopentenone prostaglandins (CyPGs), Δ12-PGJ2 and 15d-PGJ2, are derived from the polyunsaturated fatty acid, arachidonic acid (ARA), via the sequential action of cyclooxygenases (COX) and PGD2 synthases (PGDS) followed by non-enzymatic conversion of PGD2 to Δ12-PGJ2 and 15d-PGJ2 (4, 5). We have recently demonstrated the ability of Δ12-PGJ2 and 15d-PGJ2 to selectively activate p53-dependent pathway of apoptosis in leukemic stem cells (LSCs), without affecting the (normal) HSCs in a murine model of CML and in an unrelated model of Friend virus-induced erythroleukemia (6). The importance of COX pathway in leukemia has been highlighted in a few epidemiological studies where increasing incidence of leukemia were associated with NSAID consumption (7, 8), which led us to study strategies to increase endogenous CyPGs for treatment of leukemia. Previous studies have shown supplementation of mice or macrophages with selenium, a micronutrient that functions through insertion into redox-active selenoproteins, led to enhanced production of CyPGs as opposed to pro-inflammatory PGE2, TXA2, or PGF2α through a process termed “eicosanoid class switching” (9, 10). Therefore, we hypothesized that supraphysiological levels of selenium may play a key role in treating leukemia through the ability of endogenous CyPGs to target LSCs. In fact, low serum selenium levels have been noted in leukemic patients (11–16). On the other hand, use of selenium-cystine (as diseleno-dialanine) in leukemic patients decreased total leukocyte count, immature leukocytes in CML and AML patients (17). Anti-leukemic effect of sodium selenite (Na2SeO3) was suggested to activate p53 to cause apoptosis (18–24). However, the mechanism by which selenium activates p53 is not well understood.
In addition to being the guardian of the genome, p53 also regulates glycolysis and aerobic respiration through oxidative phosphorylation (OXPHOS) in CSCs (25). This process is accompanied by an increase in the expression of Tigar (TPp53 inducible glycolysis and apoptosis regulator), Rm2b (Ribonucleotide-diphosphate reductase subunit RM2B), and Sco2 (Synthesis of cytochrome oxidase-2) that induce cell cycle arrest followed by ROS-dependent apoptosis of irreparable cells (26–28). However, p53 also up-regulated an antioxidant response through a sestrin-dependent Nrf-2 activation pathway (29). Thus, by establishing complex regulatory networks involving temporally segregated responses, p53 can cooperate with redox changes to impact cellular metabolism and survival involving intricate control of intracellular ROS levels resulting in diverse outcomes. In fact, the sensitivity of LSCs to agents that target aberrant antioxidant (glutathione) metabolism (30) further highlight the importance of intracellular ROS as a means of inducing apoptosis in LSCs.
Here we show that selenium-dependent modulation of the ARA pathway is central to the ablation of LSCs in-vivo. Treatment of selenium-supplemented mice with indomethacin, an NSAID, blunted the protective effects of selenium implicating endogenous COX metabolites in the activation of p53 and apoptosis of LSCs. In addition, treatment of blood samples from human AML and blast-crisis CML patients with lipid extracts from selenium-supplemented macrophages induced apoptosis in CD34+CD38−CD123+ LSCs, that was blocked by NSAIDs. The pro-apoptotic effects of selenium were, in part, related to exacerbated oxidative stress in LSCs that involved NADPH oxidases, particularly Nox1. In contrast, selenium treatment did not affect normal HSCs suggesting that LSCs are uniquely sensitive to changes in intracellular ROS. These studies suggest a new mechanism that underlies the selenium-dependent effects on the viability of LSCs, which may open new opportunities for leukemia therapy.
Materials and Methods
Differential selenium status in mice
Three-week old male BALB/c mice (n = 7/group) for polycythemic Friend virus (FVP) infection or C57BL/6 mice (for BCR-ABL+LSC transplantation; n=5–7 per group) were weaned on semi-purified diets (AIN76A; Harlan Teklad, WI) that were either selenium-deficient (<0.01 ppm Se; Se-D), selenium adequate (0.08 ppm Se as selenite; Se-A), or selenium supplemented (0.4 ppm Se as selenite; Se-S) for 8 weeks as described earlier (9). In the FVP infection model, selenium was also used in the form of an organo-Se compound, methylseleninic acid (MSA) that was previously shown to be effective in a few in-vivo cancer models (31). Diets were provided ad libitum and mice were maintained on Milli-Q™ water throughout the experimental period. Erythrocytes obtained from the retro-orbital sinus of mice, maintained on specific diets for three months, were used to test the expression of GPX1 as a surrogate marker to confirm the selenium status (data not shown). All procedures were preapproved by the Institutional Animal Use and Care Committee (IACUC) and Institutional Biosafety Committee (IBC) at The Pennsylvania State University.
Induction of Friend erythroleukemia in mice
After completion of the diet-feeding schedule, Balb/c mice were injected with FVP via the retro-orbital sinus. On day 15 post-infection, splenomegaly and changes in the hematological parameters were assessed as described earlier (32).
Expression of BCR-ABL fusion protein in HSC and induction of CML in mice
Retroviral stocks were generated by transfecting HEK293T cells with MIGR-BCR-ABL-IRESGFP or MIGR-IRESGFP (MSCV) empty plasmid (kind gift from Dr. Warren Pear, University of Pennsylvania, Philadelphia) using Fugene 6 transfection reagent (Roche). Isolation and transduction of HSCs with these viruses were performed as described earlier (3, 6). LSCs (GFP+Kit+Sca1+Lin−) were isolated from spleen and bone marrow using FACS (BD Influx Cell sorter) as described earlier (3, 6).
Serial transplantation assays
BCR-ABL+LSCs isolated from the bone marrow of a C57BL/6 CD45.1 donor (as described above) were transplanted into Se-A mice on a CD45.2 background. The LSCs (CD45.1+) isolated from the bone marrow of primary transplants were used for secondary transplants in CD45.2+ mice maintained on Se-A or Se-S diets. The LSCs sorted from the bone marrow (using flow cytometry) from Se-A or total bone marrow (unsorted) in the case of Se-S mice were used in a tertiary transplant into CD45.2+ Se-A mice. Leukocytosis and the presence of LSCs in the bone marrow and spleen were examined using flow cytometry.
Histology and TUNEL staining
Formalin-fixed spleens were paraffin embedded, sectioned, and used for H&E or TUNEL staining to examine gross anatomical changes or apoptosis, respectively, following induction of leukemia as described in Supplementary Methods.
Gene expression analyses
Quantitative RT-PCR (qPCR) with Taqman probes (from Life Technologies) was performed to examine the gene expression in sorted BCR-ABL+LSCs and HSCs as described in Supplementary Methods. Data was analyzed according to the method of Livak and Schmittgen (33) with normalization to 18S rRNA.
Inhibition of COX-dependent ARA metabolism
Balb/c or C57BL/6 mice were administered indomethacin (0.00325 % w/v), a non-selective COX inhibitor, in drinking water for two weeks prior to FV-infection or LSC (BCR-ABL+HSC) transplantation, respectively, and continued for two additional weeks. Apoptosis of LSCs in the spleen and bone marrow were examined by flow cytometry. For ex vivo experiments, splenocytes containing LSCs or MSCV+HSC transplanted Se-D mice were cultured in defined IMDM (with a basal level of 7 nM selenium) with increasing amounts of selenium (as selenite; 0–500 nM) in the presence or absence of DMSO, indomethacin (20 μM; a general COX inhibitor), HQL-79 (25 μM; H-PGDS inhibitor), CAY10371 (25 μM; mPGES-1 inhibitor) for 36 h. Apoptosis of LSCs and HSCs were examined using flow cytometry. To examine the effect of selenium-dependent eicosanoid switching in macrophages on the viability of LSCs, total lipid extracts (LE) of the culture media from RAW264.7 murine macrophages (1 × 106 cells) treated with inhibitors, including NSAIDs (ibuprofen, 20 μM; naproxen, 50 μM; and aspirin, 2.5 mM) in the presence or absence of selenium (selenite; 250–500 nM; 3 days) were isolated as described earlier (6, 9, 10). LE was added to GFP+Kit+Sca1+Lin− sorted murine CML LSCs, or to peripheral blood samples from an AML patient as well as a blast-crisis CML patient for 6 h. Apoptosis of murine CML and human LSCs (AML and CML patient-derived; CD34+CD38−CD123+) was examined using Annexin V staining by flow cytometry. Human samples were obtained from the University of Rochester, Rochester, NY. All procedures were preapproved by the IBC and Institutional Review Board (IRB) at University of Rochester, Rochester, NY. Western immunoblotting was performed with sorted murine CML LSCs or FVP spleen before and after treatments as described in Supplementary Methods.
Detection of oxidative stress in LSCs and HSCs
CD34+Kit+Sca-1+Lin−GFP+ LSCs and CD34+Kit+Sca-1+Lin−GFP+ HSCs were isolated from the spleen of LSC or HSC transplanted mice (primary donors) as described earlier. 5×105 cells were plated in 1 ml of media containing IMDM supplemented with 15 % FBS (ATCC; final selenium concentration of 7 nM) containing 1 % v/v BSA (Sigma), insulin (Sigma; 10 μg/ml), transferrin (Sigma; 200 μg/ml), L-glutamine (Cellgro; 2 mM), SHH (Peprotech; 50 ng/ml), SCF (Peprotech; 50 ng/ml), GDF15 (Peprotech; 30 ng/ml), and recombinant murine IL-3 (Peprotech; 10 ng/ml). Following incubation with compounds for 24 h at 37 °C, 1μl of CellROX® Deep Red Reagent (Invitrogen) was added to LSCs in 1 mL media to a final concentration of 50 nM CellROX® reagent and incubated for 1 h at 37 °C. Cells were washed and re-suspended in DPBS containing 2 % FBS, 1% penicillin-streptomycin (Cellgro). Oxidized CellROX® was analyzed on a BD Accuri™ C6 flow cytometer in FL-4 channel.
Effect of NADPH oxidase inhibitors and antioxidants on LSCs
Sorted CD34+Kit+Sca1+Lin− (GFP+) cells were treated with selenium (as selenite; 250 nM) in the presence of NADPH oxidase-1 inhibitors, DPI (diphenyliodonium; 100 nM) or ML-171 (2-acetylphenothiazine; 1 μM) for 24 h. Similarly, LSCs were treated with NAc (1 mM) or ebselen (2 mM) for 24 h and the apoptosis of GFP+ cells was estimated by flow cytometry with annexin V positive staining. DPI, ML-171, and ebselen were prepared in DMSO. Final concentration of DMSO was 0.1 % v/v. All data shown were corrected for vehicle-dependent effects that were minimal.
Statistical analyses
The results are expressed as biological mean ± s.e.m. and the differences between groups were analyzed using Student’s t test or one-way ANOVA followed by Tukey’s post-hoc test using GraphPad Prism for comparisons between various groups. The criterion for statistical significance was p<0.05.
Results
Selenium supplementation alleviates splenomegaly and development of leukemia
We investigated if increased levels of selenium in the diet affected the development of leukemia in a CML model. In Se-A recipients, transplantation of BCR-ABL+LSCs led to splenomegaly and leukocytosis in the peripheral blood that is characteristic of leukemia (Fig. 1A, B). The CML mice on Se-D or Se-A diets did not survive beyond 2–3 weeks post transplantation compared to MSCV-GFP+ HSC transplanted mice (data not shown). In contrast, transplantation of BCR-ABL+LSCs into mice maintained on a Se-S diet failed to cause splenomegaly or leukocytosis (Fig. 1A, B). Analysis of bone marrow and spleen showed that the Se-S group lacked detectable LSCs (GFP+Kit+Sca1+Lin−) (Fig. 1C), which was consistent with the lack of splenomegaly and leukocytosis (Fig. 1A, B). Similar results were also obtained with the FVP infection model, where mice on the Se-S diet were resistant to FVP-induced erythroleukemia; while those on SeD and Se-A diet groups succumbed to the disease (Supplementary Fig. S1). Changes in peripheral blood values of FVP infected animals such as increased HCT (data not shown), leukocytes, reticulocytes, and decreased platelet levels in Se-D and Se-A diet were consistent with the onset of leukemia (Supplementary Fig. S1). Complete blood count (CBC) analysis performed in the FVP infected mice on Se-S diet exhibited significantly reduced WBCs and reticulocytes when compared to FVP-infected Se-D and Se-A mice. This effect was observed with both selenium supplementation in the form of inorganic selenite and organic (and bioavailable) MSA. Flow cytometric analysis indicated that selenium supplementation with selenite (0.4 ppm) or MSA (3 ppm) significantly ablated FVP-LSCs in the spleen (Supplementary Fig. S1) and bone marrow (data not shown). Furthermore, histological examination of the splenic sections suggested that selenium supplementation also resulted in a complete return to normal splenic histoarchitecture in the FVP-infected group (Supplementary Fig. 2). Together, these results support the notion that supraphysiological levels of selenium specifically affect the proliferation of LSCs, but not HSCs.
Figure 1.

Selenium supplementation negatively affects leukemia development in a murine CML model. HSCs were transduced with MSCV expressing either GFP alone (control) or BCR-ABL(GFP) followed by transplantation into mice maintained on Se-A or Se-S diets. A, effect of dietary selenium on splenomegaly in CML mice. B, WBC in peripheral blood of CML mice. Range of normal levels of WBC in mice is indicated by dotted lines. Red and blue lines indicate upper and lower limits of normal levels, respectively. C, LSCs (as a % of total number of cells) in the spleen and bone marrow of mice, respectively. All data shown are mean ± s.e.m. of n= 6 per diet group. * p<0.05. D, TUNEL assay of the splenic sections (i–iv) from BCR-ABL+LSC transplanted mice on Se-D, Se-A, or Se-S diets; untransplanted Se-S splenic section used as a negative control. Representative images with TUNEL positive staining is shown. Total area of TUNEL+ staining is shown below each panel.
Supplementation of mice with selenium increases apoptosis of LSCs
To demonstrate supranutritional levels of selenium in the diet led to apoptosis of LSCs, we examined splenic sections from the three diet-groups transplanted with LSCs using TUNEL assays. Increased TUNEL positive staining in Se-S mice transplanted with BCR-ABL+LSC was seen when compared to Se-D or Se-A groups (Fig. 1D). TUNEL staining of the MSCV-GFP+ transplanted HSC controls did not show any changes between the diet groups (data not shown). Increased TUNEL staining of the splenic sections from FVP-infected Se-S mice were also seen when compared to the FVP-infected mice fed Se-D or Se-A diets (Supplementary Fig. S3). In addition, we examined the sensitivity of LSCs from Se-D spleen in an ex-vivo analysis of apoptosis in response to exogenous selenium at concentrations that reflect in-vivo diet-derived levels. Inorganic selenium (as sodium selenite) was added at concentrations varying from 0–500 nM to total splenocytes isolated from Se-D mice transplanted with BCR-ABL-GFP+LSCs or MSCV-GFP+-HSCs. A dose-dependent decrease in GFP+ cells was seen in the BCR-ABL-GFP+LSCs; while the GFP+ cells in MSCV-GFP+ HSC transplanted mice treated with identical doses of selenium did not exhibit a similar response (Fig. 2A). Interestingly, treatment of these splenocytes containing LSCs with various forms of organoselenium compounds, such as MSA, Se-Met, and p-XSC, showed differences in the ability of these selenocompounds to target LSCs (Fig. 2B). Although not as effective as inorganic selenite, MSA was relatively more effective than Se-Met and p-XSC to cause apoptosis of LSCs. Taken together, these findings demonstrate that the ability of selenium to target LSCs for apoptosis is dependent on the form and dose of selenium.
Figure 2.
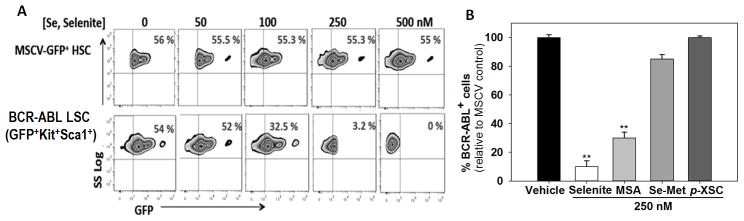
Exogenous treatment of splenocytes from CML mice with selenium causes selective apoptosis of LSCs. A, treatment of splenocytes containing HSCs (expressing MSCV-GFP) or LSCs (expressing BCR-ABL-GFP) with graded levels of selenium (in the form of sodium selenite) ex-vivo for 36 h. B, treatment of splenocytes from MSCV-GFP+HSCs or BCR-ABL+LSC transplanted mice with various forms of selenium. Splenocytes from respective mice were treated with 250 nM of sodium selenite, methylseleninic acid (MSA), seleno-L-methionine (L-SeMet), or 1,4-phenylenebis(methylene)selenocyanate (p-XSC) for 24 h and LSCs and HSCs were analyzed by flow cytometry. All data shown are mean ± s.e.m. of three independent experiments. ** p<0.01.
Serial transplantation of LSCs from Se-S mice fails to cause leukemia
Serial transplantation assays were used to further demonstrate that Se-S mice did not contain residual LSCs. In this assay, we generated LSCs using congenic CD45.1 mice (GFP+Kit+Sca1+Lin−). These LSCs from CD45.1 mice were transplanted into Se-A or Se-S CD45.2 mice and the progression of the disease was followed (Fig. 3A). Only the secondary transplants into Se-A recipients led to the development of the disease; while bone marrow transplants from Se-A donors into Se-S recipients failed to cause leukemia. Flow cytometric analysis of the spleen in these secondary recipients showed total absence of LSCs only in the Se-S group (Fig. 3B). Splenomegaly and leukocytosis was observed only in the Se-A recipients, but not in the Se-S group (Fig. 3C,D). LSCs in the bone marrow of secondary transplant Se-S recipients could not be detected, which is in contrast to those recipients that were on Se-A diets (Fig. 3E). Furthermore, we performed tertiary bone marrow transplants into Se-A recipients, where complete bone marrow was used. As expected, transplantation from Se-A (secondary) donors led to the disease, as seen in the form of LSCs in the bone marrow and spleen (Fig. 3F); while total bone marrow from Se-S donor (secondary) failed to cause leukemia in tertiary Se-A recipients (Fig. 3G,H). These studies suggest that supraphysiological levels (0.4 ppm) of selenium limit the survival of LSCs.
Figure 3.

Selenium status of mice is a critical regulator of LSC viability as seen by serial transplantation experiments. A, Schematic illustration of serial transplantation of BCR-ABL+LSCs. B, flow cytometric analysis of splenic LSCs (GFP+Kit+Sca1+Lin−) from the secondary transplant groups. C, splenomegaly, D, WBC counts on days 16 and 32 post transplantation. Range of normal levels of WBC in mice is indicated by dotted lines. a, p<0.05 compared to Se-A on day 16; b, p<0.05 compared to Se-S on day 16; c, p<0.05 compared to SeS day 32. E, GFP+ cells in the bone marrow of secondary transplant recipients. F, flow-cytometric analysis of spleen and bone marrow for the presence of LSCs. G, splenomegaly. H, WBC in peripheral blood of tertiary transplant recipients. All data shown are mean ± s.e.m. and representative of n= 5 in each group of donor and recipient mice. * p<0.05;
Endogenous CyPGs mediate the selenium-dependent eradication of LSC
Treatment of splenocytes from BCR-ABL+LSC transplanted Se-D mice with selenite ablated the GFP+Kit+Sca1+Lin− LSCs; however, co-treatment with HQL-79 or indomethacin completely blocked the pro-apoptotic effect of selenium (Fig. 4A,B). In contrast, inhibition of mPGES-1 had no effect on the selenium-dependent apoptosis of LSCs (Fig. 4A). To further validate the anti-leukemic role of endogenous eicosanoids, Se-S mice were pretreated with indomethacin followed by transplantation of LSCs. Consistent with the in-vitro results, transplantation of BCR-ABL+LSCs into these indomethacin-treated Se-S mice blocked the protective role of selenium resulting in increased WBC, lymphocytes, and neutrophils (Fig. 4C,D). Flow cytometric analysis of the bone marrow of these mice showed a substantial number of GFP+LSCs that were not observed in the BCR-ABL+LSC transplanted Se-S mice treated with the vehicle (Fig. 4E). The effect of indomethacin was, however, abrogated by intraperitoneal injection of 15d-PGJ2 (at 0.025 mg/kg/day for 1 week). 15d-PGJ2 was able to significantly decrease the LSCs in the bone marrow and spleen (data not shown). Similarly, the anti-leukemic effects of selenium were completely blunted by indomethacin in the Se-S group infected with FVP. Indomethacin-treated Se-S mice infected with FVP exhibited increased HCT, WBC, and neutrophils in the peripheral blood along with an increase in M34+Kit+Sca1+Lin− LSCs (Supplementary Fig. S4). Finally, intraperitoneal administration by 15d-PGJ2 (at 0.025 mg/kg/day for 1 week) to the indomethacin-treated FVP-infected Se-S mice rescued these mice and led to a significant decrease in LSCs in the bone marrow, while treatment of leukemic FVP-infected mice with 16,16-dimethyl-PGE2 (at 0.025 mg/kg/day for 1 week) had no effect on the viability of LSCs (Supplementary Fig. S4). These data demonstrate a key role for the COX-PGDS axis in the pro-apoptotic effects of selenium in LSCs.
Figure 4.

Essential role of the COX/H-PGDS pathway in the anti-leukemic activity of selenium. A, effect of exogenous selenium (as selenite; 250 nM) on the cKit+Sca1+GFP+ LSCs in a total splenocyte culture from BCR-ABL+LSC transplanted mice in-vitro in the presence or absence of indomethacin, HQL-79, or CAY10526. * p <0.05. B, flow cytometric analysis of sorted BCR-ABL+LSCs treated ex vivo with lipid extracts from RAW264.7 macrophages cultured in the absence or presence of selenite (250 nM), indomethacin, and HQL-79. C, splenomegaly in Se-S mice pretreated with vehicle or 0.00325 % (v/v) indomethacin in drinking water for four weeks (two weeks prior to and two weeks post) CML LSC transplantation. Data shown (mean ± s.e.m.) of at least n= 5 per group; * p <0.05. D, leukocytosis in peripheral blood of CML mice on Se-S diets upon treatment with indomethacin. Mean ± s.e.m. of n= 5 per group; * p <0.05. E, flow cytometric analysis of the spleen for LSCs (Lin−GFP+) from CML mice on Se-S diets upon treatment with indomethacin. Mean ± s.e.m. of n= 5 per group. F, apoptosis of LSCs (CD34+CD38−CD123+) from an AML patient upon treatment with lipid extracts for 24 h from macrophages cultured with various concentrations of selenite (100–500 nM) or selenite (500 nM) with naproxen, indomethacin, ibuprofen, or HQL-79. Mean ± s.e.m of triplicate assay per patient. a, p<0.05 compared to Se-deficient group; b, p<0.05 compared to Se 500 nM group without any NSAID treatment. G, blast-crisis CML patient upon treatment for 24 h with lipid extracts of culture media as described above Δ12-PGJ2 (100 nM) was used as a positive control for the apoptosis of LSCs. Results are mean ± s.e.m. of triplicate assays with each patient sample.
Lipid extracts from selenium-treated macrophages induces apoptosis in CML and AML patient samples
We next tested whether selenium treatment of macrophages could induce the production of lipid mediators that would affect LSCs in human blast-crisis CML and AML patient-derived samples. Only the LE isolated from macrophages cultured in selenium-supplemented media led to significant apoptosis of CD34+CD38−CD123+ LSCs. In contrast, LE isolated from HQL-79-treated or NSAID-treated RAW 264.7 macrophages failed to induce apoptosis of LSCs in both the AML (Fig. 4F) and blast-crisis CML samples (Fig. 4G). Furthermore, treatment of human AML and blast-crisis CML samples with Δ12-PGJ2 (100 nM) significantly reduced their colony forming ability in methylcellulose media (data not shown). Taken together, these findings demonstrate that selenium-dependent shunting of the ARA pathway leading to the production of CyPGs effectively target murine and human LSCs for apoptosis.
Selenium induces oxidative stress in LSCs
ROS production was examined in sorted BCR-ABL+LSCs and MSCV-GFP+HSCs upon treatment with selenium or lipid extracts from selenium-treated macrophages. When compared to HSCs, BCR-ABL GFP+ LSCs expressed lower levels of ROS (Fig. 5A). Addition of selenium in the form of selenite (250–500 nM) or lipid extracts from selenium-treated macrophages significantly increased ROS in LSCs. In contrast, co-treatment of selenium (500 nM) with NSAIDs (indomethacin and aspirin) or lipid extracts from NSAID (indomethacin, aspirin, ibuprofen, or naproxen)-treated selenium supplemented (500 nM) macrophages decreased ROS in the LSCs (Fig. 5B). Interestingly, addition of increasing amounts of selenite failed to increase ROS in HSCs and co-treatment of selenium (500 nM) with NSAIDs had little effect on ROS levels in HSCs (Fig. 5C). Treatment of BCR-ABL+LSCs with Δ12-PGJ2 or 15d-PGJ2 (at 25 nM) increased intracellular ROS in LSCs to levels that were seen in LSCs treated with H2O2 or daunorubicin (DNR) (Fig. 5D). LSCs treated with selenium showed a significant increase in the expression of NADPH oxidase Nox1, but not Nox2 and, Nox3 was decreased (Supplementary Fig. S5). Treatment of BCR-ABL+LSCs with NAc, an antioxidant prodrug, or NOX inhibitors such as DPI or ML171, blocked the pro-apoptotic property of selenium (Fig. 5E). Increased levels of ROS in LSCs was accompanied by dose-dependent increase in the expression of two important antioxidant genes, Sod2 and Cat; while a similar treatment failed to increase such an antioxidant response in HSCs (Fig. 5F). A similar trend was also observed in LSCs from FV-infected spleen where the expression of Cat and Sod2 were upregulated in the Se-S diet fed groups (Supplementary Fig. S6). BCR-ABL+LSCs treated with selenium exhibited increased expression of phospholipid glutathione peroxidase (Gpx4) (Supplementary Fig. S5). Treatment of BCR-ABL+ LSCs with ebselen, a glutathione peroxidase mimetic, abrogated the selenium-mediated apoptosis (Fig. 5E). Indomethacin treatment of Se-S BCR-ABL+LSCs or lipid extracts from indomethacin-treated macrophages cultured with selenium abrogated the increase in Sod2 and Cat. In addition to these antioxidant genes, we analyzed the expression of a few p53 target genes, particularly those that regulate glycolysis and oxidative phosphorylation (OXPHOS), in LSCs and HSCs following treatment with selenium. As shown in Fig. 5G, the expression of p21, Sco2, Tigar, and Rm2b were significantly increased in LSCs treated with selenite when compared to untreated LSCs or HSCs cultured with or without selenite treatment for 6 h ex-vivo. On the other hand, expression of Sesn1, Sesn2, and Gls2 were decreased in LSCs when compared to HSCs and treatment of LSCs with selenite failed to increase their expression. These studies suggest that selenium exacerbates the production of intracellular ROS, in part, by suppressing pathways that offer antioxidant protection while activating mitochondrial OXPHOS-dependent ROS production that subsequently leads to apoptosis of LSCs.
Figure 5.

Exacerbated production of ROS by supraphysiological selenium in BCR-ABL+LSCs. A, ROS levels in HSC and BCR-ABL+LSC prior to addition of selenium upon incubation with CellROX. Mean ± s.e.m of n= 3 independent assays; **** p<0.001. B, Changes in intracellular ROS in BCR-ABL+LSCs treated with various concentrations of exogenous selenium in the absence or presence of aspirin or indomethacin. LSCs were also treated with lipid extracts (LE) derived from the media supernatant of RAW264.7 macrophages cultured in the presence of various concentrations of selenium and HQL-79 or NSAIDs. All results shown are representative of n= 5–6 independent experiments per group. a, b represent P<0.001 compared to 0 nM Se and P <0.01 compared to 100 nM Se; c, P <0.01 compared to 500 nM Se; d, P <0.01 compared to 0 nM Se LE; e, P <0.05 compared to 500 nM Se LE. C, effect of addition of selenium to HSCs in the presence or absence of indomethacin, aspirin or HQL-79. Mean ± s.e.m of n=3 independent assays. D, Effects of Δ12-PGJ2 (25 nM) and 15d-PGJ2 (25 nM) on intracellular ROS in LSCs. LSCs were treated with CyPGs for 24 h; H2O2 (1 μM) for 30 min and DNR (1 μM) for 60 min. H2O2 and DNR were used as positive controls. E, effect of DPI (100 nM), ML171 (1μM, N-acetylcysteine (NAC; 1 mM), and ebselen (2 mM) on the apoptosis of sorted BCR-ABL+LSCs cultured in the presence of 250 nM selenium (as selenite) for 24 h. Cells were subjected to flow cytometric analysis with annexin V staining. *, a, p<0.01 compared to untreated (UT) or vehicle treated selenium group, respectively. F, dose-dependent effects of selenium in the presence or absence of indomethacin (INDO) on the expression of Sod2 and Cat in sorted HSCs and BCR-ABL+LSCs treated for 6 h. G, Selenium-dependent modulation of p21, Sesn1, Sesn2, Sco2, Tigar, and Rm2b in BCR-ABL+LSCs and HSCs. Cells were treated for 6 h following which mRNA was isolated and subjected to qPCR analysis. Data was normalized to 18 S rRNA and untreated HSCs were used to calculate fold changes. All data shown are mean ± s.e.m. of at least n=6 independent experiments. *, **, ***, **** represent p< 0.05, 0.01, 0.005, 0.001, respectively.
p53 pathway is activated by selenium in LSCs
Based on the increased activation of p21 and increased oxidative stress-dependent DNA damage by selenium in primary LSCs (Fig. 5G) and other cell lines (23), we examined the activation of ATM-p53 axis in LSCs. Irrespective of the leukemia model used, mice on Se-S diet exhibited a significant increase in the expression of p53 when compared to the Se-D group (Fig. 6A; Supplementary Fig. S7A). BCR-ABL GFP+LSCs treated with either lipid extracts from selenium-treated macrophage media or cultured directly in the presence of selenium (250–500 nM) led to increased expression of p53 transcript levels, which was effectively abrogated by indomethacin (Fig. 6B), HQL-79, and other NSAIDs (Fig. 6C). In contrast to the response in LSCs, treatment of HSCs with selenium or lipid extracts had no effect on p53 expression (Fig. 6B). Furthermore, pretreatment of splenocytes from BCR-ABL GFP+ LSC transplanted mice with an ATM kinase inhibitor (KU55933; 10 nM) followed by treatment with selenite (250 nM) blocked the pro-apoptotic effect of selenium (Fig. 6D). As a consequence of ATM kinase activation, phosphorylated Chk-2 and P-p53 (Ser15) were increased in sorted BCR-ABL+LSCs supplemented with selenium (100 and 500 nM) (Fig. 6E). A similar trend was also observed in the splenocytes of Se-S and Se-MSA fed groups infected with FVP (Supplementary Fig. 7B). Increase in the levels of activated caspase-3 and subsequent cleavage of PARP was seen in the selenite-treated LSCs. Treatment of AML patient cells with lipid extracts from selenium-treated macrophages upregulated p53 expression, while those from NSAID-treated macrophages blocked the effect of selenium (Fig. 6F). These results suggest that selenium-dependent metabolic shunting of the ARA pathway sensitizes murine CML and human CML and AML LSCs for apoptosis via the activation of the ATM-p53 axis.
Figure 6.

Activation of the ATM-p53 axis is critical for apoptosis in LSCs. A, western immunoblot showing p53 expression in splenocytes from BCR-ABL+LSC and MSCV-HSC transplanted mice. GPX1 expression was used to confirm selenium status. B, expression of p53 in sorted LSCs and HSCs upon treatment with exogenous selenium (as selenite; solid gray line) or lipid extracts (dotted line) in the presence or absence of indomethacin (INDO). Representative of n= 3–4 independent experiments. C, effect of other NSAIDs on selenium-induced p53 expression as seen by semi-quantitative PCR. Lanes 1–8 represent untreated, 250 nM selenite, 500 nM selenite (as selenite), LE from macrophages (500 nM selenite), LE from 500 nM selenite treated macrophages with INDO, HQL-79, naproxen, and ibuprofen, respectively. D, ATM inhibitor (KU55933; 10 nM) blocks the pro-apoptotic effect of selenite (250 nM) in BCR-ABL+LSCs. Live GFP+ cells following treatment with DMSO (vehicle) or KU55933 are shown in selenite-treated LSCs for 24 h. Mean ± s.e.m. of n= 3 independent experiment; ***p<0.005. E, densitometric analysis of immunoblotting data demonstrating the activation of Chk2-p53 axis in BCR-ABL+LSCs treated with selenite (0–500 nM) for 6 h. All data shown are mean ± s.e.m. of n=5. *, **, ***, **** represent p< 0.05, 0.01, 0.005, 0.001, respectively. F, activation of p53 in peripheral blood sample of AML patient upon treatment with selenium in the presence or absence of NSAIDs. All NSAID treatments were performed on LSCs in the presence of selenite (500 nM selenium). Mean ± s.e.m. * p<0.05.
Discussion
The anticarcinogenic activity of selenium has been demonstrated in many cancer cell lines. However, there are no reports that specifically focus on the targeting of CSCs by selenium at concentrations that are achievable through dietary supplementation without any apparent toxic effects. Here we demonstrate a novel mechanism where selenium supplementation, at supraphysiological and non-toxic doses, enhances the production of endogenous PGD2-derived CyPGs to trigger selective apoptosis of LSCs by increasing intracellular oxidative stress.
Previous studies have suggested that BCR-ABL expression was associated with a temporally regulated increase in oxidative stress by modulating upstream signaling events that had a bearing on cell fate decisions (34–37). This increase was also associated with induction of self-mutations to encode imatinib resistance. In contrast, comparison of the baseline ROS levels in LSCs and HSCs from Se-D mice indicated that the HSCs produced relatively higher ROS compared to the LSCs. The explanation for the discrepancy could be attributed to the use of a model system where the selenium levels are well defined as opposed to the use of samples from cell lines or donors with unknown selenium status (36), which could have a significant bearing on the intracellular ROS levels. In addition, BCR-ABL expressing HSCs used here are primary cells that differ from many hematopoietic cell lines previously used to suggest an association between ROS and BCR-ABL expression (34, 35). Regardless of the baseline differences in ROS, treatment of LSCs with selenite or lipid extracts exacerbated oxidative stress. While it is not clear how endogenous CyPGs increase ROS, exogenous addition of high concentrations (μM) of 15d-PGJ2 have been shown to increase intracellular iron, possibly as a by stander effect of electrophilic stress, to exacerbate ROS in CG3 human thyroid papillary cancer cells (38). This angle is worthy of further investigation to examine if a similar mechanism holds good even with low endogenous levels of CyPGs and/or increased cellular selenium. Needless to say, the mechanisms by which selenium or endogenous CyPGs activate ATM-p53 axis of apoptosis in LSCs are currently being investigated.
Our studies suggest that selenium in the form of selenoproteins and/or metabolites of selenium may play a critical role in LSC apoptosis. Increased LSC expression of Sod2, Cat, and Gpx4 to compensate for increased ROS corroborates well with the increased expression of Nox1 in LSCs treated with supra-physiological doses of selenium as reported earlier in other cancer cells (39). In addition, the ability of two NOX inhibitors, DPI and ML171, and glutathione precursor pro-drug NAc and ebselen to inhibit selenium-dependent apoptosis further suggests that LSCs are endowed with a suboptimal antioxidant response system that favors apoptosis. It also appears that excess selenium overrides these antioxidant defenses leading to increased ROS-dependent genomic instability and apoptosis through an unknown mechanism. This idea is consistent with the decrease in the expression of Sesn1, Sesn2, and Gls2 coupled to an increase in the expression of Sco2, Tigar, and Rm2b (P53r2). These observations point to the ability of selenium to favor a decrease in aerobic glycolysis in LSCs through the increased expression of Tigar, which may effectively inhibit glycolysis to activate NADPH production through the pentose phosphate pathway. Increased oxygen consumption and mitochondrial respiration as a consequence of upregulated Sco2 and Rm2b could also contribute to higher levels of ROS to effectively reverse the Warburg effect to favor apoptosis.
Our studies showed that organic forms of selenium, such as Se-Met and pXSC, were not as effective as MSA in LSC apoptosis. Inorganic selenite was even more effective than MSA in-vivo (FV model) and in ex-vivo assays with CML LSCs. This is in contrast to a mammary hyperplastic epithelial tumor model where MSA was shown to be more efficacious (31). Such model-specific differences in sensitivity could be due to the inability of these compounds to skew pathways of ARA metabolism to the same extent as selenite (9).
While our data presented here implicates macrophages as one of the key producers of PGD2-derived CyPGs, the likelihood of LSCs also contributing endogenous Δ12-PGJ2 (and 15d-PGJ2) in a selenoprotein-dependent manner cannot be ruled out. In fact, increased production of Δ12-PGJ2 was accompanied by an increase in the expression of Hpgds and prominent selenoproteins, Gpx1, Gpx4, and Txnrd1 in LSCs (Supplementary Fig. S8), which is in agreement with our previous reports in macrophages (9). Although much smaller in magnitude, Δ12-PGJ2 produced at ~6 nM/1×106 LSCs in response to selenite (500 nM) still raises the possibility of an autocrine mechanism of apoptosis of BCR-ABL+LSCs and FVP-LSCs (6). However, with the possibility of LSCs being outnumbered by monocytes, macrophages, and T-cells, which express COX-1/2 and H-PGDS (5, 40, 41) in the spleen and bone marrow microenvironment, the role of the immune component as CyPG producers in response to selenium cannot be ignored. Regardless of the origin of CyPGs, the ability of selenoproteins to increase sensitivity of only the LSCs to endogenous CyPGs is intriguing and needs to be probed further.
The abrogation of increased sensitivity of LSCs to selenium with NSAIDs and HQL-79 suggests that the endogenous prostanoids, specifically PGD2-derived CyPGs, mediate the selective eradication of LSCs. Interestingly, the role of COX in leukemia has been implicated in epidemiological studies. An increased risk of adult leukemia was associated with the use of aspirin and no-aspirin NSAIDs (7, 8). Hart et al (1984) reported that indomethacin increased mortality of mice bearing BCL1 leukemia (42). Thus, our studies suggest a confounding role for NSAIDs in clinical trials, particularly those involving supra-physiological levels of selenium as in the Selenium and Vitamin E Cancer Prevention Trial (SELECT) for prostate cancer (43, 44), where NSAID users were not excluded from the study.
In conclusion, we report the ability of selenium to selectively eradicate LSCs in two well-studied murine models of leukemia and also in AML and blast-crisis CML patient samples. Our data supports endogenous eicosanoids, such as CyPGs, to be critical in the pro-apoptotic function of selenium. The selectivity of endogenous CyPGs to target the LSCs for apoptosis highlights the importance of redox-dependent pathways as major determinants of LSC viability. While these studies suggest the use of selenium supplementation as an adjunct therapy for CML, the success of this therapy will ultimately rest on the efficient expression of selenoproteins and their ability to skew ARA metabolism to upregulate CyPG production.
Supplementary Material
Acknowledgments
Grant Support:
These studies were supported, in part, by grants (DK077152; CA162665 to KSP; DK 080040 to RFP) from the National Institutes of Health.
These studies were supported, in part, by grants (DK077152; CA0177562 to KSP; DK 080040 to RFP) from the National Institutes of Health. The authors thank the Penn State Flow Cytometry Core Facility for help with cell sorting, all past and current members of the Prabhu laboratory for their assistance, and Dr. Shantu Amin, Penn State College of Medicine, Hershey, PA, for providing p-XSC.
Footnotes
Conflict of Interest: K.S.P. and R.F.P. are inventors on a patent [Δ12-Prostaglandin J3] assigned to Penn State Research Foundation and licensed to OncOmega Pharmaceuticals, Inc. U.H.G., N.K., and S.H. are inventors on a patent [Δ12-Prostaglandin J3] assigned to Penn State Research Foundation and licensed to OncOmega Pharmaceuticals, Inc. K.S.P. and R.F. P. are scientific founders of OncOmega Pharmaceuticals, Inc. and own equity in the company. K.S.P and R.F.P’s interests were reviewed and are managed by the Penn State Office for Research Protections in accordance with their conflict of interest policies.
References
- 1.Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nature reviews Cancer. 2008;8:341–50. doi: 10.1038/nrc2368. [DOI] [PubMed] [Google Scholar]
- 2.Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: Hitting a moving target. Cancer Lett. 2013;338:15–22. doi: 10.1016/j.canlet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–92. [PubMed] [Google Scholar]
- 4.Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, et al. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc Natl Acad Sci U S A. 2007;104:20979–84. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urade Y, Hayaishi O. Prostaglandin D synthase: structure and function. Vitam Horm. 2000;58:89–120. doi: 10.1016/s0083-6729(00)58022-4. [DOI] [PubMed] [Google Scholar]
- 6.Hegde S, Kaushal N, Ravindra KC, Chiaro C, Hafer KT, Gandhi UH, et al. {Delta}12-prostaglandin J3, an omega-3 fatty acid-derived metabolite, selectively ablates leukemia stem cells in mice. Blood. 2011;118:6909–19. doi: 10.1182/blood-2010-11-317750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasum CM, Blair CK, Folsom AR, Ross JA. Non-steroidal anti-inflammatory drug use and risk of adult leukemia. Cancer Epidemiol Biomarkers Prev. 2003;12:534–7. [PubMed] [Google Scholar]
- 8.Walter RB, Milano F, Brasky TM, White E. Long-Term Use of Acetaminophen, Aspirin, and Other Nonsteroidal Anti-Inflammatory Drugs and Risk of Hematologic Malignancies: Results From the Prospective Vitamins and Lifestyle (VITAL) Study. J Clin Oncol. 2011;29:2424–31. doi: 10.1200/JCO.2011.34.6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandhi UH, Kaushal N, Ravindra KC, Hegde S, Nelson SM, Narayan V, et al. Selenoprotein-dependent up-regulation of hematopoietic prostaglandin D2 synthase in macrophages is mediated through the activation of peroxisome proliferator-activated receptor (PPAR) gamma. J Biol Chem. 2011;286:27471–82. doi: 10.1074/jbc.M111.260547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vunta H, Davis F, Palempalli UD, Bhat D, Arner RJ, Thompson JT, et al. The anti-inflammatory effects of selenium are mediated through 15-deoxy-Delta12,14-prostaglandin J2 in macrophages. J Biol Chem. 2007;282:17964–73. doi: 10.1074/jbc.M703075200. [DOI] [PubMed] [Google Scholar]
- 11.Asfour IA, El-Kholy NM, Ayoub MS, Ahmed MB, Bakarman AA. Selenium and Glutathione Peroxidase Status in Adult Egyptian Patients with Acute Myeloid Leukemia. Biol Trace Elem Res. 2009;132:85–92. doi: 10.1007/s12011-009-8401-2. [DOI] [PubMed] [Google Scholar]
- 12.Beguin Y, Bours V, Delbrouck JM, Robaye G, Roelandts I, Bury J, et al. Relationship of serum selenium levels to tumor activity in acute non-lymphocytic leukemia. Carcinogenesis. 1989;10:2089–91. doi: 10.1093/carcin/10.11.2089. [DOI] [PubMed] [Google Scholar]
- 13.Hadjibabaie M, Iravani M, Shamshiri AR, Zaker Z, Mousavi A, Alimoghaddam K, et al. The prevalence of low selenium levels in adult patients undergoing bone marrow transplantation: a brief communication. Nutr Cancer. 2008;60:837–9. doi: 10.1080/01635580802196107. [DOI] [PubMed] [Google Scholar]
- 14.Hadjibabaie M, Iravani M, Taghizadeh M, Ataie-Jafari A, Shamshiri AR, Mousavi SA, et al. Evaluation of nutritional status in patients undergoing hematopoietic SCT. Bone Marrow Transplant. 2008;42:469–73. doi: 10.1038/bmt.2008.188. [DOI] [PubMed] [Google Scholar]
- 15.Pazirandeh A, Assadi Nejad M, Vossogh P. Determination of selenium in blood serum of children with acute leukemia and effect of chemotherapy on serum selenium level. J Trace Elem Med Biol. 1999;13:242–6. doi: 10.1016/s0946-672x(99)80043-1. [DOI] [PubMed] [Google Scholar]
- 16.Zuo XL, Chen JM, Zhou X, Li XZ, Mei GY. Levels of selenium, zinc, copper, and antioxidant enzyme activity in patients with leukemia. Biol Trace Elem Res. 2006;114:41–53. doi: 10.1385/BTER:114:1:41. [DOI] [PubMed] [Google Scholar]
- 17.Weisberger AS, Suhrland LG. Studies on analogues of L-cysteine and L-cystine. III. The effect of selenium cystine on leukemia. Blood. 1956;11:19–30. [PubMed] [Google Scholar]
- 18.Thompson HJ, Meeker LD, Kokoska S. Effect of an inorganic and organic form of dietary selenium on the promotional stage of mammary carcinogenesis in the rat. Cancer Res. 1984;44:2803–6. [PubMed] [Google Scholar]
- 19.Jiang XR, Macey MG, Lin HX, Newland AC. The anti-leukaemic effects and the mechanism of sodium selenite. Leuk Res. 1992;16:347–52. doi: 10.1016/0145-2126(92)90136-u. [DOI] [PubMed] [Google Scholar]
- 20.Combs GF, Jr, Gray WP. Chemopreventive agents: selenium. Pharmacol Ther. 1998;79:179–92. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 21.Zuo L, Li J, Yang Y, Wang X, Shen T, Xu CM, et al. Sodium selenite induces apoptosis in acute promyelocytic leukemia-derived NB4 cells by a caspase-3-dependent mechanism and a redox pathway different from that of arsenic trioxide. Ann Hematol. 2004;83:751–8. doi: 10.1007/s00277-004-0920-5. [DOI] [PubMed] [Google Scholar]
- 22.Qi Y, Schoene NW, Lartey FM, Cheng WH. Selenium compounds activate ATM-dependent DNA damage response via the mismatch repair protein hMLH1 in colorectal cancer cells. J Biol Chem. 2010;285:33010–7. doi: 10.1074/jbc.M110.137406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu M, Kang MM, Schoene NW, Cheng WH. Selenium compounds activate early barriers of tumorigenesis. J Biol Chem. 2010;285:12055–62. doi: 10.1074/jbc.M109.088781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan L, Han B, Li J, Li Z, Huang F, Yang Y, et al. Exposure of human leukemia NB4 cells to increasing concentrations of selenite switches the signaling from pro-survival to pro-apoptosis. Ann Hematol. 2009;88:733–42. doi: 10.1007/s00277-008-0676-4. [DOI] [PubMed] [Google Scholar]
- 25.Puzio-Kuter AM. The Role of p53 in Metabolic Regulation. Genes & cancer. 2011;2:385–91. doi: 10.1177/1947601911409738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liebermann DA, Hoffman B, Vesely D. p53 induced growth arrest versus apoptosis and its modulation by survival cytokines. Cell Cycle. 2007;6:166–70. doi: 10.4161/cc.6.2.3789. [DOI] [PubMed] [Google Scholar]
- 27.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 28.Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, Okada Y. Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell death and differentiation. 2005;12:1390–7. doi: 10.1038/sj.cdd.4401661. [DOI] [PubMed] [Google Scholar]
- 29.Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell metabolism. 2013;17:73–84. doi: 10.1016/j.cmet.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, et al. Targeting Aberrant Glutathione Metabolism to Eradicate Human Acute Myelogenous Leukemia Cells. J Biol Chem. 2013 doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ip C, Thompson HJ, Zhu Z, Ganther HE. In vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000;60:2882–6. [PubMed] [Google Scholar]
- 32.Subramanian A, Hegde S, Porayette P, Yon M, Hankey P, Paulson RF. Friend virus utilizes the BMP4-dependent stress erythropoiesis pathway to induce erythroleukemia. J Virol. 2008;82:382–93. doi: 10.1128/JVI.02487-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–8. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 35.Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105:1717–23. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 36.Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–27. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nieborowska-Skorska M, Hoser G, Hochhaus A, Stoklosa T, Skorski T. Anti-oxidant vitamin E prevents accumulation of imatinib-resistant BCR-ABL1 kinase mutations in CML-CP xenografts in NSG mice. Leukemia. 2013 doi: 10.1038/leu.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen SY, Lu FJ, Gau RJ, Yang ML, Huang TS. 15-Deoxy-delta12,14-prostaglandin J2 induces apoptosis of a thyroid papillary cancer cell line (CG3 cells) through increasing intracellular iron and oxidative stress. Anti-cancer drugs. 2002;13:759–65. doi: 10.1097/00001813-200208000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Hail N, Jr, Cortes M, Drake EN, Spallholz JE. Cancer chemoprevention: a radical perspective. Free Radic Biol Med. 2008;45:97–110. doi: 10.1016/j.freeradbiomed.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Urade Y, Eguchi N. Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediat. 2002;68–69:375–82. doi: 10.1016/s0090-6980(02)00042-4. [DOI] [PubMed] [Google Scholar]
- 41.Urade Y, Ujihara M, Horiguchi Y, Ikai K, Hayaishi O. The major source of endogenous prostaglandin D2 production is likely antigen-presenting cells. Localization of glutathione-requiring prostaglandin D synthetase in histiocytes, dendritic, and Kupffer cells in various rat tissues. J Immunol. 1989;143:2982–9. [PubMed] [Google Scholar]
- 42.Hart DA. Increased sensitivity to indomethacin of mice bearing the BCL1-leukemia. Cancer Lett. 1984;23:145–50. doi: 10.1016/0304-3835(84)90147-2. [DOI] [PubMed] [Google Scholar]
- 43.Klein EA, Thompson IM, Lippman SM, Goodman PJ, Albanes D, Taylor PR, et al. SELECT: the selenium and vitamin E cancer prevention trial. Urol Oncol. 2003;21:59–65. doi: 10.1016/s1078-1439(02)00301-0. [DOI] [PubMed] [Google Scholar]
- 44.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.