

Introduction
Reduced delivery of glucose and oxygen during brain ischemia can cause energy failure (ATP depletion), which can in turn trigger processes leading to cell death. Glucose can support glycolytic ATP production even in the absence of oxygen, and it would seem to follow that increased glucose in the residual blood flow to ischemic brain should increase cell survival. This was tested experimentally in a rat model of is ischemia-reperfusion by Pulsinelli and colleagues several decades ago 1. Glucose had a striking effect, but in the direction opposite to what might be expected: rats in which circulating blood glucose was increased by intravenous infusion had far more extensive brain injury and mortality than normoglycemic rats. This experiment has subsequently been revisited dozens of times, using different animal species, stroke models, glucose concentrations, timing of glucose elevations, and permutations of these factors. The net result is one of the most robust in all of stroke research: hyperglycemia almost always exacerbates brain injury 2, 3. Clinical experience mirrors the animal literature in these respects, as both retrospective studies and patient registries show a striking correlation between elevated admission glucose concentrations and poor outcomes 4. Hyperglycemia is similarly associated with increased hemorrhage formation and poor outcome in patients treated with tPA 5–7.
Random blood glucose levels are roughly 4.4 to 6.1 mmol/L in both humans and rodents, with rodents being the species most commonly used for experimental stroke. The definition of hyperglycemia varies somewhat in stroke studies, ranging from 6.1 to greater than 10 mmol/L glucose. Using these definitions, hyperglycemia is found at presentation in 30–60% of all stroke patients 8, 9. Some of these patients are diabetic, but in most the acute, post-stroke hyperglycemia is a sympathomimetic stress response.
Despite the frequency of hyperglycemia in stroke, and 30-plus years of congruent experimental and clinical observations linking hyperglycemia to poor outcomes, it remains uncertain whether hyperglycemia should be corrected in acute stroke patients. This uncertainty stems in part from a concern that the hyperglycemic stress response is to some extent adaptive; that some regions of ischemic brain may require increased glucose delivery to survive ischemia. A related concern stems from the practical difficulty in preventing an overcorrection of hyperglycemia in acutely ill patients, and providers are understandably reticent to lower blood glucose when an overshoot could be catastrophic. Against this background, we aim to summarize what is now known about the mechanisms by which hyperglycemia can exacerbate ischemic brain injury in the acute stroke setting. These mechanisms are then placed in the context of relevant clinical observations and clinical trials that address this complex issue.
Dual roles of glucose in energy metabolism and oxidative stress
The central nervous system is unique in that it requires a continuous supply of glucose for normal function. All other tissues, including the heart, can readily metabolize fatty acids, amino acids, and ketone bodies in place of glucose substrate, but the blood brain barrier prevents rapid influx of these alternative substrates under most conditions 10. Glucose is metabolized almost completely to CO2 in normal brain. There can be transient local metabolism of glucose to lactate in response to local brain activity, but the lactate so produced is subsequently metabolized oxidatively, or else escapes into the venous system 11. Blood glucose concentrations are normally 4 – 6 mmol/L, essentially all of which can be extracted by brain. Normal, fully oxygenated blood carries approximately 9 mmol/L O2, of which only a portion can be extracted 10. Since 6 moles of O2 are required to oxidize each mole of glucose, this leaves a large molar excess of glucose available for anaerobic metabolism to lactate when blood delivery of oxygen falls short of demand (Fig. 1). Anaerobic glucose metabolism can rapidly generate ATP, as occurs normally in exercising muscle. However, this process also produces lactic acid, and under ischemic conditions the lactic acid accumulates and reduces tissue pH (Figs. 1, 2). This effect is magnified during hyperglycemia.
Figure 1.
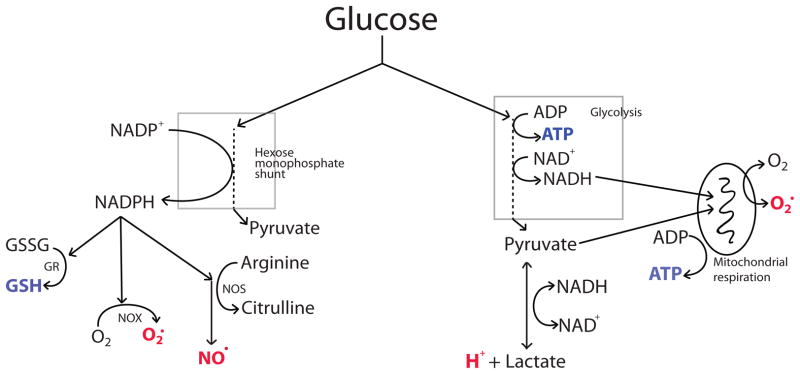
Metabolic fates of glucose relevant to ischemic injury.
Products thought to have favorable effects on stroke outcome are shown in blue font, and products thought to have deleterious effects are in red font. Glucose metabolized by the hexose monophosphate provides reducing equivalents for producing NADPH. NADPH in turn can be used by glutathione reductase (GR) to regenerate glutathione (GSH) from glutathione disulfide (GSSG), and for the production of nitric oxide (NO·) by nitric oxide synthase or superoxide (O2·) by NADPH oxidase (NOX). ATP is produced by glycolytic production of glucose to pyruvate and NADH. Pyruvate and NADH are normally oxidized to CO2 and NAD+ by mitochondrial respiration to generate additional ATP. Under ischemic conditions, oxidative metabolism cannot occur and NAD+ is instead regenerated by the formation of lactic acid. During reperfusion, damaged mitochondria may produce superoxide by donating glucose-derived reducing equivalents to molecular oxygen.
Figure 2.

Differing effects of hyperglycemia on ischemic core, penumbra, and reperfusion. Complete or near-complete ischemia in core regions with poor collateral circulation leads to glucose and oxygen depletion, accompanied by ATP depletion and mild acidosis.
Hyperglycemia supports metabolism and exacerbates the acidosis to only a minor degree in core regions, because only the glucose present at onset of ischemia is metabolized. Penumbral regions with residual blood flow through collateral circulation receive continued glucose but not oxygen delivery, due to the molar excess of glucose in arterial blood. Glycolysis fueled by the continued glucose delivery can attenuate ATP depletion, but also generates lactic acidosis in proportion to blood glucose levels. During reperfusion pH is normalized and ATP recovers where tissue is still viable, but with increased glucose delivery there is increased production of reactive oxygen species.
Independent of its function role in energy metabolism, glucose is also required for both the quenching and production of reactive oxygen species in the CNS. This stems from the fact that glucose is the exclusive substrate NADPH production through the hexose monophosphate shunt (Fig. 1). NADPH produced by this pathway is used by the enzyme glutathione reductase to convert oxidized glutathione (GSSG) back to reduced glutathione (GSH), the major thiol anti-oxidant in brain. GSH production is impaired during hypoglycemia, 12, and this may limit the capacity of brain to counter oxidative stress. It is unknown whether GSH production can be accelerated by hyperglycemia.
Somewhat paradoxically, NADPH is also used by cells to generate reactive oxygen species: NADPH used by nitric oxide synthase to generate nitric oxide (NO·), and by NADPH oxidase to generate superoxide (O2·) (Fig. 1). These reactive oxygen species are important mediators of ischemic brain injury. Glucose supply to the hexose monophosphate shunt can be rate-limiting for O2· production, and O2· production in brain can be accelerated by hyperglycemia 13. O2 · can also be generated from the electron transport chain of damaged mitochondria during ischemia-reperfusion 14; however, this process is also ultimately glucose-dependent because glucose is the source of almost all reducing equivalents passing through the mirochondrial electron transport chain in CNS tissues (Fig. 1).
Effects of blood glucose concentrations on ischemic brain injury
These multiple roles for glucose in brain lead to multiple and sometimes opposing effects of glucose on ischemic brain injury (Fig. 1). On one hand, low blood glucose concentrations (hypoglycemia) may exacerbate ischemic injury by reducing glucose delivery and hastening energy failure, particularly when oxygen supply is compromised or mitochondria are damaged. Hypoglycemia does not ordinarily cause neuronal death unless blood levels fall very low, below 1 mmol/L, but given the increased reliance on ischemic tissue for anaerobic energy production; it likely that much smaller reductions in blood glucose may exacerbate injury in ischemic brain. However, the only published study directly addressing this issue examined retinal rather than brain pathology 15.
On the other hand, elevated blood glucose concentrations can exacerbate cell injury by multiple mechanisms. Acidosis has long been recognized as one such mechanism, but even acidosis has complex and competing effects on ischemic cell survival. Normal brain pH is approximately 7.2. During ischemia, anaerobic metabolism of glucose to lactic acid can reduce this to roughly 6.6 under normoglycemic conditions, and below 6.0 under hyperglycemic conditions 16. Modest levels of acidosis, to roughly pH 6.7, inhibit the production of superoxide by NADPH oxidase and are robustly neuroprotective in ischemia 17, 18. More severe reductions in pH are thought to be exacerbate ischemic brain injury by causing protein denaturation, activation of acid-sensing ion channels, and release of ferrous iron 19, 20. It should be noted, however, that despite these potential injury mechanisms there has been no direct demonstration that acidosis has a causal role in hyperglycemic exacerbation of brain injury, as this would require evidence that the deleterious effects of hyperglycemia can be negated by normalizing brain pH.
The requisite role of glucose in the production of superoxide and nitric oxide provides an additional mechanism of glucose-mediated injury that may be particularly important during reperfusion 21. The formation of these reactive oxygen species requires oxygen and therefore cannot occur under the anaerobic conditions of severe or complete ischemia. With reperfusion there is both an influx of oxygen and a normalization of pH, which releases acid-mediated inhibition of NADPH oxidase 17. Superoxide production by NADPH oxidase during reperfusion is increased by hyperglycemia and decreased by reducing cellular glucose utilization, NADPH oxidase activity, or brain pH 13, 17, 22.
Hyperglycemia may also promote ischemic injury by other mechanisms, including enhanced glucose-sodium exchange and formation of abnormal protein glycosylation and advanced glycation products 23–25, but a definitive role for these processes in acute ischemic injury has not yet been established. Hyperglycemia is also strongly associated with an intensified post-ischemic inflammatory response, but it has been difficult to unravel whether the increased inflammation is a cause or result of increased ischemic brain injury. Interestingly, it has also been suggested that glucose-induced cortisol elevations, rather than glucose per se, exacerbate ischemic brain injury in experimental stroke 26. Cortisol can indeed potentiate neuronal death, but the magnitude of glucose-induced cortisol elevations has been disputed 24, and this explanation would not account for the effects of interventions that negate effects of hyperglycemia.
Metabolic heterogeneity in brain ischemia
The opposing effects of glucose on ischemic brain injury are further complicated by the metabolic heterogeneity of ischemic tissue. The classic concept of an ischemic core and penumbra has stood the test of time, with the “core” defined as tissue with zero or near-zero blood flow in which energy failure occurs within minutes. The term “ischemic penumbra” was originally introduced to describe regions with reduced blood flow that are electrically silent but viable if blood flow is restored 27, but the term is now applied more generally to ischemic brain regions that are metabolically compromised but potentially salvageable 28. The flow thresholds at which these conditions occurs is somewhat variable between brain regions and with duration of ischemia.
The bioenergetic state and response to hyperglycemia in these regions is quite different (Fig. 2). At one extreme, a core region of focal ischemia may have no residual blood flow at all, in which case circulating blood glucose concentrations are of little consequence. This may occur in areas with poor collateral circulation, such as lacunar strokes affecting deep white matter regions, or in the core areas of very large cortical ischemic territories. Interestingly, this is the one setting in which hyperglycemia has repeatedly been shown not to exacerbate experimental ischemic brain injury, and may in fact have a beneficial effect 2, 29. The beneficial effect may stem from the ability of glucose to fuel the very high energy demand imposed by spreading depression near ischemic core regions 30.
Where collateral circulation exists, as is more commonly the case, a “penumbral” region of reduced but non-zero blood flow is formed between non-ischemic tissue and an ischemic (or in the absence of a core). The molar excess of glucose over oxygen in arterial blood dictates that glucose delivery will continue to these ischemic regions even after all extractable oxygen removed, permitting ATP production by glucose metabolism to lactic acid. ATP consumption is slowed in these penumbral regions by cessation of electrical activity, but there remains a residual ATP demand for continued cell viability. Anaerobic metabolism of glucose to lactic acid produces only 1/16th as much ATP per molecule of glucose as normal oxidative metabolism, and consequently tissue viability in these regions can be maintained only by increasing the rate of glucose utilization to values higher than in non-ischemic tissues 30, 31.
The ischemic penumbra is unstable and dynamic, with both regional and temporal fluctuations in blood flow 28, 31. Hyperglycemia has complex effects on metabolism in the region. Where blood flow is only modestly reduced, lactic acid can be cleared and the additional ATP production fueled by augmented glucose delivery may prevent release of excitotoxic glutamate and other sequelae of energy failure 28. Conversely, where (or when) ischemia is more severe, lactic acid accumulates and pH falls in proportion to blood glucose levels 32.
Effects of hyperglycemia on vascular injury
Ischemic injury to the cerebral vasculature may be particularly dependent on circulating glucose concentrations. In animal models of ischemia-reperfusion, hyperglycemic has frequently been associated with a striking “no reflow” of blood into the microvasculature, along with evidence of increased blood-brain barrier disruption 33. It is possible that these effects on vasculature are also a manifestation of increased parenchymal injury, but evidence also exists for direct effects of hyperglycemia on cerebrovascular tone and endothelium, resulting in increased edema formation, increasing hemorrhage, and reduced microvascular reflow. Several interrelated mechanisms have been identified by which glucose can induce these changes, including increased endothelial protein kinase C activation, amplified inflammatory responses, and increased superoxide generation 22, 34–36. Hyperglycemia also increases the rate of tPA-induced hemorrhage in a model of ischemia-reperfusion, and the reversal of this effect by inhibitors of NADPH oxidase further suggest that glucose-fueled superoxide production contributes to vascular injury 22.
Correlations between experimental and clinical observations
Animal models of stroke differ in important ways from clinical stroke in that the subjects are almost young, healthy, male, and under general anesthesia. In addition, animal models of stress-induced hyperglycemia almost always employ exogenous glucose administration, which elevates insulin secretion, whereas stress-induced hyperglycemia results from an increase in circulating catecholamines, which suppress insulin secretion. These factors could in principle skew the experimental stroke literature, but despite these limitations there is a very strong agreement between experimental and clinical observations. Clinical studies show a robust association between elevated admission hyperglycemia and negative outcome measures such as infarct size, mortality, disability, and poor recovery. This association is observed in ischemic stroke with or without thrombolysis and in patients with intracerebral hemorrhage, and it remains significant in studies using logistic regression analysis to control for a number of confounding factors 4, 8, 37.
Recent clinical studies using imaging end points have further confirmed this relationship. A study using transcranial Doppler, MRI, and MRS showed that hyperglycemia is a strong predictor of infarct growth and poor outcome, even when statistically accounting for original infarct size, size of the perfusion mismatch deficit, NIHSS on admission, and time to vessel reperfusion 38. A subsequent study similarly showed that, for patients with evidence of diffusion/perfusion mismatch, admission hyperglycemia is independently associated with infarct size, progression of the ischemic penumbra to infarct, and lactate peaks in the penumbra 39. Interestingly, for those subjects with very little diffusion/perfusion mismatch (indicating a minimal penumbra), there was no relationship between hyperglycemia and lactate peaks or outcome measures 39, and in a separate study insulin treatment tended to increase rather than decrease ultimate infarct size in hyperglycemic patients with complete arterial occlusion. 40. These results are consistent with studies in animal stroke models showing little or no detrimental effect of hyperglycemia in complete ischemia 29. They also agree with the clinical observations that hyperglycemia is not detrimental and may even be beneficial in lacunar strokes 41, 42, which occur most frequently in end-arterial vascular territories with poor collateral flow. Also in parallel with results of animal studies, evaluations of hemorrhagic risk after thrombolytic therapy identify hyperglycemia to be strongly associated with both hemorrhage and poor outcomes 6, 43, particularly in the setting of vessel recanalization 44.
Nevertheless, despite well-established injury mechanisms and a general agreement between the experimental and clinical literature, there remains some evidence against a causal role of hyperglycemia in exacerbating ischemic injury. One recent study suggests that diabetes and stroke severity may be more important factors than admission blood glucose levels per se, 45, and two studies that measured serum cortisol found that these levels predicted poor stroke outcomes better than blood glucose levels 46, 47.
Prospective trials
A Cochrane review of seven trials completed before June 2010 all concluded that treatment with intravenous insulin to maintain normoglycemia after ischemic stroke provides no benefit in terms of functional improvement, ultimate functional outcome, or death, 4. Two subsequent studies arrived at similar conclusions 40, 48, but the concern remains that even the largest of these studies were underpowered. 7. The prospective studies also noted a significant increase in the number of hypoglycemic episodes in insulin-treated patients. Although glucose levels rarely fell to levels normally considered dangerous, it is possible that even modest reductions in circulating glucose concentrations can have a negative impact on energy metabolism in ischemic brain for reasons discussed above. An ongoing multi-center clinical trial with targeted enrollment of 1400 patients will assess the effects of aggressive glucose control starting within 12 hours of stroke symptom onset 7.
Conclusions
The experimental literature identifies fundamental complexities in the role of glucose and ischemic injury. Hyperglycemia can enhance glucose delivery and preserve ATP levels in ischemic tissue, but at the cost of deleterious lactic acidosis and oxidative stress. In an individual case it currently not possible to predict which of these factors will prevail, and the heterogeneous and dynamic nature of ischemic brain lesions makes it likely that both negative and positive effects may occur within a given lesion over time. The preponderance of evidence suggests that the deleterious effects of hyperglycemia will dominate in most settings, but there is clinical and experimental evidence that lesions with complete ischemia are the least likely to be exacerbated by hyperglycemia, and may even be helped.
These opposing effects of hyperglycemia on ischemic brain likewise contribute to the difficulty in determining in any given patient whether aggressive glucose management will be helpful. Future advances in spectroscopic and other imaging modalities may provide information on collateral circulation and metabolic state of penumbral tissues that may help distinguish those patients who are at high risk from ongoing hyperglycemia from those in whom reductions in blood glucose may be particularly hazardous. Perhaps the greatest promise lies in interventions targeting specific deleterious processes induced by hyperglycemia, as these could in principle obviate the need for normalizing blood glucose concentrations in acute stroke.
In contrast to this somewhat mixed effects of hyperglycemia on ischemic brain, the experimental literature indicates a univalently negative effect of hyperglycemia in the setting of reperfusion. The clinical experience likewise indicates a particularly negative effect of hyperglycemia in during reperfusion; nevertheless, hyperglycemia is not presently identified as a contraindication to thrombolytic therapy (except as a potential stroke mimic). This issue may warrant re-examination - not only because thrombolytic therapy could have a net negative effect in this patient group, but also because removing this group from treated cohorts may reveal a longer window of opportunity for treatment and/or lower hemorrhage rates in non-hyperglycemic patients.
Acknowledgments
We thank Juliet E. Swanson for editorial assistance.
Funding Sources: This review was supported by NIH grants NS041421 and NS081149 to R.A.S., and by the Department of Veterans Affairs.
Footnotes
Disclosures: None
References
- 1.Pulsinelli WA, Waldman S, Rawlinson D, Plum F. Moderate hyperglycemia augments ischemic brain damage: A neuropathologic study in the rat. Neurology. 1982;32:1239–1246. doi: 10.1212/wnl.32.11.1239. [DOI] [PubMed] [Google Scholar]
- 2.Wass CT, Lanier WL. Glucose modulation of ischemic brain injury: Review and clinical recommendations. Mayo Clin Proc. 1996;71:801–812. doi: 10.1016/S0025-6196(11)64847-7. [DOI] [PubMed] [Google Scholar]
- 3.MacDougall NJ, Muir KW. Hyperglycaemia and infarct size in animal models of middle cerebral artery occlusion: Systematic review and meta-analysis. J Cereb Blood Flow Metab. 2011;31:807–818. doi: 10.1038/jcbfm.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellolio MF, Gilmore RM, Stead LG. Insulin for glycaemic control in acute ischaemic stroke. Cochrane Database Syst Rev. 2011:CD005346. doi: 10.1002/14651858.CD005346.pub3. [DOI] [PubMed] [Google Scholar]
- 5.Demchuk AM, Morgenstern LB, Krieger DW, Linda Chi T, Hu W, Wein TH, et al. Serum glucose level and diabetes predict tissue plasminogen activator-related intracerebral hemorrhage in acute ischemic stroke. Stroke. 1999;30:34–39. doi: 10.1161/01.str.30.1.34. [DOI] [PubMed] [Google Scholar]
- 6.Derex L, Nighoghossian N. Intracerebral haemorrhage after thrombolysis for acute ischaemic stroke: An update. J Neurol Neurosurg Psychiatry. 2008;79:1093–1099. doi: 10.1136/jnnp.2007.133371. [DOI] [PubMed] [Google Scholar]
- 7.Southerland AM, Johnston KC. Considering hyperglycemia and thrombolysis in the stroke hyperglycemia insulin network effort (SHINE) trial. Ann N Y Acad Sci. 2012;1268:72–78. doi: 10.1111/j.1749-6632.2012.06731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke. 2001;32:2426–2432. doi: 10.1161/hs1001.096194. [DOI] [PubMed] [Google Scholar]
- 9.Bruno A, Durkalski VL, Hall CE, Juneja R, Barsan WG, Janis S, et al. The stroke hyperglycemia insulin network effort (SHINE) trial protocol: A randomized, blinded, efficacy trial of standard vs. Intensive hyperglycemia management in acute stroke. Int J Stroke. 2014;9:246–251. doi: 10.1111/ijs.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siesjö BK. Brain energy metabolism. New York: John Wiley & Sons; 1978. [Google Scholar]
- 11.Dienel GA. Brain lactate metabolism: The discoveries and the controversies. J Cereb Blood Flow Metab. 2012;32:1107–1138. doi: 10.1038/jcbfm.2011.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhardwaj SK, Sharma ML, Gulati G, Chhabra A, Kaushik R, Sharma P, et al. Effect of starvation and insulin-induced hypoglycemia on oxidative stress scavenger system and electron transport chain complexes from rat brain, liver, and kidney. Mol Chem Neuropathol. 1998;34:157–168. doi: 10.1007/BF02815077. [DOI] [PubMed] [Google Scholar]
- 13.Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, et al. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol. 2008;64:654–663. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, et al. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J Neurosci. 32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casson RJ, Wood JP, Osborne NN. Hypoglycaemia exacerbates ischaemic retinal injury in rats. Br J Ophthalmol. 2004;88:816–820. doi: 10.1136/bjo.2003.024661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siemkowicz E, Hansen AJ. Brain extracellular ion composition and eeg activity following 10 minutes ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–240. doi: 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- 17.Lam TI, Brennan-Minnella AM, Won SJ, Shen Y, Hefner C, Shi Y, et al. Intracellular pH reduction prevents excitotoxic and ischemic neuronal death by inhibiting NADPH oxidase. Proc Natl Acad Sci U S A. 2013;110:E4362–4368. doi: 10.1073/pnas.1313029110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon RP, Niro M, Gwinn R. Brain acidosis induced by hypercarbic ventilation attenuates focal ischemic injury. J Pharmacol Exp Ther. 1993;267:1428–1431. [PubMed] [Google Scholar]
- 19.Ying W, Han SK, Miller JW, Swanson RA. Acidosis potentiates oxidative neuronal death by multiple mechanisms. J Neurochem. 1999;73:1549–1556. doi: 10.1046/j.1471-4159.1999.0731549.x. [DOI] [PubMed] [Google Scholar]
- 20.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 21.Fabian RH, Kent TA. Hyperglycemia accentuates persistent “Functional uncoupling” of cerebral microvascular nitric oxide and superoxide following focal ischemia/reperfusion in rats. Transl Stroke Res. 2013;3:482–490. doi: 10.1007/s12975-012-0210-9. [DOI] [PubMed] [Google Scholar]
- 22.Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA. Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol. 2011;70:583–590. doi: 10.1002/ana.22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weil ZM. Ischemia-induced hyperglycemia: Consequences, neuroendocrine regulation, and a role for RAGE. Horm Behav. 2012;62:280–285. doi: 10.1016/j.yhbeh.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Martin A, Rojas S, Chamorro A, Falcon C, Bargallo N, Planas AM. Why does acute hyperglycemia worsen the outcome of transient focal cerebral ischemia? Role of corticosteroids, inflammation, and protein o-glycosylation. Stroke. 2006;37:1288–1295. doi: 10.1161/01.STR.0000217389.55009.f8. [DOI] [PubMed] [Google Scholar]
- 25.Yamazaki Y, Harada S, Tokuyama S. Post-ischemic hyperglycemia exacerbates the development of cerebral ischemic neuronal damage through the cerebral sodium-glucose transporter. Brain Res. 2012;1489:113–120. doi: 10.1016/j.brainres.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Payne RS, Tseng MT, Schurr A. The glucose paradox of cerebral ischemia: Evidence for corticosterone involvement. Brain Res. 2003;971:9–17. doi: 10.1016/s0006-8993(03)02276-5. [DOI] [PubMed] [Google Scholar]
- 27.Symon L. The relationship between CBF, evoked potentials and the clinical features in cerebral ischaemia. Acta Neurol Scand Suppl. 1980;78:175–190. [PubMed] [Google Scholar]
- 28.Obrenovitch TP. The ischaemic penumbra: Twenty years on. Cerebrovasc Brain Metab Rev. 1995;7:297–323. [PubMed] [Google Scholar]
- 29.Prado R, Ginsberg MD, Dietrich WD, Watson BD, Busto R. Hyperglycemia increases infarct size in collaterally perfused but not end-arterial vascular territories. J Cereb Blood Flow Metab. 1988;8:186–192. doi: 10.1038/jcbfm.1988.48. [DOI] [PubMed] [Google Scholar]
- 30.Nedergaard M. Mechanisms of brain damage in focal cerebral ischemia. Acta Neurol Scand. 1988;77:81–101. doi: 10.1111/j.1600-0404.1988.tb05878.x. [DOI] [PubMed] [Google Scholar]
- 31.Shiraishi K, Sharp FR, Simon RP. Sequential metabolic changes in rat brain following middle cerebral artery occlusion: A 2-deoxyglucose study. J Cereb Blood Flow Metab. 1989;9:765–773. doi: 10.1038/jcbfm.1989.110. [DOI] [PubMed] [Google Scholar]
- 32.Wagner KR, Kleinholz M, de Courten-Myers GM, Myers RE. Hyperglycemic versus normoglycemic stroke: Topography of brain metabolites, intracellular pH, and infarct size. J Cereb Blood Flow Metab. 1992;12:213–222. doi: 10.1038/jcbfm.1992.31. [DOI] [PubMed] [Google Scholar]
- 33.Huang NC, Wei J, Quast MJ. A comparison of the early development of ischemic brain damage in normoglycemic and hyperglycemic rats using magnetic resonance imaging. Exp Brain Res. 1996;109:33–42. doi: 10.1007/BF00228624. [DOI] [PubMed] [Google Scholar]
- 34.Cipolla MJ, Huang Q, Sweet JG. Inhibition of protein kinase C beta reverses increased blood-brain barrier permeability during hyperglycemic stroke and prevents edema formation in vivo. Stroke. 2011;42:3252–3257. doi: 10.1161/STROKEAHA.111.623991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martini SR, Kent TA. Hyperglycemia in acute ischemic stroke: A vascular perspective. J Cereb Blood Flow Metab. 2007;27:435–451. doi: 10.1038/sj.jcbfm.9600355. [DOI] [PubMed] [Google Scholar]
- 36.Ergul A, Li W, Elgebaly MM, Bruno A, Fagan SC. Hyperglycemia, diabetes and stroke: Focus on the cerebrovasculature. Vascul Pharmacol. 2009;51:44–49. doi: 10.1016/j.vph.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bejot Y, Aboa-Eboule C, Hervieu M, Jacquin A, Osseby GV, Rouaud O, et al. The deleterious effect of admission hyperglycemia on survival and functional outcome in patients with intracerebral hemorrhage. Stroke. 2012;43:243–245. doi: 10.1161/STROKEAHA.111.632950. [DOI] [PubMed] [Google Scholar]
- 38.Ribo M, Molina CA, Delgado P, Rubiera M, Delgado-Mederos R, Rovira A, et al. Hyperglycemia during ischemia rapidly accelerates brain damage in stroke patients treated with tPA. J Cereb Blood Flow Metab. 2007;27:1616–1622. doi: 10.1038/sj.jcbfm.9600460. [DOI] [PubMed] [Google Scholar]
- 39.Parsons MW, Barber PA, Desmond PM, Baird TA, Darby DG, Byrnes G, et al. Acute hyperglycemia adversely affects stroke outcome: A magnetic resonance imaging and spectroscopy study. Ann Neurol. 2002;52:20–28. doi: 10.1002/ana.10241. [DOI] [PubMed] [Google Scholar]
- 40.McCormick M, Hadley D, McLean JR, Macfarlane JA, Condon B, Muir KW. Randomized, controlled trial of insulin for acute poststroke hyperglycemia. Ann Neurol. 2010;67:570–578. doi: 10.1002/ana.21983. [DOI] [PubMed] [Google Scholar]
- 41.Uyttenboogaart M, Koch MW, Stewart RE, Vroomen PC, Luijckx GJ, De Keyser J. Moderate hyperglycaemia is associated with favourable outcome in acute lacunar stroke. Brain. 2007;130:1626–1630. doi: 10.1093/brain/awm087. [DOI] [PubMed] [Google Scholar]
- 42.Bruno A, Biller J, Adams HP, Jr, Clarke WR, Woolson RF, Williams LS, et al. Acute blood glucose level and outcome from ischemic stroke. Trial of ORG 10172 in acute stroke treatment (TOAST) investigators. Neurology. 1999;52:280–284. doi: 10.1212/wnl.52.2.280. [DOI] [PubMed] [Google Scholar]
- 43.Mazya M, Egido JA, Ford GA, Lees KR, Mikulik R, Toni D, et al. Predicting the risk of symptomatic intracerebral hemorrhage in ischemic stroke treated with intravenous alteplase: Safe implementation of treatments in stroke (SITS) symptomatic intracerebral hemorrhage risk score. Stroke. 2012;43:1524–1531. doi: 10.1161/STROKEAHA.111.644815. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Sabin J, Molina CA, Ribo M, Arenillas JF, Montaner J, Huertas R, et al. Impact of admission hyperglycemia on stroke outcome after thrombolysis: Risk stratification in relation to time to reperfusion. Stroke. 2004;35:2493–2498. doi: 10.1161/01.STR.0000143728.45516.c6. [DOI] [PubMed] [Google Scholar]
- 45.Rocco A, Heuschmann PU, Schellinger PD, Kohrmann M, Diedler J, Sykora M, et al. Glycosylated hemoglobin a1 predicts risk for symptomatic hemorrhage after thrombolysis for acute stroke. Stroke. 2013;44:2134–2138. doi: 10.1161/STROKEAHA.111.675918. [DOI] [PubMed] [Google Scholar]
- 46.O’Neill PA, Davies I, Fullerton KJ, Bennett D. Stress hormone and blood glucose response following acute stroke in the elderly. Stroke. 1991;22:842–847. doi: 10.1161/01.str.22.7.842. [DOI] [PubMed] [Google Scholar]
- 47.Tracey F, Crawford VL, Lawson JT, Buchanan KD, Stout RW. Hyperglycaemia and mortality from acute stroke. Q J Med. 1993;86:439–446. [PubMed] [Google Scholar]
- 48.Rosso C, Attal Y, Deltour S, Hevia-Montiel N, Lehericy S, Crozier S, et al. Hyperglycemia and the fate of apparent diffusion coefficient-defined ischemic penumbra. AJNR Am J Neuroradiol. 2011;32:852–856. doi: 10.3174/ajnr.A2407. [DOI] [PMC free article] [PubMed] [Google Scholar]