

Key Points
PS can be overlooked in the differential diagnosis of children with severe congenital anemia.
mtDNA deletion testing should be included in the genetic evaluation of patients with congenital anemia of unclear etiology.
Abstract
Pearson marrow pancreas syndrome (PS) is a multisystem disorder caused by mitochondrial DNA (mtDNA) deletions. Diamond-Blackfan anemia (DBA) is a congenital hypoproliferative anemia in which mutations in ribosomal protein genes and GATA1 have been implicated. Both syndromes share several features including early onset of severe anemia, variable nonhematologic manifestations, sporadic genetic occurrence, and occasional spontaneous hematologic improvement. Because of the overlapping features and relative rarity of PS, we hypothesized that some patients in whom the leading clinical diagnosis is DBA actually have PS. Here, we evaluated patient DNA samples submitted for DBA genetic studies and found that 8 (4.6%) of 173 genetically uncharacterized patients contained large mtDNA deletions. Only 2 (25%) of the patients had been diagnosed with PS on clinical grounds subsequent to sample submission. We conclude that PS can be overlooked, and that mtDNA deletion testing should be performed in the diagnostic evaluation of patients with congenital anemia.
Introduction
In 1979, Pearson and colleagues described 4 patients with severe anemia characterized by sideroblasts and vacuolization of marrow precursors, and pancreatic dysfunction.1 Pearson marrow pancreas syndrome (PS) is caused by large deletions in mitochondrial DNA (mtDNA), which accounts for the metabolic acidosis and variable tissue dysfunction in patients.2,3 Deleted mtDNA in patients’ cells exists in varying proportions relative to normal mtDNA, a mixture termed heteroplasmy.4 Changes in heteroplasmy are thought to underlie differences in disease manifestations and evolution in patients,5 including spontaneous hematologic improvement. The incidence of PS is unknown, with only ∼100 patients described in the literature since Pearson’s original report.6
Diamond-Blackfan anemia (DBA) is characterized by severe hyporegenerative, macrocytic anemia with erythroblastopenia,7 and variably associated congenital malformations, growth retardation, and elevations in erythrocyte adenosine deaminase and/or hemoglobin F levels.8,9 In 50% to 60% of DBA patients, mutations in ribosomal protein (RP) genes or GATA1 are found, with a high frequency of sporadic cases because of de novo mutations.10-12 The majority of patients respond to an initial course of corticosteroid (CS) therapy, and ∼20% of all patients enter a steroid- and transfusion-free “remission” by adulthood.7,13,14 DBA is more frequently occurring than PS, with an estimated incidence of 1-2/100 000 and ∼1000 cases in the literature.12,15,16
DBA and PS share important features including early onset of severe anemia, variable nonhematologic manifestations, sporadic genetic inheritance, and episodes of spontaneous hematologic improvement. Because of these features and the rarity of PS relative to DBA, we hypothesized that some patients in whom the leading clinical diagnosis is DBA actually have PS. To test this hypothesis, we retrospectively tested DNA samples from patients who enrolled in a DBA genetics research study. Prior studies from this DBA cohort have yielded the novel identification or confirmation of mutations and deletions in RP genes or GATA117-19 in 175 of 362 samples (48%), a proportion similar to that found in other DBA registries.11
Study design
Patient material
Biological samples were procured under protocols approved by the Institutional Review Board at Boston Children’s Hospital and after written informed consent in accordance with the Declaration of Helsinki. The protocol and patient cohort has been described previously.17,18
Long-range polymerase chain reaction (PCR) and deletion mapping
Peripheral blood genomic DNA (50 ng) was amplified using primers 5328F (5′-CCATCATAGCCACCATCACCCTCC-3′) and humitoDloopR (5′-CTTTATGACCCTGAAGTAGGAACC-3′) using iProof HF Master Mix (Bio-Rad). Deletion junctions were mapped using PCR and Sanger sequencing, and nucleotide positions assigned per the revised Cambridge Reference Sequence of human mtDNA (GenBank NC_12920).
Southern blot
Genomic DNA (1 μg) was digested with BbvCI, ClaI, or NheI (New England Biolabs), separated on a 0.6% agarose gel and transferred to nylon membrane. Hybridization of the 32P-labeled probe shown in Figure 1B was performed using Rapid-Hyb buffer (GE Healthcare).
Figure 1.
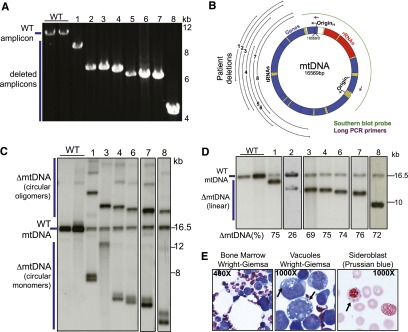
mtDNA analysis identifies 8 patients with PS in a cohort of DBA patients. (A) Long PCR, showing the preferential amplification of deleted mtDNA fragments from 8 patients in the DBA cohort, relative to amplification of wild-type (WT) mtDNA. (B) Map of the human mitochondrial genome, with genes (blue), transfer RNAs (tRNAs; yellow), ribosomal RNAs (rRNAs; red), and origins of DNA replication for heavy (H) and light (L) strands indicated. The extent of mtDNA deletions and corresponding patient numbers are shown by the black lines. Purple arrows represent location of the long PCR primers. Southern blot probe used in panels C and D is indicated in green. (C) Southern blot analysis of peripheral blood DNA from patients and normal controls, cut with BbvCI. WT mtDNA shows the expected, 16.5-kb linearized fragment. Deleted mtDNA (ΔmtDNA) remains uncut and migrates as circular monomeric and oligomeric species. The lanes shown are from the same blot, rearranged to match the order of patients in the text. Sufficient peripheral blood DNA was not available from patients 2 and 5. (D) Southern blot analysis of peripheral blood DNA from patients and normal controls cut with NheI or ClaI to linearize all species of mtDNA. The proportion of deleted mtDNA (ΔmtDNA [%]; ie, heteroplasmy) is shown for each sample. The lanes shown are from 3 different blots, arranged to match the order of patients in the text. Sufficient peripheral blood DNA was not available from patient 5. (E) Bone marrow evaluation of patient 4, showing classic features of PS. Wright-Giemsa stain of bone marrow showing mild hypocellularity and vacuoles in precursors (left; ×40 objective). Magnified image of vacuoles (arrows) in myeloid precursors (center; ×100 oil-immersion objective). Prussian blue stain (for iron) of bone marrow aspirate, showing a ringed sideroblast (arrow) (right; ×100 oil-immersion objective).
Results and discussion
We used a long PCR strategy to screen DNA samples from the DBA cohort, by amplifying an 11.2-kb region of mtDNA where pathological deletions are found.5 We detected large mtDNA deletions in 8 of 173 genetically uncharacterized samples (4.6%; Figure 1A-B), but in none of 152 patients with known DBA-associated mutations. The detected mtDNA deletions ranged in size from 2.3 kb to 7.0 kb, 5 of which were novel, and 3 of which were previously described, including the 4977-bp “common deletion”20 (Figure 1A, Table 1, and supplemental Table 1 available on the Blood Web site). Southern blot analysis using BbvCI, whose unique site is lost in all deletions, revealed that the deleted mtDNA existed as monomeric or oligomeric species (Figure 1C). On southern blots using NheI or ClaI, whose unique sites are preserved in deleted and normal mtDNA, these species resolved into single bands, indicating a single deletion event (Figure 1D). We quantified heteroplasmy and found that in all patients with contemporaneous hematologic abnormalities, the burden of deleted mtDNA was high, ranging from 69% to 76% of total mtDNA; whereas, interestingly, in one transfusion-independent patient (patient 2), it was only 26% (Figure 1D). These results establish the molecular diagnosis of PS in 8 out of 173 genetically uncharacterized patients in the DBA cohort.
Table 1.
Clinical and molecular characteristics of 8 patients with PS in the DBA cohort
| Patient | mtDNA Δ size (bp) | Knew Dx? | CS therapy: duration/outcome | Other therapy | HSCT | Reached transfusion independence? | MCV (fL) | Erythroid hypoplasia? | RS? | Vacuoles? | Outcome (age) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2309 | No | Yes: 8 wk/CMV hepatitis | Epo: no response | Yes | No | 119 | Yes | No | Yes | Deceased (29 m.o.) |
| 2 | 4403 | No | Yes: 3 y/deemed responsive | N.a. | No | Yes | N.a. | N.a. | N.a. | N.a. | Alive (3 y.o.) |
| 3 | 4419 | Yes | Yes: 4 mo/adrenal insufficiency | — | No | Yes | 126 | Yes | No | Yes | Alive (7 y.o.) |
| 4 | 4623 | No | Yes: 8 wk/no response | — | Yes | No | 136 | Yes | Yes | Yes | Deceased (25 m.o.) |
| 5 | 4977 | Yes | N.a. | N.a. | No | Yes | N.a. | N.a. | N.a. | N.a. | Alive (9 y.o.) |
| 6 | 4977 | No | No* | — | No | No | 110 | Yes | No | Yes | Alive (1 y.o.) |
| 7 | 5149 | No | Yes: 6 mo/no response | GCSF: ANC improved | No | No | 97 | Yes | No | Yes | Deceased (19 m.o.) |
| 8 | 6956 | No | No | GCSF: ANC improved | No | Yes | 105 | Yes | Yes | Yes | Alive (6 y.o.) |
ANC, absolute neutrophil count; CMV, cytomegalovirus; Δ, deletion; Dx, diagnosis; Epo, erythropoietin; GCSF, granulocyte colony stimulating factor; HSCT, hematopoietic stem cell transplantation; MCV, mean corpuscular volume; m.o., months old; N.a., information not available; RS, ringed sideroblasts; y.o., years old.
CS trial planned, but PS diagnosis made prior to intervention.
Follow-up with referring providers in the 1-month to 8-year time span since sample submission revealed that only 2 patients (patients 3 and 5) were eventually diagnosed with PS on clinical grounds, both of whom were alive and transfusion independent (Table 1; supplemental Text). Of the remaining 6 undiagnosed patients, in only 1 (patient 8) was clinical mtDNA testing performed, but in this case, mtDNA sequencing was conducted and did not detect the patient’s mtDNA deletion. Three of the 6 undiagnosed patients had died, 2 after HSCT and 1 from bacterial sepsis. Two of the other 3 undiagnosed, living patients had reached transfusion independence (Table 1).
A review of laboratory and clinical data available on 6 patients (patients 1, 3, 4, 6, 7, and 8) showed that all had macrocytic anemia and developed pancytopenia (Table 1; supplemental Table 1 and supplemental Text). On retrospective bone marrow examination, all patients had erythroid hypoplasia and vacuolated progenitors, but ringed sideroblasts were present or reported in only 2 patients (Table 1; Figure 1E). Dysplastic changes were apparent in 5 of the 6 patients, and importantly, monosomy 7 was documented in patient 8, which resolved on follow-up. These results demonstrate the variable presence of “classic hallmarks” and frequent dysplastic changes in the bone marrow examination of PS patients.
Five patients underwent a trial of CSs, with 4 showing no hematologic response, and 1 patient (patient 2) deemed responsive with improvement in hemoglobin levels, but without a trial off CSs to establish therapeutic efficacy (Table 1). Two patients (patients 7 and 8) received granulocyte colony stimulating factor because of neutropenia, with 1 patient (patient 8) treated continually until transfusion independence. Erythropoietin yielded no benefit in the single patient treated. These results show that there is no clear benefit from CS therapy in patients with PS, and a role for cytokine growth factors remains to be established.
Our findings illustrate several important points. First, PS is easily overlooked in the differential diagnosis of patients with congenital anemia. The primary reason may be lack of familiarity with this disorder, but our experience shows that both PS and DBA can share an identical presentation of severe neonatal hyporegenerative anemia, in the absence of other clinically apparent, “classic hallmarks” of PS such as pancreatic insufficiency, sideroblasts, and metabolic acidosis. Second, several pitfalls confound the diagnosis of DBA and PS. DBA remains a clinical diagnosis in many cases because the genetic characterization of DBA is incomplete and a clinically validated functional test for DBA is not available. Appropriate testing for mtDNA deletions will readily identify patients with PS, but because of heteroplasmy, sequencing of short mtDNA fragments amplified by PCR will not identify large deletions. Moreover, standard whole exome sequencing strategies do not target mtDNA. Third, establishing the diagnosis of PS as distinct from DBA and other bone marrow failure syndromes impacts disease surveillance and management, as well as family counseling regarding recurrence risk. CSs do not benefit patients with PS and may exacerbate infectious and metabolic complications. Here and elsewhere,21 myelodysplastic changes were motivating factors for HSCT, but whether these features represent a preleukemic condition in PS is uncertain. Finally, the overall burden of mtDNA deletions in this cohort (8/362 patients; 2.2%) exceeds the frequency of mutations in several DBA-associated genes including RPL26, RPS7, RPS17, RPS24, and GATA1.8,18 We propose that mtDNA deletion testing should be performed during the initial genetic evaluation of all patients with congenital anemia.
Acknowledgments
The authors thank the patients and families for participation in the research.
This work was supported by the Manton Center for Orphan Disease Research (H.T.G. and S.A.), the Diamond Blackfan Anemia Foundation (H.T.G.), the National Institutes of Health National Heart, Lung, and Blood Institute grants R01 HL107558 and K02 HL111156 (H.T.G.), the Harvard Stem Cell Institute (S.A.), and the Charles H. Hood Foundation, Inc., Boston, MA (S.A.).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.E.G., H.T.G., and S.A. designed the study; K.E.G, R.G., D.Y., and R.L.Z. performed research; K.S., M.M.-P., L.A., H.B., S.G., M.A.H., K.K., P.K., M.M., E.N., S.P., M.J.P., A. P.-P., and T.S. collected data; K.E.G., M.D.F., H.T.G., and S.A. analyzed and interpreted the data; and K.E.G. and S.A. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Suneet Agarwal, 3 Blackfan Circle, CLS 3002, Boston Children’s Hospital, Boston, MA 02115; e-mail: suneet.agarwal@childrens.harvard.edu; and Hanna T. Gazda, 3 Blackfan Circle, CLS 15023, Boston Children’s Hospital, Boston, MA 02115; e-mail: hanna.gazda@childrens.harvard.edu.
References
- 1.Pearson HA, Lobel JS, Kocoshis SA, et al. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95(6):976–984. doi: 10.1016/s0022-3476(79)80286-3. [DOI] [PubMed] [Google Scholar]
- 2.Rotig A, Colonna M, Bonnefont JP, et al. Mitochondrial DNA deletion in Pearson’s marrow/pancreas syndrome. Lancet. 1989;333(8643):902–903. doi: 10.1016/s0140-6736(89)92897-3. [DOI] [PubMed] [Google Scholar]
- 3.Rötig A, Cormier V, Blanche S, et al. Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86(5):1601–1608. doi: 10.1172/JCI114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rötig A, Bourgeron T, Chretien D, Rustin P, Munnich A. Spectrum of mitochondrial DNA rearrangements in the Pearson marrow-pancreas syndrome. Hum Mol Genet. 1995;4(8):1327–1330. doi: 10.1093/hmg/4.8.1327. [DOI] [PubMed] [Google Scholar]
- 5.Sadikovic B, Wang J, El-Hattab A, et al. Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes. PLoS ONE. 2010;5(12):e15687. doi: 10.1371/journal.pone.0015687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manea EM, Leverger G, Bellmann F, et al. Pearson syndrome in the neonatal period: two case reports and review of the literature. J Pediatr Hematol Oncol. 2009;31(12):947–951. doi: 10.1097/MPH.0b013e3181bbc4ef. [DOI] [PubMed] [Google Scholar]
- 7.Vlachos A, Ball S, Dahl N, et al. Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clinton C, Gazda HT. Diamond-Blackfan anemia. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews™ [Internet]. Seattle: University of Washington, Seattle; 1993-2013. Available from: http://www.ncbi.nlm.nih.gov/books/NBK7047/
- 9.Vlachos A, Dahl N, Dianzani I, Lipton JM. Clinical utility gene card for: Diamond-Blackfan anemia—update 2013. Eur J Hum Genet. 2013;21(10) doi: 10.1038/ejhg.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boria I, Garelli E, Gazda HT, et al. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat. 2010;31(12):1269–1279. doi: 10.1002/humu.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrar JE, Dahl N. Untangling the phenotypic heterogeneity of Diamond Blackfan anemia. Semin Hematol. 2011;48(2):124–135. doi: 10.1053/j.seminhematol.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willig TN, Niemeyer CM, Leblanc T, et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d’Hématologie et d’Immunologie Pédiatrique (SHIP), Gesellshaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI). Pediatr Res. 1999;46(5):553–561. doi: 10.1203/00006450-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Warszawski J, Bader-Meunier B, et al. Société Française d’Hématologie et d’Immunologie Pédiatrique. Diamond-blackfan anemia and growth status: the French registry. J Pediatr. 2005;147(5):669–673. doi: 10.1016/j.jpeds.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Narla A, Vlachos A, Nathan DG. Diamond Blackfan anemia treatment: past, present, and future. Semin Hematol. 2011;48(2):117–123. doi: 10.1053/j.seminhematol.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ball SE, McGuckin CP, Jenkins G, Gordon-Smith EC. Diamond-Blackfan anaemia in the U.K.: analysis of 80 cases from a 20-year birth cohort. Br J Haematol. 1996;94(4):645–653. doi: 10.1046/j.1365-2141.1996.d01-1839.x. [DOI] [PubMed] [Google Scholar]
- 16.Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007;2007(1):29–39. doi: 10.1182/asheducation-2007.1.29. [DOI] [PubMed] [Google Scholar]
- 17.Doherty L, Sheen MR, Vlachos A, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010;86(2):222–228. doi: 10.1016/j.ajhg.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gazda HT, Preti M, Sheen MR, et al. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum Mutat. 2012;33(7):1037–1044. doi: 10.1002/humu.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sankaran VG, Ghazvinian R, Do R, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122(7):2439–2443. doi: 10.1172/JCI63597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuels DC, Schon EA, Chinnery PF. Two direct repeats cause most human mtDNA deletions. Trends Genet. 2004;20(9):393–398. doi: 10.1016/j.tig.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Hoyoux C, Dresse MF, Robinet S, et al. Cord blood transplantation in a child with Pearson’s disease. Pediatr Blood Cancer. 2008;51(4):566. doi: 10.1002/pbc.21615. [DOI] [PubMed] [Google Scholar]