

Abstract
The profiles of serine hydrolases in human and mouse macrophages are similar yet different. For instance, human macrophages express high levels of carboxylesterase 1 (CES1), whereas mouse macrophages have minimal amounts of the orthologous murine CES1. On the other hand, both species' macrophages exhibit limited expression of the canonical 2-arachidonoylglycerol (2-AG) hydrolytic enzyme, MAGL. Our previous study showed carboxylesterase 1 (CES1) was partly responsible for the hydrolysis of 2-AG (50%) and prostaglandin glyceryl esters (PG-Gs) (80-95%) in human THP1 monocytes/macrophages. However, MAGL and other endocannabinoid hydrolases, FAAH, ABHD6 and ABHD12, did not have a role because of either limited or no expression. Thus, another enzyme was hypothesized to be responsible for the remaining 2-AG hydrolysis activity following chemical inhibition and immunodepletion of CES1 (previous study) or CES1 gene knockdown (this study). Here we identified two candidate serine hydrolases in THP1 cell lysates by activity-based protein profiling (ABPP)–MudPIT and western blotting: cathepsin G and palmitoyl protein thioesterase 1 (PPT1). Both proteins exhibited similar electrophoretic properties to a serine hydrolase in THP1 cells detected by gel-based ABPP at 31-32 kDa; however, only PPT1 exhibited lipolytic activity and hydrolyzed 2-AG in vitro. Interestingly, PPT1 was highly expressed in THP1 cells but was significantly less reactive than cathepsin G toward the activity-based probe, fluorophosphonate-biotin. KIAA1363, another serine hydrolase, was also identified in THP1 cells but did not have significant lipolytic activity. On the basis of chemoproteomic profiling, immunodepletion studies and chemical inhibitor profiles, we estimated that PPT1 contributed 32-40% of 2-AG hydrolysis activity in the THP1 cell line. In addition, pure recombinant PPT1 catalyzed the hydrolysis of 2-AG, PGE2-G and PGF2α-G, although the catalytic efficiency of 2-AG hydrolysis by PPT1 was ∼10-fold lower than CES1's. PPT1 was also insensitive to several chemical inhibitors that potently inhibit CES1, such as organophosphate poisons and JZL184. This is the first report to document the expression of PPT1 in a human monocyte/macrophage cell line and to show PPT1 can hydrolyze the natural substrates 2-AG and PG-Gs. These findings suggest that PPT1 may participate in endocannabinoid metabolism within specific cellular contexts, and highlights the functional redundancy often exhibited by enzymes involved in lipid metabolism.
Keywords: Palmitoyl protein thioesterase 1, carboxylesterase 1, endocannabinoid, 2-arachidonoylglycerol, THP1 monocytes/macrophages, hexadecylsulfonyl fluoride, paraoxon
Introduction
The endogenous cannabinoid system is comprised of several components including two distinct G-protein coupled receptors (CB1 and CB2), the endogenous arachidonoyl-containing ligands 2-arachidonoylglycerol (2-AG) and anandamide (AEA), and enzymes responsible for 2-AG and AEA biosynthesis and inactivation 1. The CB1 receptor is expressed primarily in the central nervous system (CNS) and to a more limited extent in peripheral tissues and immune cells. On the other hand, CB2 is mainly expressed peripherally, particularly on immune cells such as macrophages with lower quantities found in brain 2, 3. Therefore, activation of CB1 and CB2 receptors by both endogenous and exogenous cannabinoids has several well-characterized central and peripheral effects 3. The two widely studied endocannabinoids, 2-AG and AEA, are produced on demand, act locally in a paracrine and autocrine manner, and their physiological actions are terminated by hydrolytic enzymes that reduce their local concentration. The balance between biosynthesis and degradation of endocannabinoids is often altered during pathological conditions, and an imbalance can modulate the actions of these bioactive lipids 1, 4. Therefore, the local concentration of 2-AG in various niches may be an important determinant of disease development. For example, endocannabinoids released from endothelial cells, macrophages and platelets play a critical role during atherogenesis and can partially contribute to its clinical manifestations 5.
In cells 2-AG is synthesized from arachidonoyl-containing membrane lipids by a sn-1-specific diacylglycerol lipase (DAGLα or β). Following production in the CNS, 2-AG is primarily degraded (∼80%) by monoacylglycerol lipase (MAGL) 1, although α/β hydrolases ABHD6 and ABHD12 also make contributions 6-8, 9. Though much is known about 2-AG-mediated retrograde neurotransmission and how 2-AG is metabolized in the brain, far less is known about the physiological actions of 2-AG in peripheral tissues, such as macrophages, and its catabolism. We previously showed that in certain cellular contexts 2-AG can be degraded by carboxylesterase 1 (CES1) 10. CES1 is a well-known xenobiotic metabolizing enzyme expressed at high levels in human liver 11. However, it is less well appreciated that CES1 is abundantly found in human macrophages 12, 13 but not in murine macrophages 13. In human macrophages CES1 contributes to the liberation of free cholesterol from neutral lipid droplets, thus initiating the mobilization of cholesterol from macrophage foam cells for reverse cholesterol transport 12, 14. In addition to 2-AG, CES1 can also hydrolyze prostaglandin glyceryl esters (PG-Gs) 10, which are cyclooxygenase-derived oxygenated products of 2-AG 15, 16. CES1 accounted for 40-50% and 80-95% of the 2-AG and PG-G hydrolysis activity in the THP1 macrophage cell line, respectively. Furthermore, the catalytic efficiency of 2-AG hydrolysis by CES1 was similar to MAGL's. However, on the basis of western blotting, MAGL was expressed in very low amounts in the THP1 cell line 10 and was undetectable by activity-based protein profiling (ABPP)-MudPIT (vide infra). It is noteworthy that bona fide 2-AG hydrolysis enzymes FAAH, ABHD6 and ABHD12 were also undetectable in THP1 cells by either western blotting or gel-based ABPP assay10. Furthermore, the selective ABHD6 inhibitor WWL7017 did not block the 2-AG hydrolysis activity of THP1 cell lysates 10. These findings suggested that the remaining 2-AG hydrolysis activity in the cell line was not due to MAGL, FAAH, ABHD6 or ABHD12, which led us to examine other candidates.
When serine hydrolases in THP1 cells were labeled by the chemoproteomic probe fluorophosphonate-biotin (FP-biotin)18 and separated by SDS-PAGE it was found that CES1 and an uncharacterized protein (doublet at Mr ∼31-32 kDa) were the major hydrolases detected by avidin-horseradish peroxidase blotting 10, 19. We initially considered the possibility that the uncharacterized enzyme was MAGL; however, two lines of evidence argued against this10: (i) the protein was not immunoreactive toward a human MAGL antibody, and (ii) its catalytic activity was not inhibited by the potent MAGL inhibitor, JZL184, as judged by competitive gel-based ABPP in which inhibitors are evaluated for their ability to impair probe labeling of target serine hydrolases 20, nor was it inhibited by the potent serine hydrolase inhibitors paraoxon and chlorpyrifos oxon, which are organophosphate poisons. Thus, we sought to identify this uncharacterized serine hydrolase. In the current study, the identities of two unknown serine hydrolases in the THP1 cell line are reported for the first time: a serine protease (cathepsin G) and serine lipase (palmitoyl protein thioesterase 1, PPT1). Because of their similar molecular masses (∼30-34 kDa), both likely contribute to the signals detected by gel-based ABPP10, 19. Furthermore, we have confirmed by gene knockdown methods that CES1 is involved in 2-AG catabolism in intact THP1 monocytes/macrophages. Importantly, we characterized some unique hydrolytic activities for PPT1 that suggest it contributes to 2-AG hydrolysis in THP1 cells. Although PPT1 is known to hydrolyze fatty acyl groups attached to cysteine residues in proteins21, we determined that PPT1 can also catalyze the hydrolysis of 2-AG and PG-Gs, and that PPT1 may collaborate with CES1 to degrade 2-AG in THP1 monocytes/macrophages.
Experimental Procedures
Chemicals, cells, and reagents
Authentic standards of 2-AG, 2-AG-d8, arachidonic acid (AA), AA-d8, prostaglandin E2 glyceryl ester (PGE2-G), prostaglandin F2α glyceryl ester (PGF2α-G), PGE2, PGF2α, and 8-iso-PGF2α-d4 were from Cayman Chemicals (Ann Arbor, MI). p-Nitrophenyl valerate (pNPV), 4-methylumbelliferyl acetate (4-MUBA), and 4-methylumbelliferyl oleate (4-MUBO) were from Sigma (St. Louis, MO). Paraoxon (PO) was a kind gift from Dr. Howard Chambers (Mississippi State University). Hexadecylsulfonylfluoride (HDSF) was purchased from Calbiochem. Trypan blue, β-mercaptoethanol, phorbol 12-myristate 13-acetate (PMA), methyl arachidonoylfluorophosphonate (MAFP), fatty-acid free bovine serum albumin (BSA), penicillin, streptomycin, puromycin, and all buffer components were purchased from Sigma. The activity-based serine hydrolase probe, fluorophosphonate-biotin (FP-biotin), was from Toronto Research Chemicals (North York, Ontario). Streptavidin-agarose beads and avidin-horseradish peroxidase were from Sigma. HPLC grade solvents were from Thermo-Fisher.
Human THP1 monocytes, HepG2 cells, RPMI-1640 medium, Dulbecco's modified Eagle's medium (DMEM), gentamicin sulfate solution (50 mg/mL), and Hank's balanced salt solution without calcium, magnesium, or phenol red were purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Fetal bovine serum (FBS) was purchased from Invitrogen (Carlsbad, CA). Primary mouse macrophages were obtained from lavage of the peritoneal cavity of adult female B6C3F1 mice using sterile-PBS containing 3% (v/v) FBS. Washed cells (PBS) were plated overnight in DMEM supplemented with 10% (v/v) FBS and antibiotics (penicillin/streptomycin) and adherent cells harvested. Primary human monocytes were commercially obtained from Astarte Biologics (Redmond, WA).
CHO-hPPT1 cells were a kind gift from Dr. Sandra Hofmann (University of Texas Southwest Medical Center, Dallas, USA). Nucleoside-deficient minimum essential medium (Gibco), dialyzed FBS (Gemini), F-12 medium (Invitrogen), and serum-free medium (Hyclone CD4CHO) were obtained for culturing CHO-hPPT1 cells. Lentiviruses containing scrambled shRNA (non-specific) and CES1 shRNA were from Santa Cruz Biotechnology.
Recombinant human CES1 was expressed in baculovirus-infected Spodoptera frugiperda cells and purified as previously described 22. Recombinant KIAA1363 was overexpressed in COS7 cells transfected with an expression vector containing KIA1363 cDNA (Origene). Anti-CES1, anti-PPT1 antibody (ab89022), and anti-β-actin antibodies were purchased from Abcam (Cambridge, MA).
Culture conditions
THP1 monocytes were grown in suspension of RPMI-1640 medium supplemented with 10% FBS, 0.05 mM β-mercaptoethanol, and 50 μg gentamicin/mL (complete growth medium) at 37°C in an atmosphere of 95% air/5% CO2. The cells were grown at a density between 0.2×106 and 1×106 cells/mL, as recommended by ATCC. THP1 monocytes were differentiated into macrophages by the addition of PMA to the culture medium (final concentration nM) for 48-72 h. The culture medium was replaced every two days with fresh PMA and growth medium.
Preparation of cell lysates
THP1 monocytes were collected by centrifugation (500 × g for 10 min, 4°C), washed with cold phosphate-buffered saline (PBS), re-suspended in ice-cold 50 mM Tris-HCl (pH 7.4) buffer, and lysed by sonication (four 15 s bursts on ice at 30% max. power). THP1 macrophage monolayers were washed with cold PBS and scraped into cold 50 mM Tris-HCl (pH 7.4) buffer and sonicated. Protein concentrations of cell lysates were determined using the BCA reagent according to the manufacturer's instructions (Thermo-Fisher).
Primary mouse macrophages were plated in DMEM medium containing antibiotics (penicillin-streptomycin) and non-adherent cells removed after 3-4 hours. Fresh medium was added and the cells cultured overnight. Adherent cells were washed with PBS and the cells were scraped into ice-cold 50 mM Tris-HCl (pH 7.4) buffer and sonicated as above.
Protease inhibitors and detergents were typically avoided when the hydrolytic activity of cell lysates was determined. In some cases, the cells were lysed in cold RIPA buffer containing protease inhibitors (Promega, catalog number G6521) for subsequent immunoblot analysis.
Identification of serine hydrolases: On-bead digestion of serine hydrolases (ABPP– MUDPIT)
THP1 monocyte lysate (2 mg/mL protein in 50 mM Tris-HCl, pH 7.4) was incubated with the activity-based probe FP-biotin (final concentration 8 μM) for 1 h at room temperature, followed by removal of excess FP-biotin as previously described 14. This is termed the native sample. To control for non-specific/non-catalytic labeling of proteins by FP-biotin, a separate, equivalent amount of lysate protein was heated for 5 min (90°C) to denature proteins prior to addition of FP-biotin. This is termed the heated sample. After removal of excess FP-biotin, biotinylated proteins were captured by addition of washed streptavidin beads (150 μL), followed by incubation on a rotator (room temperature, 3 h). The beads were subsequently washed with 5 ml of 0.2% (w/v) SDS in PBS once, 5 ml of PBS three times and 5 ml of distilled water three times. After transferring the beads to a microfuge tube and removing the supernatant, the captured proteins were on-bead digested with trypsin according to standard protocols 23 and the tryptic peptides were desalted and analyzed by LTQ LC-MS/MS. Peptides were separated on a 75-μm i.d. × 15 cm reverse phase C18 column (Thermo) controlled by an Ultimate 3000 nanoflow HPLC (Dionex) and eluted using a 55-min gradient from 2%-55% solvent B (99.9% acetonitrile, 0.1% formic acid) at a flow rate of 0.3 μl/min, and further introduced into an LTQ-OrbiTrap Velos mass spectrometer (Thermo Fisher). The mass spectrometer was operated in LTQ data-dependent mode, automatically switching between MS and MS/MS. Full scan MS spectra (300-2,000 amu) were analyzed in a profile mode by the LTQ analyzer. The seven most intense ions in a scan were selected for collision-induced fragmentation (CID) in the LTQ at normalized collision energy of 35% and activation time of 40 ms. The acquired data were analyzed by Proteome Discoverer 1.4 (Thermo Fisher) using SEQUEST algorithm and a human Uniprot database along with the reversed decoy database. Searches were done using a precursor mass tolerance of 1.8 Da and fragment tolerance of 0.5 Da and the following dynamic modifications were included: oxidation on methionine, N-terminal acetylation, and carbamidomethylation on cysteine. The results were filtered using normalized XCorr values for different charge states 24 and were accepted as valid identifications only if the XCorr values were >1.5, >2.5, >3.75 and >4 for singly, doubly, triply and quadruply charged peptides, respectively. Moreover, the maximum delta Cn was 0.15 for peptides. Proteins with a minimum of two peptides detected using the above settings were considered for the final list of proteins. The proteins were further grouped, where PSMs with only high confidence were used, and the strict maximum parsimony principle was applied to the protein groups. Normalized spectral counts for each identified serine hydrolase were determined using the commercially available software Scaffold 4.0.7.
Identification of palmitoyl protein thioesterase 1: ABPP–In-gel digest–MUDPIT
THP1 monocyte lysate (1 mg/mL protein in 50 mM Tris-HCl, pH 7.4) was incubated with an activity-based probe (FP-biotin, final concentration 4 μM) for 1 h at room temperature followed by removal of excess FP-biotin. To control for non-specific/non-catalytic labeling of proteins by FP-biotin, a separate equivalent amount of lysate protein was heated for 5 min (95°C) to denature proteins prior to addition of FP-biotin. The biotinylated proteins were captured by addition of streptavidin beads (100 μL) and the beads washed as previously described 14. Proteins were eluted from the beads by addition of 50 μL of 2× SDS-PAGE loading buffer (reducing) and heating (95°C, 5 min). After separation of the affinity enriched proteins by 10% SDS-PAGE, the gel was stained with Coommasie blue (Biosafe, Bio-Rad) for 4 h at room temperature. The gel bands, ranging in size between 31-43 kDa, were excised with a razor blade. Proteins were in-gel digested with trypsin according to standard protocols and the resulting peptides were desalted and analyzed by LTQ LC-MS/MS as described above.
Expression of PPT1 in THP1 monocytes and macrophages and other cells
Whole-cell lysates (typically prepared in 50 mM Tris-HCl, pH 7.4) from THP1 monocyte/macrophage, primary murine macrophages, primary human monocytes, or HepG2 cells were separated by SDS-PAGE. Following electrophoretic transfer, PVDF membranes were probed with either rabbit anti-CES1 (1:4000 v/v) or mouse anti-PPT1 (1 μg/mL) in 3% non-fat milk overnight at 4°C. Membranes were washed followed by incubation with secondary antibodies conjugated to horseradish peroxidase (1:20,000 v/v). Following addition of luminol reagent, chemiluminescence was recorded using X-OMAT photographic film (Eastman Kodak Co., Rochester, NY). Increasing amounts of recombinant CES1 and PPT1 (10, 25, 50 and ng) were used on gels as calibrants for semi-quantitative immunoblots.
In addition, THP1 monocyte lysate was treated with FP-biotin and biotinylated proteins enriched using streptavidin beads, as described above. After separation of eluted proteins by SDS-PAGE and electrophoretic transfer to a PVDF membrane, the biotinylated proteins were detected using either avidin-horseradish peroxidase or anti-PPT antibodies.
Transduction of shRNA-containing lentiviral particles into THP1 monocytes
THP1 monocytes were plated into a 12-well plate (5×105 cells/well) in complete RPMI medium (RPMI 1640 supplemented with 10% FBS, 0.05 M 2-mercaptoethanol, and 0.05 mg/mL gentamicin) containing 8 μg/mL polybrene. Two wells were inoculated with control lentiviral particles (0.1 MOI, scrambled shRNA, Santa Cruz Biotechnology, sc-108080) and two wells inoculated with CES1 shRNA lentiviral particles (0.1 MOI, CES1 shRNA, Santa Cruz Biotechnology, sc-62096-V). The lentivirus-treated cells were cultured overnight and the medium was subsequently replaced with complete RPMI medium (without polybrene) and cells incubated for additional 24 h. The cells were then split 1:2 and incubated for an additional 24-48 h. To select stable clones of THP1 cells expressing shRNA, the culture medium was replaced with complete RPMI medium containing 5 μg/mL puromycin dihydrochloride. The medium was replaced with fresh puromycin-containing medium every 3-4 days for a three week period. Only puromycin-resistant cells were used for subsequent experiments. Knockdown of CES1 in the THP1 cells was verified by pNPV activity assay and CES1 immunoblot analysis. Lysates of parental THP1 cells (control) and CES1-deficient THP1 cells were used to compare 2-AG hydrolysis rates.
Intact monocytes (both control and CES1-deficient; 1.2 × cells per well) were treated with ionomycin (3 μM) in serum-free RPMI medium (2 mL) to stimulate in situ biosynthesis of 2-AG; the levels of 2-AG released into the culture medium were determined after 1 h, as described previously 10.
Preparation of recombinant human PPT1 from a CHO cell line
CHO-hPPT1 cells 25 were cultured in nucleoside-deficient minimum essential medium containing 30 μM methotrexate, 10% dialyzed FBS and units/ml penicillin, μg/ml streptomycin, and 0.25 μg/ml amphotericin B. For production of recombinant PPT1 protein, confluent adherent CHO-hPPT1 cells were split 1:2 and grown in F-12 medium (Invitrogen) supplemented with 10% regular FBS until confluent. The cells were washed with serum-free medium three times and subsequently cultured in serum-free Hyclone medium (CDM4CHO, SH30557) for 12-14 days, with medium collected (and stored 4°C) every two days and the cells were replenished with fresh medium. Typically, eight mm dishes each containing 5 mL of medium were used. The pooled media was collected by centrifugation at × g (4°C), filtered through a 0.2 μm filter unit to remove debris and floating cells, and concentrated approximately 40-fold using a stirred cell equipped with a YM-10 membrane (Millipore) at 4°C. The concentrated protein was subsequently exchanged into 50 mM Tris-HCl (pH 7.4) buffer containing 0.1 mM EDTA and 20% (v/v) glycerol by repeated rounds of concentration and dilution through an YM-3 centrifugal filter. The protein concentration was measured and aliquots were snap-frozen in liquid nitrogen and stored at -80°C. Protein purity and identity were verified by coomassie blue staining of SDS-PAGE gels and immunoblot analysis using a polyclonal mouse anti-PPT1 antibody. Using 4-MUBO as substrate (final concentration 75 μM), the specific activity for the recombinant human PPT1 protein was determined to be 336 nmol/min/mg protein, which is ∼10-fold higher than the specific activity of recombinant human CES1 using the same substrate 14. In terms of yield, ∼260 mL of conditioned serum-free medium from CHO-hPPT1 cells yielded ∼64 mg of crude PPT1 protein.
Hydrolysis of 2-AG and PG-Gs by recombinant human PPT1 or CES
Hydrolysis reactions using recombinant protein (5 μg PPT1 or 0.2 μg CES1 per reaction) were performed in 50 mM Tris-HCl (pH 7.4) buffer supplemented with 0.01% (w/v) fatty acid-free bovine serum albumin (BSA) and lipid glyceryl substrate varying from 0-μM (for PG-Gs) and 0-μM (for 2-AG) in a total reaction volume of μL. After preincubation of buffer and substrate for 5 min at 37°C, reactions were initiated by addition of recombinant enzyme or buffer alone (non-enzymatic control). Reactions were quenched after 30 min with an equal volume of cold acetonitrile containing internal standards, 8-iso-PGF2α-d4 (140 pmol for PG-G hydrolysis reaction) or AA-d8 (500 pmol for 2-AG hydrolysis reaction). Quenched reactions were placed on ice for 15 min, then centrifuged at 16,000 × g (4°C, 5 min) prior to transferring supernatants into HPLC vials. Supernatants were analyzed for the hydrolysis products PGE2, PGF2α, or AA by LC-MS/MS and quantification using the stable-isotope procedure. Kinetic parameters of the hydrolysis reactions (kcat, Km, and kcat/Km) were determined by performing nonlinear regression analysis using the Michaelis-Menten equation with Sigma Plot v. 8.02.
Residual 2-AG hydrolysis activity of recombinant human PPT1 following JZL184, PO and HDSF treatments: comparison to recombinant human CES1
Inhibition reactions were performed in 50 mM Tris-HCl (pH 7.4) buffer containing 0.01% (w/v) fatty acid-free BSA, 10 μM 2-AG, recombinant enzyme, and various amounts of inhibitor in a total volume of μL. Recombinant PPT1 enzyme (5 μg) was added to reactions containing 1 μM or 10 μM JZL184, 100 μM or μM PO, and 200 μM of HDSF. After preincubation (30 min, 37°C) of enzyme and inhibitors, hydrolytic reactions were initiated by addition of 2-AG (final concentration 10 μM). After 30 min, reactions were quenched with an equal volume of acetonitrile containing internal standard (AA-d8,500 pmol), placed on ice, and centrifuged at 16,000 × g for 5 min (4°C). Supernatants were analyzed for AA by LC-MS/MS.
Treatment of THP1 macrophages with PO and HDSF, followed by addition of 2-AG
THP1 monocytes (3 × 106 cells) were seeded into a 6-well plate in complete growth medium and differentiated into macrophages by the addition of PMA (70 nM) for 48-72 h. After cell differentiation, the culture medium was removed and the cells washed with Hank's buffer. Cells were then overlaid with serum-free culture medium containing PO (10 μM), HDSF (200 μM) or vehicle (ethanol, 0.1% v/v) and incubated for 30 min. 2-AG was then directly added to the culture medium (final concentration 10 μM) and incubation continued for another 60 min. The cells were collected and the culture medium removed for analysis. The cells were washed three times with ice-cold PBS, resuspended in 50 mM Tris-HCl (pH 7.4) buffer, and lysates prepared by sonication. PPT1 activity of the lysates was determined using either pNPV or 4-MUBO as substrate 26 to verify inhibition by PO and HDSF. The culture medium was spiked with internal standard (AA-d8, 500 pmol) and extracted with three volumes of ethyl acetate containing 0.1% acetic acid. The ethyl acetate layer was recovered and evaporated to dryness under nitrogen. The residues were redissolved in 100 μL of acetonitrile/water (1:1, v/v), filtered through a microfuge filter unit (0.22 μm), and transferred to LC vials for LC-MS/MS analysis.
2-AG hydrolysis by immunodepleted THP1 monocyte lysates
THP1 monocyte lysate (1 mg protein/mL) was solubilized in 50 mM Tris-HCl (pH 7.4) buffer containing 10% (v/v) fatty acid free BSA (mixed on a rotator for 1 h at 4°C), then incubated overnight at 4°C with preimmune IgG antibodies (non-specific control) or anti-PPT1 IgG antibodies. The mixtures were subsequently transferred to protein A-agarose beads, which had been pre-washed three times with IP wash buffer (50 mM Tris-HCl, pH 7.4 supplemented with 300 mM NaCl, 5 mM EDTA and 0.1% v/v Triton X-100), and incubated for 4 h on rotator (at 4°C). The beads were pelleted in a microcentrifuge and the resulting immunodepleted supernatants were used for immunoblot analysis and hydrolysis reactions. For 2-AG hydrolysis reactions, triplicate reactions were performed in 50 mM Tris-HCl (pH 7.4) buffer containing 2-AG (final concentration 10 μM) in a total reaction volume of 50 μL. After preincubation of buffer/substrate for 5 min at 37°C, reactions were initiated by addition of 25 μg control or immunodepleted THP1 cell lysates. Reactions were quenched after a 30 min incubation with an equal volume of acetonitrile containing internal standard AA-d8 (500 pmol) and placed on ice for 15 min. Samples were centrifuged at 16,000 × g for 5 min (4°C) to remove precipitated protein prior to transferring supernatants into HPLC vials. AA content in supernatant was determined by LC-MS/MS.
LC-MS/MS analytical procedures
Analysis of products obtained from recombinant enzyme-catalyzed reactions and THP1 cell/medium extracts was performed on a Thermo UPLC-MS/MS (Thermo Fisher Scientific, San Jose, CA). Samples (10 μL) were injected into an Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7μm) equipped with a VanGuard pre-column (2.1 × 5 mm, 1.7 μm). For arachidonic acid and prostaglandins (AA, AA-d8, PGE2, PGF2α, and 8-iso-PGF2α-d4), the mobile phases used were a blend of solvent A (0.1% v/v acetic acid in water) and solvent B (0.1% v/v acetic acid in acetonitrile) and the gradient program was 0 min (20% A, 80% B), 0.25 min (20% A, 80% B), 2.0 min (0% A, 100% B), 3.0 min (0% A, 100% B), 3.5 min (20% A, 80% B) and 5.0 min (20% A, 80% B). For neutral glyceryl ester analytes (2-AG, 2-AG-d8), the mobile phases were a blend of solvent A (2 mM ammonium acetate/0.1% acetic acid in water) and solvent B (2 mM ammonium acetate/0.1% acetic acid in methanol) and the gradient program was 0 min (95% A, 5% B), 0.5 min (95% A, 5% B), 5 min (5% A, 95% B), 6 min (5% A, 95% B), 7 min (95% A, 5% B) and 8 min (95% A, 5% B). The flow rate was 0.4 mL/min and the column eluate directed into the mass spectrometer using heated electrospray ionization in either negative or positive ion mode. Single-reaction monitoring (SRM) of analytes were as follows: AA, [M-H]− m/z 303.1>259.4; AA-d8, [M-H]− m/z 311.2>267.2; PGE2, [M-H]− m/z 351.2>271.2; PGF2α, [M-H]− m/z 353.2>193.1; 8-iso-PGF2α-d4 [M-H]− m/z 357.2>197.1; 2AG, [M+NH4]+ m/z 396.3>287.3; and 2AG-d8, [M+NH4]+ m/z 404.3>293.5. Scan times were 0.2 s per SRM and scan width was 0.01 m/z. Collision energies and tube lens voltage were optimized using autotune software for each analyte by post-column infusion of the individual compounds into a 50% A/50% B blend of mobile phase pumping at a flow rate 0.4 mL/min. Internal standards AA-d8, 8-iso-PGF2α-d4, and 2-AG-d8 were used for quantification. Calibration standards containing each analyte were routinely prepared in serum-free RPMI culture medium at four different concentration levels (1, 0.1, 0.01, and 0.001 μM). The spiked culture medium was fortified with internal standards (8-iso-PGF2α-d4 and AA-d8, 7 pmol and pmol, respectively) and extracted with ethyl acetate, as described above.
Results
Identification of cathepsin G and PPT1 in THP1 monocytes/macrophages
On the basis of chemical proteomic–LC-MS/MS and immunoblotting analyses (Fig. 1), we identified several serine hydrolases in human THP1 cells including cathepsin G (CTSG) and palmitoyl protein thioesterase 1 (PPT1). Ten catalytically active serine hydrolases were identified by ABPP-MudPIT (Fig. 1A; Supplementary Table 1). One of the 10, the serine protease cathepsin G, has a molecular mass similar to the unknown protein derived signals at ∼31-32 kDa in activity gels (Fig. 1B, left; and ref. 10). In addition, subsequent to FP-biotin treatment of THP1 cell lysate, streptavidin bead-enriched proteins were subjected to electrophoresis and coomassie stained bands indicated by a rectangle (∼30-34 kDa; Fig. 1B, right) were digested with trypsin. Several proteins were identified by LC-MS/MS analysis of the tryptic peptides using the SEQUEST search algorithm, including the serine hydrolase PPT1 (Supplementary Fig. 1). Interestingly, PPT1 was not identified by the ABPP-MudPIT approach (Fig. 1A), but only after trypsin digestion of gel bands in the 30-34 kDa region. Thus, the proteomic analyses taken as a whole suggested that PPT1 and cathepsin G were both candidate enzymes that accounted for the 31-32 kDa signals on activity gels. It is noteworthy that the validated 2-AG hydrolytic enzymes MAGL, FAAH, ABHD6 and ABHD12 were not detected by either ABPP-MudPIT or in-gel digestion and LC-MS/MS analysis of peptides.
Figure 1. Identification of serine hydrolases including PPT1 in THP1 cells.
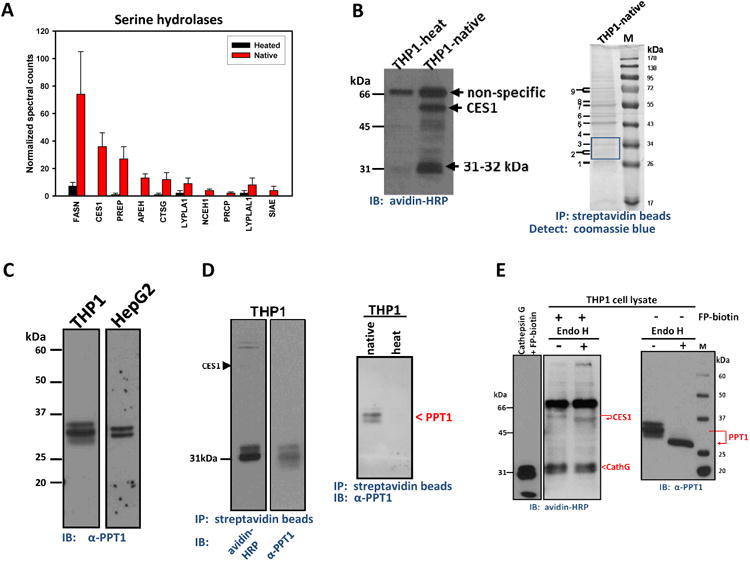
(A) ABPP-MudPIT of serine hydrolases in THP1 monocytes. The relative amounts of each serine hydrolase were semi-quantified by spectral counting of tryptic peptides. Heated and Native indicates the cell lysate was heat denatured (90°C, 5 min), or not, prior to treatment with FP-biotin (8 μM, 1 h, room temp), respectively. Data represent mean ± SEM of duplicate analyses. The full list of identified serine hydrolases (including abbreviations and molecular masses) are given in Supplementary Table 1. (B) Left, serine hydrolase activity profile of THP1 monocytes determined using FP-biotin. THP1-heat and THP1-native indicate that lysates were either heated or not heated prior to reaction with FP-biotin. Right, THP1 monocyte lysate was treated with FP-biotin followed by enrichment of labeled proteins using streptavidin beads. Captured proteins were separated by SDS-PAGE and detected with coomassie blue. M, molecular weight marker; IP, immunoprecipitation; IB, immunoblot. (C) PPT1 immunoblot of whole THP1 monocyte and HepG2 cell lysates separated by SDS-PAGE. (D) Left, FP-biotin–treated THP1 cell lysates were enriched on streptavidin beads and the captured proteins separated by SDS-PAGE. PVDF membranes were probed with either avidin-horseradish peroxidase (HRP) or anti-PPT1 antibodies. Note that three bands are detected ∼31 kDa in the anti-PPT1 blot, whereas two bands are detected in the avidin-HRP blot. This indicates that detection of PPT1 with antibodies is more sensitive than avidin blotting. Right, native versus heat indicate that FP-biotin only reacts with active PPT1 in THP1 cell lysate prior to pull-down on streptavidin beads; PPT1 was detected after SDS-PAGE by anti-PPT1 antibodies. (E) THP1 monocyte lysates pretreated with FP-biotin (left) or not (right) were subsequently treated overnight with or without Endoglycosidase H and the resulting samples separated by SDS-PAGE, followed by avidin-HRP (left) or PPT1 immunoblot (right). Also shown (far left) is pure cathepsin G treated with FP-biotin (8 μM, 1 h, room temp) prior to SDS-PAGE and avidin blotting.
On the basis of western blot analysis, PPT1 was abundantly expressed in THP1 cells (Fig. 1C). Immunoblots of whole THP1 monocyte lysates following SDS-PAGE revealed three separate immunoreactive bands that reacted with anti-PPT1 antibodies, while a doublet band pattern was observed in the HepG2 cell line. In addition, cellular PPT1 protein was covalently labeled with the chemoproteomic probe FP-biotin, affinity captured on streptavidin beads, and subsequently detected by immunoblot using either avidin-horseradish peroxidase or anti-PPT1 antibodies following separation of proteins by SDS-PAGE (Fig. 1D). Multiple protein forms detected by PPT1 antibodies suggested that PPT1 was glycosylated and its heat-sensitive reactivity toward FP-biotin was consistent with it being a member of the serine hydrolase family (Fig. 1D).
It was subsequently determined that pure recombinant human PPT1 was not very reactive toward FP-biotin. For instance, pretreatment of recombinant PPT1 with 10 μM FP-biotin inhibited just 18% of PPT1's hydrolytic activity toward the substrate 4-MUBO. Moreover, FP-biotin–treated recombinant PPT1 protein was weakly detected by gel-based ABPP (data not shown). Meanwhile, ∼30% of the 2-AG hydrolytic activity in THP1 whole-cell lysate was insensitive to pretreatment with FP-biotin (Supplementary Fig. 2). Together these findings were consistent with extensive data obtained on mouse brain serine hydrolases indicating that PPT1, although expressed in high levels in brain 27, was not strongly labeled by FP activity-based probes 28, although a small amount of PPT1 was detected in conditioned media obtained from the mouse macrophage RAW264.7 cell line28. In contrast, we found that cathepsin G was readily labeled by FP-biotin and detected by gel-based ABPP (Fig. 1E, left). This result was consistent with the large amounts of cathepsin G detected in mouse peritoneal macrophages by ABPP-MudPIT 29. Therefore, because PPT1 and cathepsin G were detected in THP1 cells by proteomic methods, it is likely that both proteins co-migrate on SDS-PAGE and that the strongly labeled cathepsin G masked the weaker PPT1 signals on activity gels (Fig. 1B, left). Supporting evidence for this includes: (i) endoglycosidase H treatment of FP-biotin–treated THP1 cell lysate did not alter the migration rate of the 31-32 kDa signals on an activity gel, whereas it did alter the migration behavior of the glycosylated proteins CES1 and PPT1 (Fig. 1E); (ii) although the sulfonyl fluoride inhibitor HDSF inhibits PPT1 (vide infra), it did not diminish the 31-32 kDa signals on activity gels (Supplementary Fig. 3A) nor did it inhibit pure cathepsin G activity at 200 μM (Supplementary Fig. 3B). Further, the potent MAGL inhibitor JZL184 and the organophosphate poison paraoxon (PO) also had no effect on this enzyme activity (Supplementary Fig. 3A). On the other hand, the general serine hydrolase inhibitor, MAFP, reduced the intensity of the 31-32 kDa signals in THP1 cell lysate (Supplementary Fig. 3C) and pure cathepsin G (Supplementary Fig. 3D). MAFP also blocked pure recombinant PPT1 hydrolysis activity toward 4-MUBO (data not shown), indicating it could inhibit both cathepsin G and PPT1.
Treatment of THP1 monocyte lysate with endoglycosidase H, which recognize and hydrolyze N-type glycans attached to asparagine residues, caused three PPT1 immunoreactive bands with Mr 31, 33 and 35 kDa to collapse into a single, faster migrating immunoreactive band of Mr ∼29 kDa (Fig. 1E). This result is consistent with the fact that human PPT1 has three asparagine-linked glycosylation sites at amino acid positions 197, 212, and 232 30. The rank order of abundance for each glycosylated PPT1 form was diglycosylated > triglycosylated > monoglycosylated.
Knockdown of CES1 expression in THP1 monocytes
Previous findings which suggested CES1 was a 2-AG hydrolytic enzyme 10 were verified by knocking down CES1 expression in THP1 cells (Fig. 2). CES1 protein expression was markedly reduced in THP1 cells using a lentiviral CES1 shRNA system, as determined by both immunoblot analysis (Fig. 2A) and esterase activity (Fig. 2B) of control and CES1-deficient cells. Interestingly, PPT1 protein expression appeared to be slightly enhanced in the CES1-deficient cells. Hydrolysis of 2-AG by cell lysates obtained from control and CES1-deficient (CES1 KD) THP1 monocytes demonstrated that CES1 knockdown reduced 2-AG hydrolysis activity by 32% relative to control (Fig. 2C). Further, treatment of CES1-deficient lysate with μM HDSF inhibited nearly all the remaining 2-AG hydrolysis activity (Fig. 2C; 97% reduction compared to control). Moreover, stimulation of living CES1-deficient THP1 monocytes with ionomycin, which stimulates biosynthesis of 2-AG, caused 2-AG levels to be elevated nearly 3-fold compared to control cells (Fig. 2D). Further in vivo relevance for CES1 in 2-AG metabolism was evident when THP1 macrophages were activated by exposure to lipopolysaccharide (to induce COX-2), followed by addition of exogenous 2-AG in the presence or absence of benzil, a CES1 inhibitor 31 (Supplementary Fig. 4A). Upon 2-AG hydrolysis, AA is oxygenated by COX-2 to yield prostaglandins in specific tissues 32, 33. Crucial to this mechanism is the regulation of 2-AG hydrolysis. Thus, we speculated that inhibition of CES1-mediated 2-AG hydrolysis in intact macrophages would partially reduce the pool of precursor AA available to COX-2. In line with this, pretreatment with benzil significantly reduced the levels of PGF2α and PGE2 synthesized by macrophages (Supplementary Fig. 4A). Inhibition of the 2-AG biosynthetic enzyme, DAGLβ, by RHC-80267 also resulted in decreased levels of prostaglandins following stimulation of cellular 2-AG biosynthesis by ionomycin (Supplementary Fig. 4B), which is consistent with the partially reduced 2-AG and prostaglandin production that was observed following DAGLβ blockade in murine peritoneal macrophages by Hsu et al.29.
Figure 2. CES1 knockdown in THP1 cells using lentiviral shRNA.

(A) Immunoblot analysis for CES1 and PPT1 in control THP1 monocytes (THP1/control) and CES1-deficient THP1 monocytes (THP1/CES1 KD). Coomassie blue detection of proteins on PVDF membrane and β-actin immunoblot verified equal protein loading (50 μg per lane). Recombinant CES1 protein was used as a molecular weight marker and positive control. pNPV hydrolytic activity (B) and 2-AG hydrolytic activity (C) of whole-cell lysates obtained from THP1 monocytes (control) and CES1-deficient THP1 monocytes (CES1 KD). Inhibition of remaining 2-AG hydrolytic activity in CES1-deficient THP1 monocytes (CES1KD+HDSF) was done by incubating lysate with 200μM HDSF for 30 min prior to adding 2-AG. (D) Intact control and CES1-deficient THP1 monocytes were treated with ionomycin (3μM), followed by determination of 2-AG levels in the culture supernatant after 60 min. pNPV hydrolysis assay of cell lysates confirmed knockdown of CES1 protein (data not shown). (E) 2-AG hydrolysis activity of pure cathepsin G and PPT1 proteins. N.E., non-enzymatic hydrolysis. Cath G, cahthepsin G. Data in each panel represent the mean ± SD of 3 independent experiments; *, p<0.05, Student's t-test. mAbs, milliabsorbance units.
To examine the possibility that cathepsin G and PPT1 also contribute to 2-AG hydrolytic activity, we used pure cathepsin G and PPT1 proteins and determined whether they hydrolyzed 2-AG in vitro. As indicated in Fig. 2E, PPT1 catalyzed the hydrolysis of 2-AG but cathepsin G did not. In light of the fact that MAGL, FAAH, ABHD6 and ABHD12 were either undetectable or expressed at very low levels in THP1 cells, we followed up on the possibility that PPT1 contributed to the 2-AG hydrolysis activity that was not accounted for by CES1.
Expression levels of PPT1 in human THP1 monocytes/macrophages and other cells
First we determined the amount of PPT1 expressed in THP1 cells in order to compare with levels of CES1. Semi-quantitative immunoblotting analysis performed on THP1 whole-cell lysates indicated that PPT1 was abundant in both THP1 monocytes and macrophages, with expression levels of 75±3 pmol/mg and 28±8 pmol/mg lysate protein, respectively (Fig. 3A). On a molar basis PPT1 was more abundant than CES1 in THP1 monocytes and macrophages; PPT1 levels were 3-fold and 1.5-fold greater than CES1, respectively. In addition, the amount of PPT1 in THP1 macrophages was estimated to be ∼34-fold greater than the canonical 2-AG hydrolytic enzyme, MAGL, based on our previously published data 10.
Figure 3. Expression levels of PPT1 in monocytes and macrophages.
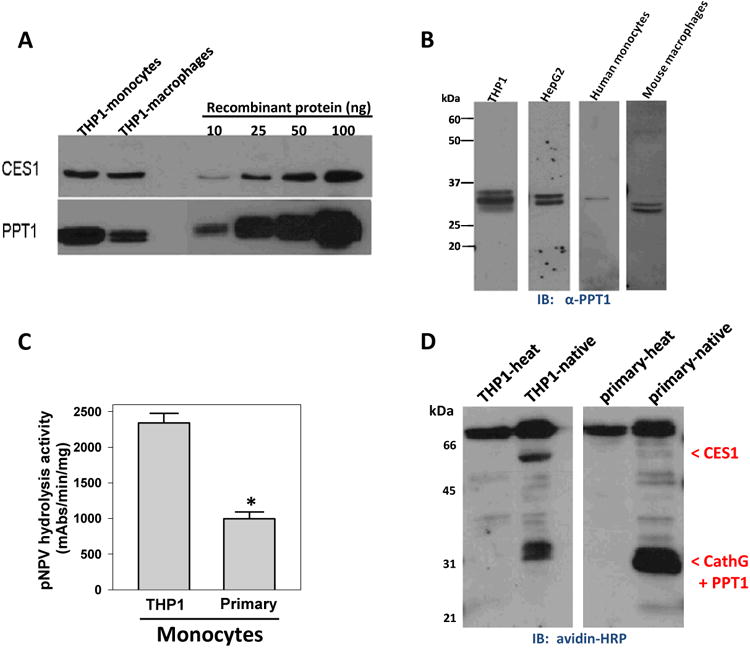
(A) Semi-quantification of PPT1 and CES1 in THP1 monocytes and macrophages by immunoblot analysis. (B) PPT1 immunoblot of cell lysate proteins obtained from THP1 monocytes, HepG2, human primary monocytes and mouse primary macrophages. HepG2 blot in Fig. 1C is reproduced here for comparative purposes only. Note, we verified that anti-PPT1 does not cross-react with pure human cathepsin G (data not shown). (C) Esterase activity of THP1 monocytes and primary human monocytes determined using pNPV. Data are mean ± SD of 3 independent experiments; *, p<0.05, Student's t-test. (D) Serine hydrolase activity profile of THP1 monocytes and primary human monocytes determined using FP-biotin. Locations of CES1, PPT1 and cathepsin G are indicated.
In addition to THP1 cells, we determined that PPT1 was expressed in HepG2, human primary monocytes and mouse peritoneal macrophages (Fig. 3B); indicating PPT1 is distributed in multiple cell types. Another notable finding was that CES1 expression was less abundant in primary human monocytes compared to THP1 cells on the basis of esterase activity (Fig. 3C), gel-based ABPP (Fig. 3D) and immunoblot analysis (data not shown). Furthermore, gel-based ABPP indicated the 31-32 kDa activity signals, which are likely caused by co-migration of cathepsin G and PPT1, were clearly observable in both primary human monocytes and THP1 cells (Fig. 3D).
Expression and isolation of recombinant human PPT
Having determined that PPT1 is abundantly expressed in THP1 cells, we next examined the biochemical activities of this serine lipase in more detail using recombinant human PPT1 protein. The coomassie-stained gel and western blot of the PPT1 protein produced by CHO-hPPT1 cells showed a single diffuse band around 34 kDa (Fig. 4A), which is consistent with the protein being glycosylated in a heterogeneous manner. On the basis of the coomassie-stained gel (Fig. 4A), the recombinant PPT1 protein preparation was estimated to be >90% pure. Furthermore, mass spectrometric analysis of tryptic peptides derived from the recombinant protein confirmed its identity was PPT1 and indicated it was not contaminated by another serine hydrolase such as MAGL (data not shown). Recombinant PPT1 could hydrolyze both small acyl ester substrates (4-MUBA) and fatty acyl ester substrates (4-MUBO) (Fig. 4B); however, it was more effective (28-fold) at hydrolyzing substrates with a fatty acyl moiety, such as the oleoyl group in 4-MUBO, compared to acetyl-containing substrates such as 4-MUBA. The kinetic parameters for 4-MUBO hydrolysis by PPT1 (assuming a molecular mass of 34 kDa) were kcat, 2.79 ± 0.05 min−1; Km, 21.8 ± 0.4 μM (mean ± range, n=2 determinations). These results demonstrated that PPT1, although termed a thioesterase, also acts as an esterase and lipase.
Figure 4. Production of recombinant PPT1 in stably overexpressing CHO cells.
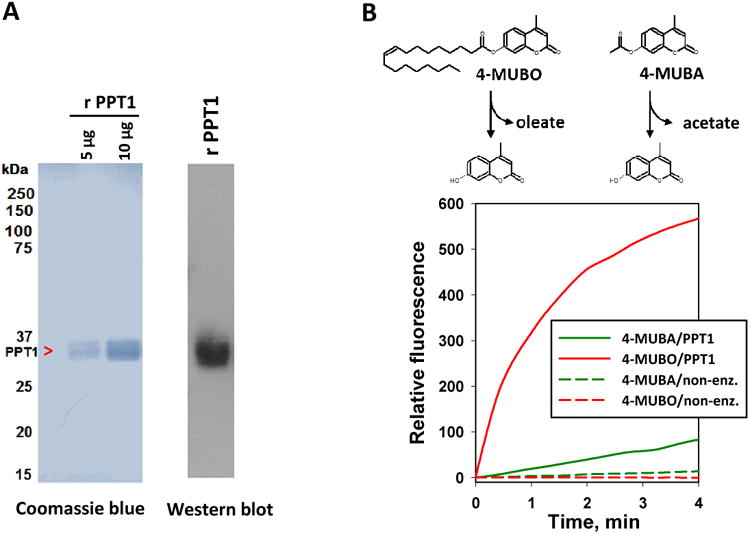
(A) Coomassie blue staining (left) and immunoblot analysis (right) of recombinant (r) PPT1 protein produced by CHO-hPPT1 cells. (B) Activity progress curves for 4-MUBO and 4-MUBA hydrolysis by recombinant PPT1. Data represent the mean fluorescence obtained from at least 3 independent reactions for each condition. The variation (SD) in the activity progress curves for each condition was <10%.
Hydrolysis of 2-AG and PG-Gs by recombinant human PPT1: Comparison to CES
Because recombinant human PPT1 could hydrolyze the ester-containing 2-AG (Fig. 2E), we next characterized the kinetics of the reaction (Fig. 5A) and compared the catalytic efficiency of PPT1 to CES1 (Fig. 5A, inset; Table 1). In addition to 2-AG, PPT1 also hydrolyzed PGE2-G and PGF2α-G to their respective free prostaglandins (Fig. 5B). The extent of 2-AG and PG-G hydrolysis by recombinant PPT1 was protein concentration-dependent (data not shown), time-dependent (2-AG hydrolysis shown in Fig. 5C), and concentration-dependent (Fig. 5 A, B). Comparison of steady-state Michaelis-Menten kinetic parameters for 2-AG hydrolysis reactions catalyzed by human PPT1 and CES1 are reported in Table 1. The turnover number (kcat) for PPT1 is 246-fold lower than for CES1, whereas the catalytic efficiency (kcat/Km) for PPT1 was only 13-fold lower than CES1's. Km values for the substrate-enzyme pairs differed by 20-fold; however, because deacylation of the acyl-enzyme intermediate is usually rate-limiting for the hydrolysis of esters (i.e., k2 ≫ k3)34, Km is a complex function of individual rate constants and can be difficult to interpret with respect to substrate affinity 34. In addition, the kcat/Km value for PPT1-catalyzed hydrolysis of 2-AG (0.093 min−1 μM−1) was lower than the comparable parameters previously reported for rat and human MAGLs (22-fold and 8-fold, respectively) 35, but similar to the catalytic efficiency of 4-MUBO hydrolysis by PPT1 (0.13 min−1 μM−1) giving confidence in its estimate. Thus, on the basis of its Michaelis constant and turnover number, the intrinsic 2-AG hydrolytic activity of PPT1 appears to be maximal at physiological concentrations of 2-AG (∼1-10 μM).
Figure 5. Rates of PPT1-catalyzed hydrolysis of 2-AG and PG-Gs.
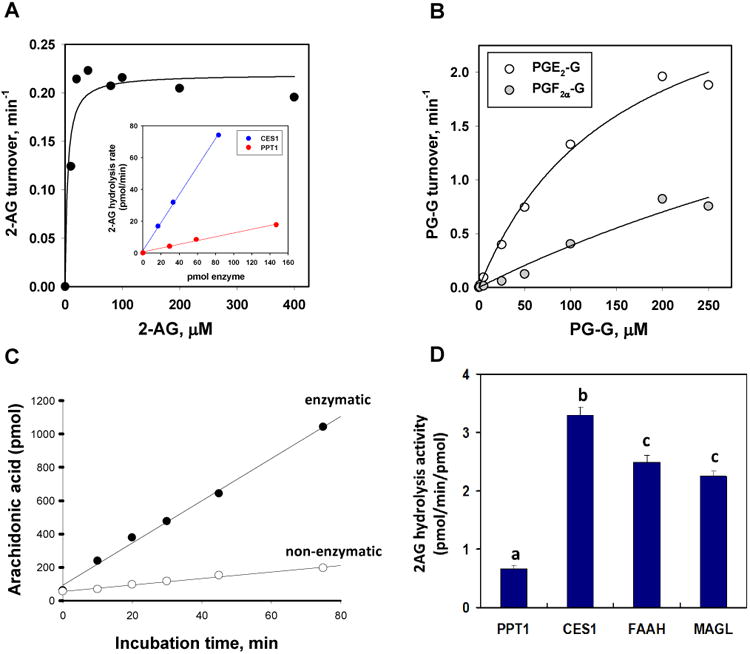
Substrate concentration versus velocity plots for recombinant PPT1-catalyzed hydrolysis of 2-AG (A) and PGF2α-G and PGE2-G (B). Data are representative of at least 3 independent experiments. Inset in A, 2-AG hydrolysis rate (final 2-AG concentration 10μM) as a function of either CES1 or PPT1 amounts. The slopes for the two lines differ by 7.4-fold. (C) Time course of AA formation when 2-AG (10μM) was incubated in the presence (enzymatic) or absence (non-enzymatic) of PPT1. (D) Comparison of specific activities for enzymes that hydrolyze 10μM 2-AG (30 min, 37°C, reaction buffer: 50 mM Tris-HCl, pH 7.4). Data represent the mean ±SD of duplicate reactions. Different lower case alphabetical letters indicate statistical differences between groups (p<0.05, one-way ANOVA and Tukey's test). Data for FAAH and MAGL are from 10.
Table 1. Steady-State Kinetic Parameters for the Hydrolysis of 2-AG and PG-Gs by Recombinant Human PPT1 and CES1a.
| PPT1 | |||
|---|---|---|---|
|
|
|||
| Substrate | kcat(min−1) | Km(μM) | kcat/Km(min−1 μM−1) |
| 2-AG | 0.24±0.02 | 2.5±4.0 | 0.093±0.002 |
| PGF2a-G | 3.7±0.04 | 845±0.0 | 0.0043±0.0004 |
| PGE2-G | 3.2±0.0 | 152±0.0 | 0.021±0.003 |
|
| |||
| CES1 | |||
|
|
|||
| Substrate | kcat(min−1) | Km(μM) | kcat/Km(min−1μM−1) |
|
| |||
| 2-AG | 59±4.0 | 49±11 | 1.2±0.0 |
| PGF2a-G | 29±3.5 | 93±43 | 0.49±0.18 |
| PGE2-G | 90±16 | 250±60 | 0.37±0.05 |
Parameters for PPT1 and CES1 are the average ± S.D. of 3 independent experiments. Kinetic parameters for CES1 are from Xie et al.10
Prostaglandin glyceryl esters, e.g. PGE2-G and PGF2α-G, are formed by COX-mediated oxygenation of 2-AG and are prostaglandins that retain the esterified glycerol moiety 15. Recombinant PPT1 could hydrolyze both PGE2-G and PGF2α-G, and analysis of substrate concentration-velocity plots yielded Michaelis-Menten parameters for PPT1- and CES1-mediated PG-G hydrolysis reactions reported in Table 1. As judged by kcat/Km values, hydrolysis rates of PGE2-G and PGF2α-G by CES1 were not significantly different. On the other hand, PGE2-G was hydrolyzed 5 times faster than PGF2α-G by PPT1. When CES1 and PPT1 enzymes are compared, PGE2-G and PGF2α-G were hydrolyzed 17 and times faster by CES1 relative to PPT1, respectively (Table 1).
The enzymes MAGL, FAAH, CES1 and CES2 have been previously reported to hydrolyze 2-AG and PG-Gs 10, 35, but this is the first time that these lipid mediators are reported to be substrates for PPT1. We next compared recombinant forms of human MAGL, FAAH, CES1, and PPT1 for their ability to hydrolyze a fixed amount of 2-AG (final concentration 10 μM). As shown in Fig. 5D, CES1, FAAH and MAGL hydrolyzed 2-AG in vitro at roughly similar rates per mole of enzyme, whereas PPT1 hydrolyzed 2-AG at a significantly slower rate compared to the other enzymes.
Hydrolysis of 2-AG by PPT1: Inhibition by PO, JZL184 and HDSF
To compare the extent of inhibition of PPT1- and CES1-catalyzed hydrolytic activity by the small molecule inhibitors PO, JZL184 and HDSF, the recombinant enzymes were pretreated with each compound for 30 min, followed by addition of 2-AG substrate (final concentration 10 μM) and incubation for an additional 30 min. As was expected36, Fig. 6A shows that PO potently inhibited CES1; however, PPT1 activity was much less sensitive to PO treatment. This result is reminiscent of the fact that the well-known serine hydrolase inhibitor PMSF also did not block PPT1 enzymatic activity 37, which was attributed to the steric bulk of the phenyl group of PMSF prohibiting access of the electrophilic sulfonyl fluoride moiety to the narrow catalytic gorge of PPT1. PO would offer similar steric constraints as PMSF due to its bulky p-nitrophenyl leaving group. In a similar vein, differential inhibition of CES1 and PPT1 was also obtained for JZL184; CES1 activity was blocked 10 but PPT1 activity was not (Supplementary Fig. 3A). By contrast, 200 μM HDSF inhibited CES1 and PPT1 activities by nearly 100% (Fig. 6A). HDSF inhibited recombinant PPT1 in a time- and concentration-dependent manner, suggesting a covalent mechanism of inhibition (Fig. 6B). It is important to note again that HDSF had no effect on cathepsin G on the basis of competitive ABPP (Supplementary Fig. 3A, B). In addition, another serine hydrolase, KIAA1363 (also abbreviated NCEH), was detected in THP1 cells by ABPP-MudPIT (Fig. 1A). However, even though recombinant KIAA1363 protein exhibits esterase activity it does not have lipolytic activity toward 4-MUBO (Supplementary Fig. 5A,B), nor can it hydrolyze 2-AG 6,28. Furthermore, based on competitive ABPP, μM HDSF had no effect on KIAA1363 activity in THP1 cell lysates (Supplementary Fig. 5C) and mouse brain extracts (data not shown). Thus, KIAA1363 was probably not the serine hydrolase enzyme we sought that metabolizes 2-AG.
Figure 6. Inhibition of PPT1 by small molecule inhibitors.
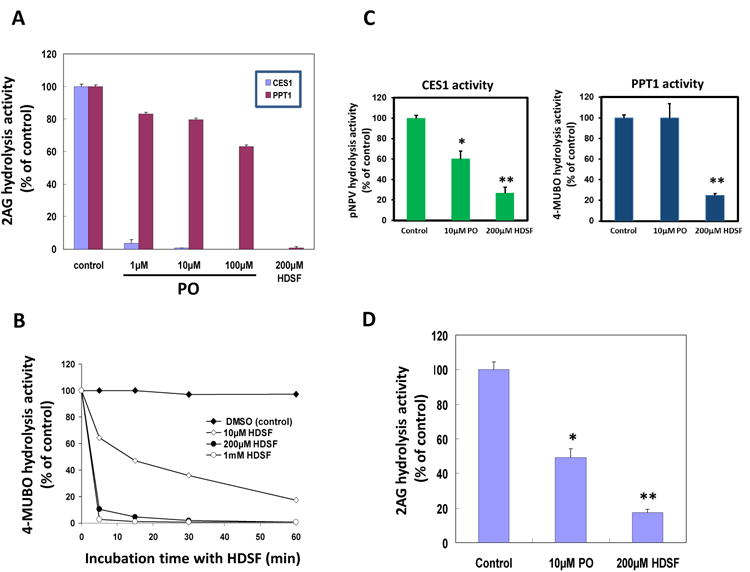
(A) 2-AG hydrolytic activity of recombinant PPT1 pretreated with 1μM, 10μM and 100μM PO or 200μM HDSF for 30 min, followed by incubation with 10μM 2-AG for another 30 min. (B) Time-dependent and concentration-dependent 4-MUBO hydrolysis activity curve of recombinant PPT1 pretreated with HDSF. Recombinant PPT1 was incubated with 10μM, 100μM or 1mM of HDSF for different times. (C) Hydrolysis of pNPV (left) and 4-MUBO (right) by cell lysates obtained from THP1 macrophages that had been pretreated with either PO (10μM) or HDSF (200μM) for 30 min. (D) Hydrolysis of 2-AG by intact THP1 macrophages pretreated with either PO (10μM) or HDSF (200μM) for 30 min, followed by 60 min incubation with 10μM 2-AG. Data represent the mean ± SD of 3 independent experiments; *, p<0.05; **, p<0.01 (Student's t-test).
We previously investigated the extent of inhibition of CES1 and unknown 31-32 kDa serine hydrolase in whole THP1 monocyte lysates by PO, CPO and JZL184 using competitive gel-based ABPP 10. The results indicated CES1 was significantly more sensitive than the unknown 31-32 kDa enzyme10, with CES1 exhibiting large kinact/Ki values with these inhibitors36,38. In contrast, high concentrations of PO and CPO (∼100 μM) were needed to modestly impair PPT1 activity (Fig. 6A for PO), while JZL184 at concentrations as high as μM had no effect10 (and data not shown).
On the basis of these inhibition findings, we designed an experiment to determine the relative contributions of PPT1 and CES1 to 2-AG hydrolysis activity in intact living THP1 monocytes/macrophages. THP1 macrophages were pretreated for 30 min with 10 μM PO to chemically knock down CES1 activity but leave PPT1 activity essentially intact; alternatively, 200 μM HDSF was used to chemically knock down both CES1 and PPT1 activities (Fig. 6C). (It was pre-determined this concentration of HDSF did not adversely affect cell viability.) Although the concentration of HDSF used to treat the cells was high, it was necessary in order to inactivate PPT1 in intact cells (as judged by a reduction in 4-MUBO hydrolysis activity). Furthermore, MAGL, FAAH, ABHD6 and ABHD12 are not present in THP1 cells (Fig. 1A) and therefore can be ruled out as off-target enzymes for either PO or HDSF. Following pretreatment with each inhibitor, the cells were exposed to exogenous 2-AG (final concentration 10 μM) and the extent of 2-AG hydrolytic metabolism was determined by quantifying the yield of AA. It was discovered that pretreatment with PO and HDSF inhibited 51% and 83% of the 2-AG hydrolytic activity in THP1 macrophages, respectively (Fig. 6D). From these data, we estimate that CES1 and PPT1 contribute ∼51% and ∼32% of the 2-AG hydrolytic activity in THP1 macrophages, respectively.
Immunodepletion of PPT1 from THP1 cell lysates and 2-AG hydrolysis
Immunoprecipitation of PPT1 from THP1 monocyte lysates using anti-PPT1 antibodies was successfully accomplished on the basis of immunoblots of non-specific antibody and anti-PPT1 antibody treated samples (Fig. 7A), and the significantly reduced 4-MUBO hydrolytic activity (Fig. 7B). Importantly, the hydrolytic activity of the PPT1-depleted lysate toward 2-AG was also markedly reduced by ∼40% (Fig. 7C). These results are consistent with a role for PPT1 in the hydrolysis of 2-AG by THP1 cells. Furthermore, we previously showed that immunodepletion of CES1 from THP1 cell lysates caused ∼60% reduction in 2-AG hydrolytic activity 10, thus complementing the PPT1 immunodepletion results reported here.
Figure 7. Immunoprecipitation of PPT1 from THP1 monocyte lysates reduces the hydrolysis of 2-AG.
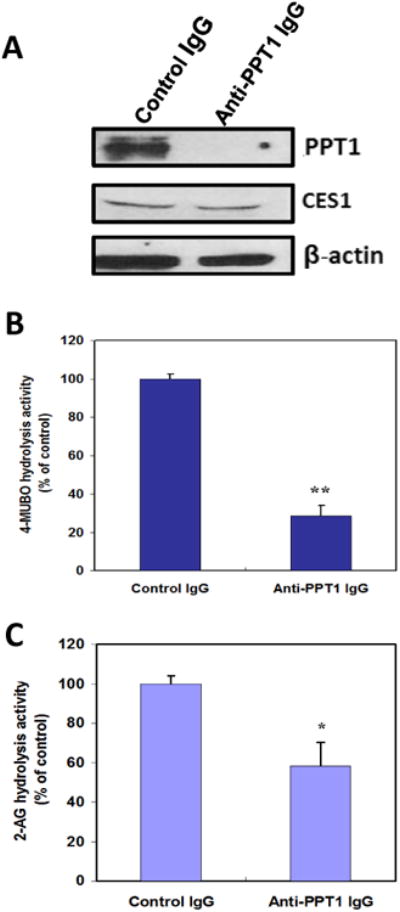
(A) PPT1 was immunoprecipitated from cell lysates using anti-PPT1 IgG antibodies. As a negative control, lysates were incubated with non-specific IgG antibodies. Following incubation of whole cell lysates with antibodies, the antibody-antigen complexes were attached to protein A beads and pelleted by centrifugation. Equivalent amounts of the supernatants were subjected to SDS-PAGE. Immunodepleted supernatants were also subjected to 4-MUBO hydrolysis activity assay (B), or incubated with 10μM of 2-AG for 30 min to determine residual 2-AG hydrolytic activity (C). Data represent the mean ± SD of 3 independent experiments; *, p<0.05; **, p<0.01 (Student's t-test).
Discussion
Noteworthy findings of this study include identification of serine hydrolases in the human THP1 monocyte/macrophage cell line by ABPP–MudPIT and ABPP–in-gel digest–MudPIT approaches. One of the identified hydrolases was a serine lipase termed PPT1 (EC 3.1.2.22). It was abundantly expressed in this cell line but was minimally reactive toward the activity-based probe FP-biotin. PPT1 is a monomeric lysosomal hydrolase that metabolizes S-acylated proteins by hydrolyzing thioester bonds that connect long-chain fatty acids of varying lengths (14-18 carbons) to the thiol of cysteine 39. In addition, PPT1 can hydrolyze palmitoyl-CoA and S-palmitoyl thioglucoside, and was shown to cleave palmitate from H-Ras proteins 40. PPT1 is targeted to lysosomes through the classical mannose 6-phosphate receptor pathway. It was first purified from bovine brain 41 and its crystal structure revealed a canonical α/β-hydrolase fold typical of lipases, with an essential catalytic triad composed of Ser115-His289-Asp233 30. Bovine PPT1 is 95% identical to human PPT1 at the amino acid level 42; the polypeptide consists of amino acids and a signal sequence of 25 amino acids that is co-translationally cleaved. We and others found that PPT1 migrates as a doublet or triplet during SDS-PAGE (Fig. 1C) 43, which collapses to a single ∼29 kDa protein upon deglycosylation (Fig. 1E). PPT1 has three conserved asparagine-linked glycosylation sites at amino acid positions 197, 212, and 232, which are all utilized in vivo. The glycan moieties are endoglycosidase H sensitive, demonstrating that PPT1 possesses only N-type glycosylation. Proper glycosylation is required for the stability and activity of the enzyme 30 and glycosylation site mutants of PPT1 have greatly reduced enzyme activity 44. Interestingly, despite localization of PPT1 in lysosomes, its enzymatic activity is optimal at neutral pH not pH 4-5 40 (data not shown). In mammals, PPT1 is found in many different tissues at varying quantities, with highest levels in brain, eye, and spleen 43, 45. In addition to being found in lysosomal compartments of neurons, the protein is also expressed in pre-synaptic compartments 46–48. The expression of PPT1 is under developmental control in the CNS and it is well known that loss of PPT1 activity causes a lysosomal storage disorder, which leads to neuronal cell death causing infantile neuronal ceroid lipofuscinoses 49, 50.
This is the first report that PPT1 is expressed in human THP1 cells and primary monocytes/macrophages. The result is consistent with PPT1 being detected by immunohistochemical approaches in macrophages within human liver, lung and bowel, but having little or no expression in tissue parenchymal cells 51. Of the hydrolases currently known to metabolize the endocannabinoid 2-AG, it was interesting that only CES1 was detected in THP1 cells by ABPP-MudPIT, while MAGL, FAAH, ABHD6 and ABHD12 were undetectable. PPT1 was also expressed in higher amounts than CES1 in THP1 monocytes/macrophages (Fig. 3). An unexpected and noteworthy finding was that PPT1 could hydrolyze 2-AG, which is a bioactive lipid with an important role in regulating inflammation in macrophages 33. In addition, PG-Gs, which are COX-mediated oxygenation products of 2-AG, were also hydrolyzed by PPT1. The catalytic efficiency for 2-AG hydrolysis by recombinant PPT1 is lower (13-fold) than CES1's (Table 1). Thus, examination of kinetic parameters suggested that PPT1 is unlikely the major 2-AG hydrolytic enzyme in monocytes/macrophages, but it could have an important auxiliary role in regulating the levels of this endocannabinoid in cell types that do not express MAGL, ABHD6 and ABHD12. The catalytic efficiency for the PPT1-2AG pair is also in line with the modest catalytic efficiencies observed for enzymes involved in secondary metabolic pathways 52. Because PPT1 is primarily found within lysosomes and has a low Km for 2-AG, it may also regulate distinct pools of lipid glyceryl esters that are either synthesized in or transported to this subcellular compartment. The catalytic efficiency of 2-AG hydrolysis by pure PPT1 is ∼10-fold lower than the thioesters palmitoyl-CoA and 4-methylumbelliferyl-6-thiopalmitoyl-β-D-glucoside 53, which is consistent with the fact that O-acyl esters are hydrolyzed slower than thioesters.
Three lines of evidence indicate that CES1 is the major 2-AG hydrolytic enzyme in THP1 cells and PPT1 has an auxiliary role. (i) 51% of the 2-AG hydrolytic activity was inhibited by PO, which can inhibit CES1 but not PPT1, whereas 83% was inhibited by HDSF, which can inhibit both CES1 and PPT1 (Fig. 6) but not cathepsin G and KIAA1363, which are also expressed in THP1 cells (Supplementary Figs. 3 and 5). Although high concentrations of HDSF are needed to inhibit PPT1 and it is likely HDSF has several off-target enzymes, it is reasonable to rule out MAGL, FAAH, ABHD6 and ABHD12 as off-targets in the context of the THP1 cell line because of their lack of expression. Therefore, 51% and 32% of the 2-AG hydrolytic activity can be attributed to CES1 and PPT1, respectively. These results are consistent with our previous report 10, which concluded that CES1 accounted for 40-50% of the 2-AG hydrolytic activity in THP1 cells based on the inhibition profile of CES1 chemical inhibitors and CES1 immunodepletion from THP1 cell lysate. (ii) Knockdown of CES1 expression in THP1 cells reduced 2-AG hydrolytic activity of THP1/CES1 KD cells by 32% relative to the control THP1 cells (Fig. 2A-C). Although the reduction in 2-AG hydrolytic activity in the CES1-deficient cells did not reach 51%, this could be explained by the fact that PPT1 protein expression appeared to be upregulated in the CES1-deficient cell line compared to the control THP1 cell line. Enhanced PPT1 expression may be a compensatory mechanism following knockdown of CES1 expression, thus contributing to the 2-AG hydrolytic activity of the CES1-deficient cells. (iii) Immunodepletion of PPT1 from THP1 cell lysates reduced 2-AG hydrolytic activity by ∼40% (Fig. 7), which is similar to the amount estimated using chemical inhibitor HDSF (32%). This result provided strong evidence that PPT1 may have a collaborative role in 2-AG catabolism.
It was also shown in this study that 4-MUBO is an excellent substrate for PPT1 (Fig. 4B), and pretreatment of intact macrophages with 200 μM HDSF inhibited nearly 80% of the 4-MUBO hydrolytic activity, whereas 10 μM PO had no effect (Fig. 6C). Because 200 μM HDSF completely inhibited both PPT1 and CES1, whereas 10 μM PO completely inhibited CES1 but only minimally affected PPT1 activity (Fig. 6A), we interpreted these results to indicate PPT1 was primarily responsible for the 4-MUBO hydrolytic activity of THP1 cells. This conclusion is supported by the markedly reduced 4-MUBO hydrolysis activity of PPT1-depleted cell lysates following immunoprecipitation (Fig. 7B). These findings also provided insight into enigmatic results previously reported by us14, which showed that high concentrations of organophosphate poisons (oxons) minimally impacted the 4-MUBO-hydrolytic activity of THP1 cell lysates. In addition, we observed that 200 μM PMSF inhibited only ∼20% of the 4-MUBO hydrolytic activity of whole-cell lysates (data not shown), which is in line with the fact that PMSF is a poor inhibitor of PPT1 37. Thus, we conclude that PPT1 accounts for the organophosphate- and PMSF-insensitive 4-MUBO hydrolytic activity that we previously observed14.
In summary, this study confirms and extends several findings of our previous reports 10,14, including validation of an in vivo role for CES1 in 2-AG metabolism (Fig. 2; Supplementary Fig. 4). It is also the first to identify and quantify palmitoyl protein thioesterase 1 (PPT1) in human THP1 monocytes/macrophages, and the first to show PPT1 can hydrolyze the endogenous cannabinoid 2-arachidonoylglycerol (2-AG) and two prostaglandin glyceryl esters, PGE2-G and PGF2α-G, in vitro. These findings suggest that PPT1 may also have a role in degrading endocannabinoids in certain cellular niches, especially in cell types lacking or having limited expression of MAGL, FAAH, ABHD6, ABHD12 and CES1. Furthermore, we characterized the hydrolytic efficiency of PPT1 using both 2-AG and PG-G substrates and compared the kinetic parameters to CES1, MAGL and FAAH. We also investigated the relative contributions of PPT1 and CES1 to the hydrolysis of 2-AG in human THP1 cells using chemical inhibitors, immunoprecipitation of PPT1, and knockdown of CES1 expression using a lentiviral shRNA system. Collectively, the results suggest that PPT1 has the ability to recognize and hydrolyze lipid glyceryl esters in human THP1 cells, and that PPT1 accounts for ∼32% of the 2-AG hydrolytic activity while CES1 contributes ∼51%.
There are limitations to the current work that should be addressed in future studies, including the need to better characterize the full complement of serine hydrolases in primary human monocytes/macrophages; a need to develop small molecules to overcome the paucity of selective PPT1 inhibitors; and a necessity to validate the 2-AG hydrolytic activity of PPT1 in vivo (e.g., in intact cells and whole animals using knockdown/knockout approaches) by determining whether this enzymatic activity controls the efficacy of 2-AG signaling at cannabinoid receptors. Human macrophages are notoriously difficult to introduce shRNA or siRNA constructs into; we are currently attempting to modulate PPT1 expression in cell lines with viral vectors. However, despite being unable to knock down PPT1 expression in THP1 cells, we did successfully immunodeplete PPT1 from cell lysates and demonstrate a subsequent reduction (∼40%) in 2-AG hydrolysis activity (Fig. 7), which complemented the ∼60% reduction in 2-AG hydrolysis activity caused by immunodepletion of CES1 10. Notwithstanding these limitations, the current findings highlight the overlapping substrate specificity and functional redundancy often exhibited by lipases involved in lipid metabolism, and the auxiliary roles certain enzymes such as PPT1 may have in cells. It also potentially opens new avenues of research related to endocannabinoid biochemistry.
Supplementary Material
Acknowledgments
We thank Dr. Sandra L. Hofmann (UT Southwestern) for the kind gift of hPPT1-CHO cells and Dr. Jui-Yun Lu (UT Southwestern) for helpful technical advice. We are also grateful to Dr. Phil Potter (St. Jude Children's Research Hospital) for providing pure recombinant CES1 protein and Dr. Wei Tan (MSU) for assistance with peritoneal macrophage isolation. We also thank the Jiangsu Academy of Agricultural Sciences (JAAS) and MSU College of Veterinary Medicine for scholarship support to Dr. Ran Wang.
Funding: This study was supported by NIH 1R15ES015348-02 (M.K.R.) and the protein mass spectrometric work was supported by the Mississippi Agricultural and Forestry Experiment Station Special Research Initiatives (Edelmann) (160000-018100-027100-401120).
Abbreviations
- AA
arachidonic acid
- 2-AG
2-arachidonoylglycerol
- ABPP
activity-based protein profiling
- CES1
carboxylesterase 1
- CPO
chlorpyrifos oxon
- HDSF
hexadecylsulfonylfluoride
- 4-MUBA
4-methylumbelliferyl acetate
- 4-MUBO
4- methylumbelliferyl oleate
- MUDPIT
multi-dimensional protein identification technology
- PPT1
palmitoyl protein thioesterase 1
- PO
paraoxon
- PGE2-G
prostaglandin E2 glyceryl ester
- PGF2α-G
prostaglandin F2α glyceryl ester
- PMSF
phenylmethylsulfonyl fluoride
Footnotes
Supporting Information Available: Supporting materials may be accessed free of charge online at http://pubs.acs.org. Supplementary table and figures include the list of identified serine hydrolases in THP1 monocytes, CID fragmentation spectra of one of the identified PPT1 peptides, inhibition of 2-AG hydrolytic activity of THP1 monocyte lysates by FP-biotin, competitive ABPP demonstrating the lack of cathepsin G inhibition in THP1 cell lysates by HDSF, hydrolysis of 2-AG in THP1 macrophages regulates prostaglandin biosynthesis, overexpression of human KIAA1363 in COS-7 cells and its esterase and lipase activities.
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes: The authors declare no competing financial interest.
References
- 1.De Petrocellis L, Cascio MG, Di Marzo V. The endocannabinoid system: a general view and latest additions. Br J Pharmacol. 2004;141:765–774. doi: 10.1038/sj.bjp.0705666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 3.Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, Friedman H. The cannabinoid system and immune modulation. Journal of leukocyte biology. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- 4.Di Marzo V. The endocannabinoid system: its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacological research : the official journal of the Italian Pharmacological Society. 2009;60:77–84. doi: 10.1016/j.phrs.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Mach F, Steffens S. The role of the endocannabinoid system in atherosclerosis. Journal of neuroendocrinology. 2008;20(Suppl 1):53–57. doi: 10.1111/j.1365-2826.2008.01685.x. [DOI] [PubMed] [Google Scholar]
- 6.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chemistry & biology. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, Lopez Rodriguez ML, Bajjalieh S, Stella N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J Neurosci. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navia-Paldanius D, Savinainen JR, Laitinen JT. Biochemical and pharmacological characterization of human alpha/beta-hydrolase domain containing 6 (ABHD6) and 12 (ABHD12) Journal of lipid research. 2012;53:2413–2424. doi: 10.1194/jlr.M030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie S, Borazjani A, Hatfield MJ, Edwards CC, Potter PM, Ross MK. Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: role of carboxylesterases 1 and 2. Chemical research in toxicology. 2010;23:1890–1904. doi: 10.1021/tx1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross MK, Borazjani A, Wang R, Crow JA, Xie S. Examination of the carboxylesterase phenotype in human liver. Archives of biochemistry and biophysics. 2012;522:44–56. doi: 10.1016/j.abb.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh S. Cholesteryl ester hydrolase in human monocyte/macrophage: cloning, sequencing, and expression of full-length cDNA. Physiol Genomics. 2000;2:1–8. doi: 10.1152/physiolgenomics.2000.2.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Quiroga AD, Lehner R. Role of endoplasmic reticulum neutral lipid hydrolases. Trends Endocrinol Metab. 2011;22:218–225. doi: 10.1016/j.tem.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Crow JA, Middleton BL, Borazjani A, Hatfield MJ, Potter PM, Ross MK. Inhibition of carboxylesterase 1 is associated with cholesteryl ester retention in human THP-1 monocyte/macrophages. Biochim Biophys Acta. 2008;1781:643–654. doi: 10.1016/j.bbalip.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouzer CA, Marnett LJ. Glycerylprostaglandin synthesis by resident peritoneal macrophages in response to a zymosan stimulus. J Biol Chem. 2005;280:26690–26700. doi: 10.1074/jbc.M501021200. [DOI] [PubMed] [Google Scholar]
- 16.Rouzer CA, Marnett LJ. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev. 2011;111:5899–5921. doi: 10.1021/cr2002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borazjani A, Edelmann MJ, Hardin KL, Herring KL, Allen Crow J, Ross MK. Catabolism of 4-hydroxy-2-trans-nonenal by THP1 monocytes/macrophages and inactivation of carboxylesterases by this lipid electrophile. Chem Biol Interact. 2011;194:1–12. doi: 10.1016/j.cbi.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moellering RE, Cravatt BF. How chemoproteomics can enable drug discovery and development. Chemistry & biology. 2012;19:11–22. doi: 10.1016/j.chembiol.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu JY, Hofmann SL. Thematic review series: lipid posttranslational modifications. Lysosomal metabolism of lipid-modified proteins. J Lipid Res. 2006;47:1352–1357. doi: 10.1194/jlr.R600010-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Morton CL, Potter PM. Comparison of Escherichia coli, Saccharomyces cerevisiae, Pichia pastoris, Spodoptera frugiperda, and COS7 cells for recombinant gene expression. Application to a rabbit liver carboxylesterase. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 23.Speers AE, Cravatt BF. Activity-Based Protein Profiling (ABPP) and Click Chemistry (CC)-ABPP by MudPIT Mass Spectrometry. Curr Protoc Chem Biol. 2009;1:29–41. doi: 10.1002/9780470559277.ch090138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacCoss MJ, Wu CC, Yates JR., 3rd Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal Chem. 2002;74:5593–5599. doi: 10.1021/ac025826t. [DOI] [PubMed] [Google Scholar]
- 25.Lu JY, Hu J, Hofmann SL. Human recombinant palmitoyl-protein thioesterase-1 (PPT1) for preclinical evaluation of enzyme replacement therapy for infantile neuronal ceroid lipofuscinosis. Molecular genetics and metabolism. 2010;99:374–378. doi: 10.1016/j.ymgme.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross MK, Borazjani A. Enzymatic activity of human carboxylesterases. Current protocols in toxicology/editorial board, Mahin D Maines Chapter 4. 2007;Unit 4:24. doi: 10.1002/0471140856.tx0424s33. [DOI] [PubMed] [Google Scholar]
- 27.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol. 2012;8:999–1007. doi: 10.1038/nchembio.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellizzi JJ, 3rd, Widom J, Kemp C, Lu JY, Das AK, Hofmann SL, Clardy J. The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4573–4578. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wadkins RM, Hyatt JL, Wei X, Yoon KJ, Wierdl M, Edwards CC, Morton CL, Obenauer JC, Damodaran K, Beroza P, Danks MK, Potter PM. Identification and characterization of novel benzil (diphenylethane-1,2-dione) analogues as inhibitors of mammalian carboxylesterases. J Med Chem. 2005;48:2906–2915. doi: 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- 32.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alhouayek M, Masquelier J, Muccioli GG. Controlling 2-arachidonoylglycerol metabolism as an anti-inflammatory strategy. Drug Discov Today. 2013 doi: 10.1016/j.drudis.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 35.Vila A, Rosengarth A, Piomelli D, Cravatt B, Marnett LJ. Hydrolysis of prostaglandin glycerol esters by the endocannabinoid-hydrolyzing enzymes, monoacylglycerol lipase and fatty acid amide hydrolase. Biochemistry. 2007;46:9578–9585. doi: 10.1021/bi7005898. [DOI] [PubMed] [Google Scholar]
- 36.Crow JA, Bittles V, Herring KL, Borazjani A, Potter PM, Ross MK. Inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by chlorpyrifos oxon, paraoxon and methyl paraoxon. Toxicol Appl Pharmacol. 2012;258:145–150. doi: 10.1016/j.taap.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das AK, Bellizzi JJ, 3rd, Tandel S, Biehl E, Clardy J, Hofmann SL. Structural basis for the insensitivity of a serine enzyme (palmitoyl-protein thioesterase) to phenylmethylsulfonyl fluoride. J Biol Chem. 2000;275:23847–23851. doi: 10.1074/jbc.M002758200. [DOI] [PubMed] [Google Scholar]
- 38.Crow JA, Bittles V, Borazjani A, Potter PM, Ross MK. Covalent inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by the carbamates JZL184 and URB597. Biochemical pharmacology. 2012;84:1215–1222. doi: 10.1016/j.bcp.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu JY, Hofmann SL. Inefficient cleavage of palmitoyl-protein thioesterase (PPT) substrates by aminothiols: implications for treatment of infantile neuronal ceroid lipofuscinosis. Journal of inherited metabolic disease. 2006;29:119–126. doi: 10.1007/s10545-006-0225-z. [DOI] [PubMed] [Google Scholar]
- 40.Verkruyse LA, Hofmann SL. Lysosomal targeting of palmitoyl-protein thioesterase. J Biol Chem. 1996;271:15831–15836. doi: 10.1074/jbc.271.26.15831. [DOI] [PubMed] [Google Scholar]
- 41.Camp LA, Hofmann SL. Purification and properties of a palmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J Biol Chem. 1993;268:22566–22574. [PubMed] [Google Scholar]
- 42.Schriner JE, Yi W, Hofmann SL. cDNA and genomic cloning of human palmitoyl-protein thioesterase (PPT), the enzyme defective in infantile neuronal ceroid lipofuscinosis. Genomics. 1996;34:317–322. doi: 10.1006/geno.1996.0292. [DOI] [PubMed] [Google Scholar]
- 43.Isosomppi J, Heinonen O, Hiltunen JO, Greene ND, Vesa J, Uusitalo A, Mitchison HM, Saarma M, Jalanko A, Peltonen L. Developmental expression of palmitoyl protein thioesterase in normal mice, Brain research. Developmental brain research. 1999;118:1–11. doi: 10.1016/s0165-3806(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 44.Lyly A, von Schantz C, Salonen T, Kopra O, Saarela J, Jauhiainen M, Kyttala A, Jalanko A. Glycosylation, transport, and complex formation of palmitoyl protein thioesterase 1 (PPT1)--distinct characteristics in neurons. BMC cell biology. 2007;8:22. doi: 10.1186/1471-2121-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suopanki J, Tyynela J, Baumann M, Haltia M. The expression of palmitoyl-protein thioesterase is developmentally regulated in neural tissues but not in nonneural tissues. Molecular genetics and metabolism. 1999;66:290–293. doi: 10.1006/mgme.1999.2839. [DOI] [PubMed] [Google Scholar]
- 46.Heinonen O, Kyttala A, Lehmus E, Paunio T, Peltonen L, Jalanko A. Expression of palmitoyl protein thioesterase in neurons. Molecular genetics and metabolism. 2000;69:123–129. doi: 10.1006/mgme.2000.2961. [DOI] [PubMed] [Google Scholar]
- 47.Lehtovirta M, Kyttala A, Eskelinen EL, Hess M, Heinonen O, Jalanko A. Palmitoyl protein thioesterase (PPT) localizes into synaptosomes and synaptic vesicles in neurons: implications for infantile neuronal ceroid lipofuscinosis (INCL) Hum Mol Genet. 2001;10:69–75. doi: 10.1093/hmg/10.1.69. [DOI] [PubMed] [Google Scholar]
- 48.Ahtiainen L, Van Diggelen OP, Jalanko A, Kopra O. Palmitoyl protein thioesterase 1 is targeted to the axons in neurons. The Journal of comparative neurology. 2003;455:368–377. doi: 10.1002/cne.10492. [DOI] [PubMed] [Google Scholar]
- 49.Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 50.Goebel HH, Wisniewski KE. Current state of clinical and morphological features in human NCL. Brain pathology. 2004;14:61–69. doi: 10.1111/j.1750-3639.2004.tb00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Margraf LR, Boriack RL, Routheut AA, Cuppen I, Alhilali L, Bennett CJ, Bennett MJ. Tissue expression and subcellular localization of CLN3, the Batten disease protein. Molecular genetics and metabolism. 1999;66:283–289. doi: 10.1006/mgme.1999.2830. [DOI] [PubMed] [Google Scholar]
- 52.Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, Tawfik DS, Milo R. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry. 2011;50:4402–4410. doi: 10.1021/bi2002289. [DOI] [PubMed] [Google Scholar]
- 53.Das AK, Lu JY, Hofmann SL. Biochemical analysis of mutations in palmitoyl-protein thioesterase causing infantile and late-onset forms of neuronal ceroid lipofuscinosis. Hum Mol Genet. 2001;10:1431–1439. doi: 10.1093/hmg/10.13.1431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.