

Key Points
Tumor neoantigens are a promising class of immunogens based on exquisite tumor specificity and the lack of central tolerance against them.
Massively parallel DNA sequencing with class I prediction enables systematic identification of tumor neoepitopes (including from CLL).
Abstract
Genome sequencing has revealed a large number of shared and personal somatic mutations across human cancers. In principle, any genetic alteration affecting a protein-coding region has the potential to generate mutated peptides that are presented by surface HLA class I proteins that might be recognized by cytotoxic T cells. To test this possibility, we implemented a streamlined approach for the prediction and validation of such neoantigens derived from individual tumors and presented by patient-specific HLA alleles. We applied our computational pipeline to 91 chronic lymphocytic leukemias (CLLs) that underwent whole-exome sequencing (WES). We predicted ∼22 mutated HLA-binding peptides per leukemia (derived from ∼16 missense mutations) and experimentally confirmed HLA binding for ∼55% of such peptides. Two CLL patients that achieved long-term remission following allogeneic hematopoietic stem cell transplantation were monitored for CD8+ T-cell responses against predicted or confirmed HLA-binding peptides. Long-lived cytotoxic T-cell responses were detected against peptides generated from personal tumor mutations in ALMS1, C6ORF89, and FNDC3B presented on tumor cells. Finally, we applied our computational pipeline to WES data (N = 2488 samples) across 13 different cancer types and estimated dozens to thousands of predicted neoantigens per individual tumor, suggesting that neoantigens are frequent in most tumors.
Introduction
Recent progress in the development of potent vaccine adjuvants, clinically effective vaccine delivery systems, and agents that overcome tumor-induced immunosuppression strongly support the possibility that long-awaited effective therapeutic cancer vaccines are feasible.1-4 Past cancer vaccine efforts have lacked efficacy that may stem from their focus on overexpressed or selectively expressed tumor-associated native antigens as vaccine targets that require overcoming the challenging hurdles of breaking central and peripheral tolerance while risking the generation of autoimmunity.4-6 The rare examples of successful cancer vaccines in humans have targeted foreign pathogen-associated antigens7 or a mutated growth factor receptor8 or are idiotype vaccines derived from patient-specific rearranged immunoglobulins.9 These studies point to the importance of selecting immunogens distinct from self, where central/peripheral tolerance can be overcome and the risk of autoimmunity is minimal.
A hallmark of tumorigenesis is the accumulation of mutations in cancer cells. These mutations are found as both driver and passenger events10 and collectively provide an opportunity to specifically target tumor cells through the creation of tumor-specific novel immunogenic peptides (neoantigens). Neoantigens are generated from peptides encoded by gene alterations that are exclusively present in tumor but not normal tissue and therefore fulfill criteria as highly promising vaccine immunogens.11,12 Several seminal studies have suggested the immunotherapeutic potential of neoantigens: (1) mice and humans can mount T-cell responses against mutated antigens13,14; (2) mice are tumor protected by immunization with a single mutated peptide15; and (3) memory cytotoxic T lymphocyte (CTL) responses to mutated antigens are generated in patients with unexpected long-term survival or those who have undergone effective immunotherapy.16-18 Neoantigens, however, have not been used for immunotherapy due to technical difficulties in their identification and preparation.13
Two recent technologies now overcome this limitation. First, massively parallel sequencing now readily provides the comprehensive identification of tens to thousands of somatic protein-coding mutations, which may create epitopes that can be recognized immunologically in an individual- and tumor-specific fashion.10,19 Second, refinements in class I HLA prediction algorithms have enabled the reliable prediction of peptide binding for a broad range of class I HLA alleles.20,21
Herein, we report that putative neoantigens identified through sequential application of massively parallel sequencing followed by HLA-binding prediction are immunogenic in humans and can target malignant cells in a tumor-specific fashion. We focused on chronic lymphocytic leukemia (CLL), a common adult B-cell malignancy that remains largely incurable but is potentially immune responsive based on reports of its spontaneous regression and susceptibility to the graft-versus-leukemia effect.22-24 We predicted candidate leukemia neoantigens from CLL DNA sequencing data25,26 and then monitored neoantigen-specific T-cell responses in patients who had undergone allogeneic-hematopoietic stem cell transplantation (allo-HSCT).27 Our approach provides a basis for designing truly personalized immunotherapeutic vaccines in humans.
Materials and methods
Patient samples
Heparinized blood was obtained from patients enrolled on clinical research protocols at the Dana-Farber Cancer Institute (DFCI). All clinical protocols were approved by the DFCI Human Subjects Protection Committee. The study was conducted in accordance with the Declaration of Helsinki. Patient peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll/Hypaque density-gradient centrifugation, cryopreserved with 10% dimethylsulfoxide, and stored in vapor-phase liquid nitrogen until time of analysis. HLA typing was performed by standard methods (supplemental Methods, available on the Blood Web site).
Whole-exome capture sequencing data for CLL and other cancers
The somatic mutations detected in CLL have been previously reported,26 whereas those for melanoma were obtained from the dbGaP database (phs000452.v1.p1) and for the 11 other cancers, through The Cancer Genome Atlas (Sage Bionetworks' Synapse resource; https://www.synapse.org/#!Synapse:syn1729383). We genotyped the HLA-A, -B, and -C loci in 2488 samples across 13 tumor types using a 2-stage likelihood-based approach (supplemental Methods; supplemental Table 9).
Prediction of peptides derived from gene mutations with binding to personal HLA alleles
Major histocompatibility complex (MHC)-binding affinity was predicted across all possible 9- and 10-mer peptides generated from each somatic mutation and the corresponding wild-type peptides using NetMHCpan (v2.4). These tiled peptides were analyzed for their binding affinities (IC50 nM) to each class I allele in the patients’ HLA profile. An IC50 value of <150 nM was considered a predicted strong binder; between 150 and 500 nM, an intermediate to weak binder; and >500 nM, a nonbinder. We empirically confirmed predicted peptides binding to HLA molecules (IC50 <500 nM) by competitive MHC class I allele-binding experiments.28,29
Generation and detection of patient antigen-specific T cells
Autologous dendritic cells (DCs) were generated as previously described.29 For some experiments, CD40L-Tri activated and expanded CD19+ B cells were used as antigen-presenting cells (APCs) (supplemental Methods).
To generate peptide-reactive T cells from CLL patients, immunomagnetically selected CD8+ T cells (10 million) from pre- and posttransplant PBMCs (CD8+ Microbeads; Miltenyi, Auburn, CA) were cultured with autologous peptide pool-pulsed DCs (at a 40:1 ratio). Subsequently, T cells were restimulated weekly (starting on day 7) with peptide-pulsed CD40L-Tri-activated irradiated B cells (at 4:1 ratio) either once more, to detect memory T-cell responses,27,30 or thrice more, to detect naïve T-cell responses.31 All stimulations were conducted in complete medium supplemented with 10% fetal bovine serum and 5 to 10 ng/mL interleukin (IL)-7, IL-12, and IL-15 (R&D Systems, Minneapolis, MN). APCs were pulsed with peptide pools (10 μM/peptide/pool for 3 hours). T-cell specificity against peptide pools or autologous tumor was tested by interferon (IFN)-γ ELISPOT or a CD107a degranulation assay (supplemental Methods) 10 days following the last stimulation.
Statistical considerations
Two-way analysis of variance models were constructed for cytokine secretion measurements and included concentration and mutational status as fixed effects along with an interaction term as appropriate. P values for these models were adjusted for multiple comparisons post hoc (Tukey method). For other comparisons of continuous measures between groups, a Welch t test was used. All other P values reported are 2-sided and considered significant at the .05 level with appropriate adjustment for multiple comparisons. Analyses were performed in SAS, version 9.2.
Results
Pipeline for the systematic identification of tumor neoantigens
We leveraged recent advances in sequencing technologies and peptide epitope prediction to establish a 2-step process for systematic discovery of candidate tumor-specific HLA-bound neoantigens. As depicted in Figure 1, we comprehensively identified tumor-specific nonsynonymous somatic mutations from DNA sequencing data of tumors (by whole-exome sequencing [WES] or whole-genome sequencing [WGS]) with matched sequencing of germ-line DNA.32,33 Next, we used NetMHCpan, the well-validated prediction algorithm, to predict candidate tumor-specific mutated peptides with the potential to bind personal class I HLA proteins and hence for presentation to CD8+ T cells.21,34 Finally, to identify a smaller number of epitopes for deeper investigation, we selected a subset of candidate peptide antigens based on experimental validation of their binding to HLA and expression of cognate mRNAs in autologous leukemia cells.
Figure 1.
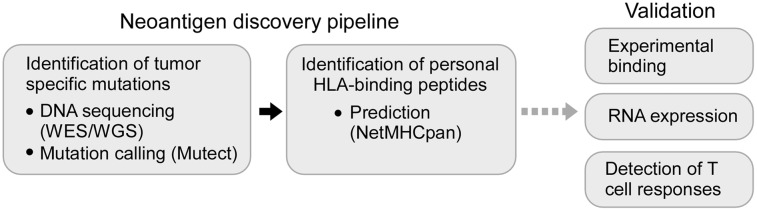
Schematic representation of a strategy to systematically discover tumor neoantigens. Tumor-specific mutations in cancer samples are detected using WES or WGS and identified through the application of mutation calling algorithms (such as Mutect).33 Next, candidate neoepitopes can be predicted using well-validated algorithms (NetMHCpan), and their identification can be refined by experimental validation for peptide-HLA binding and by confirmation of gene expression at the RNA level. These candidate neoantigens can be subsequently tested for their ability to stimulate tumor-specific T-cell responses.
Frequency of classes of somatic mutations in CLL
We applied this pipeline to a large dataset of 91 CLL samples,26 previously characterized by WES or WGS. From a total of 1838 nonsynonymous mutations previously discovered in protein-coding regions of these samples, we identified 3 general classes of mutations that could generate tumor-specific neoantigens. The most abundant class consisted of missense mutations that cause single amino acid changes and comprised 90% of somatic mutations per CLL, with 69% of 91 cases having between 10 and 25 missense mutations per sample (Figure 2A). The other 2 classes of mutations, frameshifts and splice-site mutations, have the potential to generate longer stretches of novel amino acid sequences entirely specific to the tumor (neo-open reading frames [neoORFs]), with a higher number of neoantigen peptides per alteration (compared with missense mutations). However, neoORF-generating mutations were ∼10-fold less abundant than missense mutations in CLL (Figure 2A-C). Given the prevalence of missense mutations, we focused our initial studies on the analysis of neoantigens generated from this mutation class.
Figure 2.

Frequency of classes of point mutations that have the potential to generate neoantigens in CLL. Analysis of WES and WGS data generated from 91 CLL cases26 reveals that (A) missense mutations are the most frequent class of the somatic alterations with the potential to generate neoepitopes, whereas (B) frameshifts and (C) splice-site mutations constitute rare events.
Predicted neopeptides that bind personal HLA class I alleles arising from somatic missense mutations
T-cell receptor (TCR) recognition of peptide epitopes requires the presentation of peptides by HLA molecules on the surface of APCs. We applied NetMHCpan prospectively to 31 of 91 CLL cases for which HLA typing information was available26 to predict the binding affinities of peptides generated by somatic mutation to each patient’s MHC class I alleles, because this algorithm consistently performs with high sensitivity and specificity across HLA alleles.21,35,36 Based on standard criteria in the field, we considered peptides with IC50 < 150 nM as strong binders, IC50 of 150 to 500 nM as intermediate to weak binders, and IC50 >500 nM as nonbinders.29 For the 31 cases, we found a median of 10 strong (range, 2-40) and 12 intermediate to weak binding peptides (range, 2-41) per case. In total, a median of 22 (range, 6-81) peptides per case was predicted with IC50 < 500 nM (Figure 3A; supplemental Table 1).
Figure 3.

Application of the NetMHCpan prediction algorithm to CLL cases. (A) Distribution of the number of predicted peptides with HLA binding affinity <150 (black) and 150 to 500 nM (gray) across 31 CLL patients with available HLA typing information (supplemental Table 1). (B) Peptides with predicted binding (IC50 <500 nM by NetMHCpan) from 4 patients were synthesized and tested for HLA-A and -B allele binding using a competitive MHC I allele-binding assay. The percent of predicted peptides with evidence of experimental binding (IC50 < 500 nM) are indicated. (C) The distribution of gene expression for all somatically mutated genes (n = 347) from 26 CLL patients and for the subset of gene mutations encoding neoepitopes with predicted HLA binding scores of IC50 <500 nM (n = 180). No-low, genes within the lowest quartile expression; medium, genes within the 2 middle quartiles of expression; high, genes within the highest quartile of expression.
Majority of predicted HLA-binding neopeptides directly bind HLA proteins in vitro
To experimentally validate the predicted IC50 nanomolar scores, we performed a competitive MHC I allele binding assay (supplemental Table 2)28 and focused on class I HLA-A and -B alleles. To this end, we synthesized 102 unique mutated 9- or 10-mer peptides with predicted IC50 < 500 nM (representing a total of 112 epitopes, as 10 peptides were predicted to bind to multiple alleles within the same patient), identified from 4 CLL cases (patients 1-4). Experimental binding (defined as IC50 <500 nM) was confirmed in 76.5% and 36% of the 112 peptides predicted with IC50 of < 150 or 150 to 500 nM, respectively (Figure 3B). In total, ∼55% (61 of 112) of predicted peptides were experimentally validated as binders to personal HLA alleles. Eighty percent of the 347 mutated genes (or 79% of the 180 mutations with predicted HLA binding) were expressed at medium or high expression levels (Figure 3C).
Detection of immunogenic neoepitopes personal to CLL patients
As proof of concept that an immune response against the predicted mutated peptides can develop in humans, we studied 2 patients who underwent reduced-intensity allogeneic HSCT in CLL and had achieved continuous remission for >4 years (supplemental Table 3). We reasoned that reconstitution of T cells from a healthy donor following HSCT would overcome endogenous immune defects of the host and allow priming against leukemia cells in the host in vivo. Posttransplant T cells were collected 7 (patient 1) and 4 years (patient 2) from the time of transplant.
For patient 1, we identified 25 missense mutations by WES. In total, 30 candidate epitopes (from 25 unique peptides, with 5 peptides predicted to bind to multiple HLA alleles) from 13 mutations were predicted to bind to personal HLA (13 peptides with IC50 < 150; 17 peptides with IC50 of 150-500 nM), with empirical confirmation of HLA binding for 13 candidate epitopes (10 unique peptides) derived from 8 mutations (Figure 4A). We organized all 30 predicted HLA binding peptides into 5 pools (6 peptides/pool) for T-cell priming studies (supplemental Table 4). To test T cells for neoantigen reactivity, we first expanded them using autologous APCs pulsed with candidate neoantigen peptide pools (once per week × 4). As shown in Figure 4B, reactivity was detected against pool 2 by IFN-γ ELISPOT, but not against an irrelevant HTLV1-Tax peptide. Deconvolution of the pool revealed that the mutated (mut) ALMS1 and C6ORF89 peptides within pool 2 were immunogenic (Figure 4C). ALMS1 plays a role in ciliary function, cellular quiescence, and intracellular transport,37,38 whereas C6ORF89 encodes a protein that interacts with bombesin receptor subtype 3 (involved in cell cycle progression and wound repair of bronchial epithelial cells).39,40
Figure 4.

Mutations in ALMS1 and C6ORF89 in patient 1 generate immunogenic peptides. (A) Twenty-five missense mutations were identified in patient 1 CLL cells, from which 30 putative epitopes (from 25 unique peptides; 5 peptides were predicted to bind to >1 HLA allele) from 13 mutations were predicted to bind to patient 1’s MHC class I alleles. A total of 13 peptides from 8 mutations were experimentally confirmed as HLA binding. Posttransplant T cells (7 years) from patient 1 were stimulated weekly ex vivo for 4 weeks with 5 pools of 6 mutated peptides per pool (supplemental Table 4), and subsequently tested by the IFN-γ ELISPOT assay. (B) Increased IFN-γ secretion by T cells was detected against pool 2 peptides. Negative control, irrelevant tax peptide; positive control, PHA. (C) Of pool 2 peptides, patient 1 T cells were reactive to mutated ALMS1 and C6ORF89 peptides (right, averaged results from duplicate wells are displayed). (Left) The predicted and experimental IC50 scores (nM) of mutated and wild-type ALMS1 and C6ORF89 peptides.
Detection of neoantigen-specific memory T-cell responses in patient 2
In patient 2, we tested whether personal neoantigens could contribute to memory T-cell responses in the setting of long-lived remission. From this individual, we identified 26 missense mutations. In total, 37 candidate epitopes (from 36 unique peptides; 1 peptide was predicted to bind to 2 different HLA alleles) from 16 mutations were predicted to bind to personal HLA alleles, of which 18 peptides (17 unique) from 12 mutations could be experimentally validated (15 with IC50 < 150; 3 with IC50 of 150-500 nM; Figure 5A). We studied all 18 experimentally validated HLA-binding peptides, and T-cell stimulations were performed using 3 pools of 6 peptides/pool (supplemental Methods; supplemental Table 5). We assessed memory responses by undertaking short-term stimulations of T cells in vitro (ie, 2 rounds of weekly stimulations of T cells against mutated peptide pool-pulsed autologous APCs),27,30 and identified T cells reactive against pool 1 (Figure 5B). Deconvolution of the pool revealed mut-FNDC3B as the dominant immunogenic peptide within this pool (experimental IC50 of mut- and wt-FNDC3B: 6.2 and 2.7 nM, respectively; Figure 5C). Presence of this population of neoepitope-reactive T cells was specific to patient 2 because a similar stimulation procedure applied to 3 unrelated HLA-A*02:01 healthy adult volunteers failed to generate detectable mut-FNDC3B peptide-specific T cells despite 4 rounds of weekly stimulations, although T-cell responses could be generated against the M1 (Flu peptide-GILGFVFTL) peptide (positive control) (supplemental Figure 1). The function of FNDC3B in blood malignancies is unclear, although down-regulation of its expression is known to up-regulate miR-143 expression, which differentiates prostate cancer stem cells and promotes prostate cancer metastasis.40,41
Figure 5.

Mutated FNDC3B generates a naturally immunogenic neoepitope in patient 2. (A) Twenty-six missense mutations were identified in patient 2 CLL cells, from which 37 epitopes (from 36 unique peptides; 1 peptide was predicted to bind to 2 HLA alleles) from 16 mutations were predicted to bind to patient 2’s MHC class I alleles. A total of 18 peptides from 12 mutations were experimentally confirmed to bind. Posttransplant T cells (∼4 years) from patient 2 were stimulated with autologous DCs or B cells pulsed with 3 pools of experimentally validated binding mutated peptides (18 peptides total) for 2 weeks ex vivo (supplemental Table 5). (B) Increased IFN-γ secretion was detected by ELISPOT assay in T cells stimulated with pool 1 peptides. (C) Of pool 1 peptides, increased IFN-γ secretion was detected against the mut-FNDC3B peptide (lower, averaged results from duplicate wells are displayed). (Upper) Predicted and experimental IC50 scores of mut- and wt-FNDC3B peptides. (D) T cells reactive to mut-FNDC3B demonstrate specificity to the mutated epitope but not the corresponding wild-type peptide (concentrations: 0.1-10 µg/mL) and are polyfunctional, secreting IFN-γ, GM-CSF, and IL-2 (Tukey post-hoc tests from 2-way analysis of variance modeling; mut vs wt). (E) (Left) Mut-FNDC3B-specific T cells are reactive in a class I-restricted manner and (right) recognize an endogenously processed and presented form of mutated FNDC3B, because they recognized HLA-A2 APCs transfected with a plasmid encoding a minigene of 300 bp encompassing the FNDC3B mutation (2-sided Welch t test) but not wild-type sequences beyond the background of nontransfected APCs alone. (Top right) Western blot analysis confirming expression of minigenes encoding mut- and wt-FNDC3B. (F) Patient 2 CD8+ T cells specifically recognizing mut-FNDC3B (5 μg/mL) demonstrate antitumor responses in an IFN-γ ELISPOT assay (2-sided Welch t test). (G) Specificity of T cells recognizing the mut-FNDC3B epitope detected by HLA-A2+/mut− FNDC3B tetramer-positive T cells in patient 2.
T-cell reactivity against mut-FNDC3B in patient 2 was polyfunctional (secreting IFN-γ, granulocyte macrophage–colony-stimulating factor [GM-CSF], and IL-2), highly avid, and specific to the mut-FNDC3B peptide but not its wild-type counterpart (Figure 5D). T-cell reactivity was abrogated by the presence of class I blocking antibody (W6/32), indicating that T-cell reactivity was class I restricted (Figure 5E, left). Moreover, the mut-FNDC3B peptide appeared to be naturally processed and presented because T-cell reactivity was detected against HLA-A2-expressing APCs that were transfected with a 300-bp minigene encompassing the region of gene mutation, but not the wild-type minigene or untransfected HLA-A2-expressing APCs (Figure 5E, right). Furthermore, mut-FNDC3B peptide-recognizing T cells from patient 2 were also reactive against autologous tumor cells, generating IFN-γ secretion at levels comparable to mutated peptide-pulsed targets (Figure 5F).
Long-lived neoantigen-specific antitumor T cells can be temporally tracked in CLL patient 2
Using a mut-FNDC3B/A2+-specific tetramer, we detected a discrete population of mut-FNDC3B-reactive CD8+ T cells within pool 1-stimulated T cells (2.42% of the population) compared with control PBMCs from a healthy adult HLA-A2+ volunteer (0.38%) (Figure 5G). Gene expression analysis and quantitative polymerase chain reaction (qPCR) of FNDC3B in CLL cases (including patient 2) and resting and CD40L-activated CD19+ B cells from healthy adult volunteers revealed this gene to be relatively overexpressed in patient 2 (supplemental Figure 2).
To define the relative kinetics of mut-FNDC3B-specific T cells in relationship to post-HSCT course, patient 2 T cells isolated from different time points before and after HSCT were stimulated in the same experiment for only 2 weeks and then tested for IFN-γ reactivity on ELISPOT. As shown in Figure 6A (top and middle panels), mut-FNDC3B T cell responses were not detected before or up to 3 months following HSCT. Molecular remission was first achieved 4 months following HSCT, and mut-FNDC3B-specific T cells were then first detected 6 months following HSCT, coinciding the molecular remission. Antigen-specific reactivity subsequently waned (between 12 and 20 months after HSCT) but was again strongly detected at 32 months after HSCT. Based on molecular analysis of the TCR of the mut-FNDC3B-specific T cells, we identified Vβ11 as the predominant CDR3 Vβ subfamily used by the reactive T cells (supplemental Figure 3; supplemental Table 6). Using this molecular information, we developed a clone-specific nested PCR and observed that T cells with the same specificity for mut-FNDC3B were not detected in PBMCs (n = 3) and CD8+ T cells of normal healthy volunteers (supplemental Table 7), but could be detected with similar kinetics as detection of IFN-γ secretion following HSCT in the patient (Figure 6A, bottom panel).
Figure 6.

Kinetics of the mut-FNDC3B-specific T-cell response in relation to posttransplant course and CD107a degranulation in the presence of tumor cells. (A) (Top) Molecular tumor burden was measured in patient 2 using a patient tumor-specific Taqman PCR assay based on the clonotypic IgH sequence at serial time points before and after HSCT (supplemental Methods). (Middle) Detection of mut-FNDC3B-reactive T cells in comparison with wt-FNDC3B or irrelevant peptides from peripheral blood before and after allo-HSCT by IFN-γ ELISPOT following stimulation with peptide-pulsed autologous B cells. The number of IFN-γ-secreting spots per cells at each time point was measured in triplicate (Welch t test; mut vs wt). (Bottom) Detection of mut-FNDC3B-specific TCR Vβ11 cells by nested clone-specific CDR3 PCR before and after HSCT in peripheral blood of patient 2 (supplemental Methods; supplemental Figure 3). Triangles, time points at which a sample was tested; black, amplification detected, where + indicates detectable amplification up to twofold and ++ indicates more than twofold greater amplification than the median level of all samples with detectable expression of the clone-specific Vβ11 sequence. (B) Evaluation of CD107a expression on 6m/32m mut-FNDC3B-reactive posttransplant CD8+ T cells (effectors) in the presence of patient 2 CLL cells or control donor-engrafted normal B cells in patient 2. The numbers indicate the percentage of mut-FNDC3B tetramer-positive cells that are also CD107a positive. For controls (tetramer negative T cells and nonrelated HLA-A*02:01 tumors), please see supplemental Figures 4 and 5.
Mut-FNDC3B T cells are cytolytic against autologous CLL
Consistent with a phenotype of degranulation, we observed higher levels of surface CD107a expression on mut-FNDC3B tetramer-positive T cells at 6 (by 32%) and 32 months (by 16%) after transplant following exposure to autologous leukemia cells compared with exposure to donor-engrafted normal B cells (Figure 6B). By contrast, we noted the absence of CD107a expression in the tetramer-negative CD8+ T cells (supplemental Figure 4). These results confirm that CD107a-mediated degranulation was directed against a tumor neoepitope (mut-FNDC3B) and not an alloantigen. The total percentage of CD8+ T cells expressing CD107a was ∼5% higher following stimulation with patient 2 tumor cells than with donor-engrafted normal B cells (representing background reactivity) or with 3 other HLA-A*02:01-positive CLL cells (supplemental Figure 5). These data demonstrate that the cytolytic capacity of mut-FNDC3B T cells against autologous CLL was personal and preserved over time.
Large numbers of candidate neoantigens were predicted across diverse cancers
To examine how tumor type and mutation rate could impact the overall estimate of abundance of candidate neoantigens, we applied our pipeline to publicly available WES data from 13 malignancies, whose overall somatic mutation rates have been previously reported.32 To do so, we further implemented a recently developed algorithm that enables accurate inference of HLA typing from WES data (supplemental Methods).42
We predicted neoantigen loads from missense and frameshift mutations across tumors, setting an IC50 threshold of <500 nM. Commensurate with the high mutation rate of melanoma, we observed an ∼20-fold higher number of predicted neoantigens per melanoma case (488; range, 18-5811), compared with CLL (24; range, 2-124) (Figure 7; supplemental Table 8). Our analysis supports the idea that the median number of predicted neopeptides generated from missense and frameshift events per sample is proportional to the mutation rate.
Figure 7.

Estimation of tumor neoantigen load across cancers. (A) Box plots comparing overall somatic mutation rates detected across cancers by massively parallel sequencing. AML, acute myeloid leukemia; DLBCL, diffused large B-cell lymphoma; ESO AD, esophageal adenocarcinoma; GBM, glioblastoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; clear cell RCC, clear cell renal carcinoma; papillary RCC, papillary renal cell carcinoma. The distributions (shown by box plot) of (B) the number of missense, frameshift, and splice-site mutations per case across 13 cancers, (C) the summed neoORF length generated per sample and of the predicted neopeptides with (D) IC50 <150 and (E) <500 nM generated from missense and frameshift mutations. For all panels, the left and right ends of the boxes represent the 25th and 75th percentile values, respectively, and the segment in the middle is the median. The left and right extremes of the bars extend to the minimum and maximum values.
Discussion
Each individual tumor harbors a broad spectrum of shared and personal genetic alterations that continue to evolve in response to the environment and often lead to therapeutic resistance.10 Given the uniqueness and plasticity of tumors, an optimal therapy may need to be customized based on the exact mutations present in each tumor and may need to target multiple nodes to avoid resistance.43-46 The immune system of each individual, with its vast repertoire of CTLs, possesses this potential to simultaneously target multiple, personalized mutations (neoantigens). In a seminal and elegant study of a long-term melanoma survivor, Lennerz et al found that CTLs targeting neoantigens are significantly more abundant and sustained than those against nonmutated overexpressed tumor antigens.16 Since then, many studies in human18,36,47-50 and murine51-53 solid tumors support the critical role of protective immunity targeting neoantigens specifically expressed on the surface of tumor cells. Leveraging massively parallel sequencing and algorithms that effectively predict HLA-binding peptides, we report herein a comprehensive strategy to systematically identify potential immunogenic neoantigens across cancers.
We applied our strategy to a unique group of CLL patients who developed clinically evident durable remission associated with antitumor immune responses following allo-HSCT. These graft-versus-leukemia responses have typically been attributed to allo-reactive immune responses targeting hematopoietic cells,54 but recent studies by our group and others have demonstrated the existence of graft-versus-leukemia-associated CTLs with specificity for tumor rather than allo-antigens.27,55,56 In 2 patients in clinical remission for >4 years, we now show that peripheral blood-derived T cells recognizing 3 predicted neoantigen peptides can be stimulated and observed. For 1 of these neoepitopes (mutated FNDC3B), identified following only 2 weeks of in vitro stimulation, these T cells are (1) stimulated by the mutated but not by the cognate native peptide (Figure 5D), (2) recognize autologous APCs transfected with a minigene encoding a portion of the FNDC3B gene including the mutation (Figure 5E), and (3) recognize autologous tumor cells and peptide-pulsed autologous B cells (Figure 5F). Clinically, the level of this T-cell population correlated with disease remission as measured both by ELISPOT analysis and qPCR of the TCR associated with this target (Figure 6A), and these T cells were induced to express CD107a, a marker of cytotoxic potential, following exposure to patient CLL cells (Figure 6B). All these features are consistent with the role of neoantigens in protective immunity in solid tumors as discussed above, but now are extended to a hematological malignancy with a low mutation rate. Our results and the results of others, together with our pipeline, provide a method for selecting neoantigens for future personalized vaccines broadly applicable across tumors.
More generally, we estimated the abundance of neoantigens across many tumors (and a broad array of HLA alleles) and found ∼1.5 HLA-binding peptides with IC50 < 500 nM per point mutation and ∼4 binding peptides per frameshift mutation. As expected, the estimated rate of predicted HLA binding peptides mirrored the somatic mutation rate per tumor type (Figure 7), wherein melanoma has the potential to generate the highest number of neoantigens and hematological malignancies including CLL generate less abundant neoantigens.
We recently reported the retrospective application of our pipeline to published immunogenic tumor neoantigens (ie, in which CTLs reactive to the mutated peptide were observed in patients) demonstrating that the vast majority (97%) of functional neoantigens are predicted to bind HLA with IC50 <500 nM (with ∼70% of wild-type counterpart epitopes predicted to bind at a similar affinity).36 This test using a gold standard set of neoantigens confirms that our pipeline largely classifies true positives correctly. A prospective prediction of neoepitopes followed by functional validation showed that 6% (3/48) of our predicted epitopes were associated with neoantigen-specific T-cell responses in patients—comparable to the rate of 4.8% found recently for melanoma.47 The low proportion does not necessarily imply low prediction accuracy for the algorithm. Rather, number of true neoantigens is greatly underestimated by our experiments because (1) allo-HSCT is a general cellular therapy likely to induce only a small number of neoantigen-specific T-cell memory clones; and (2) our T-cell expansion methods are not sensitive enough to detect naïve T cells that represent a much larger part of the repertoire but with much lower precursor frequencies. Beyond the issue of prediction accuracy, we have also not yet measured the frequency of CTLs that target neoORFs—a class of neoantigens that we expect to be more specific (for lack of a wild-type counterpart) and immunogenic (as a result of bypassing thymic tolerance). Future improvements in predictive algorithms, and eventually, development of direct physical methods for detecting HLA-binding peptides using mass spectrometry,57-59 will make it possible to more effectively select neoantigens presented by tumor HLA proteins. Considering the limitations described here, we conclude that there will be many more neoantigens per tumor.
Given the large number of candidate neoantigens, are there additional considerations that could help select the most useful antigens for targeting tumors? First, is it important to have high RNA or protein expression for CTLs to detect HLA-peptide complexes? Although no empirical studies have tested this question in human trials, it appears that very low levels (ie, even a single peptide-HLA protein) may be sufficient for a cell to be targeted by a CTL,60 suggesting that high expression may not be required for inclusion of a neoantigen in a vaccine. Second, although theory suggests that essential cancer genes (drivers) are less likely to develop resistance under immune pressure, the 3 neoantigens (mutated FND3CB, ALMS1, and C6ORF89) found in our study exhibit attributes of passenger mutations as they are not evolutionarily conserved among species, and the mutation site has not been previously reported in other cancers (supplemental Figures 6 and 7). We found that the 3 CLL neoantigens were clonal or near-clonal (data not shown).25,61 We thus suggest that CTLs associated with clinical responses can target tumor-specific mutations that are present in the bulk of the cancer mass (whether recurrent or not).
It is becoming increasingly evident that targeting multiple personal tumor epitopes is likely a useful therapeutic approach. In a recent study of the B16 murine melanoma model, HLA-binding neoantigens were predicted based on tumor mutations. Mice that were immunized with the corresponding mutated peptides developed CTLs specific to the mutated but not wild-type peptide and controlled disease therapeutically and prophylactically.51 In addition, recent studies in mice showing immune editing and escape to dominant CTL responses support the notion of targeting multiple epitopes to avoid resistance.53,62 Focused prospective trials on small patient cohorts should now be possible to design based on our pipeline for selecting neoantigens to directly test the hypothesis that neoantigens are true targets of rejection responses and, due to their specificity, can be targeted both efficiently and safely by personalized immunotherapy.
Acknowledgments
The authors thank all the members of the Broad Institute’s Biological Samples, Genetic Analysis, and Genome Sequencing Platforms, who made this work possible. The authors also thank the expert clinical care provided by the DFCI clinical transplant team and assistance from the DFCI Cell Manipulation Core Facility, Pasquarello Tissue Bank. The authors also thank Jaewon Choi and Jessica Wong for excellent technical assistance and Glenn Dranoff, Todd Carter, Ute Burkhardt, and Ursula Hainz for valuable discussions.
This research was supported in parts by National Institutes of Health, National Human Genome Research Institute grant U54HG003067, National Cancer Institute grant 1RO1CA155010-02, National Heart, Lung, and Blood Institute grant 5R01HL103532-03, the Blavatinik Family Foundation, and the Leukemia and Lymphoma Translational Research Program. C.J.W. is a recipient of an Innovative Research Grant for Stand-Up to Cancer/American Association of Cancer Research.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.R. directed the study design and performed experimental and data analysis with guidance from C.J.W. S.A.S., V.B., E.F.F., and D.D. developed the neoantigen discovery pipeline; J.R. provided clinical specimens for the study; E.C., W.Z., and D.B.K. contributed to the T-cell experiments; C.S., K.C., G.G., and S.G. coordinated the tumor sequencing and somatic mutation analysis; J.S. performed the experimental binding assays; K.S. and D.N. performed statistical analysis; M.R. wrote the manuscript; C.J.W., N.H., E.F.F., and E.S.L. edited the manuscript; and all authors discussed and interpreted results.
Conflict-of-interest disclosure: Patent applications have been filed on aspects of the work described in the paper entitled as follows: Compositions and Methods for Personalized Neoplasia Vaccines (N.H., E.F.F., and C.J.W.) and Methods for Identifying Tumor Specific Neo-Antigens (N.H. and C.J.W.) (PCT/US2011/036665). The remaining authors declare no competing financial interests.
Correspondence: Catherine J. Wu, Dana-Farber Cancer Institute, 450 Brookline Ave, Dana 540B, Boston, MA 02215; e-mail: cwu@partners.org.
References
- 1.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taneja SS. Re: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. J Urol. 2012;188(6):2149. [PubMed] [Google Scholar]
- 4.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Burg SH, Melief CJ. Therapeutic vaccination against human papilloma virus induced malignancies. Curr Opin Immunol. 2011;23(2):252–257. doi: 10.1016/j.coi.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Nedergaard MK, Hedegaard CJ, Poulsen HS. Targeting the epidermal growth factor receptor in solid tumor malignancies. BioDrugs. 2012;26(2):83–99. doi: 10.2165/11599760-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Baskar S, Kobrin CB, Kwak LW. Autologous lymphoma vaccines induce human T cell responses against multiple, unique epitopes. J Clin Invest. 2004;113(10):1498–1510. doi: 10.1172/JCI20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen-based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1(1):11–15. doi: 10.1158/2326-6066.CIR-13-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J. 2013;32(2):194–203. doi: 10.1038/emboj.2012.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sensi M, Anichini A. Unique tumor antigens: evidence for immune control of genome integrity and immunogenic targets for T cell-mediated patient-specific immunotherapy. Clin Cancer Res. 2006;12(17):5023–5032. doi: 10.1158/1078-0432.CCR-05-2682. [DOI] [PubMed] [Google Scholar]
- 14.Parmiani G, De Filippo A, Novellino L, Castelli C. Unique human tumor antigens: immunobiology and use in clinical trials. J Immunol. 2007;178(4):1975–1979. doi: 10.4049/jimmunol.178.4.1975. [DOI] [PubMed] [Google Scholar]
- 15.Mandelboim O, Vadai E, Fridkin M, et al. Regression of established murine carcinoma metastases following vaccination with tumour-associated antigen peptides. Nat Med. 1995;1(11):1179–1183. doi: 10.1038/nm1195-1179. [DOI] [PubMed] [Google Scholar]
- 16.Lennerz V, Fatho M, Gentilini C, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A. 2005;102(44):16013–16018. doi: 10.1073/pnas.0500090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J, Dudley ME, Rosenberg SA, Robbins PF. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother. 2005;28(1):53–62. doi: 10.1097/00002371-200501000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–e442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin HH, Ray S, Tongchusak S, Reinherz EL, Brusic V. Evaluation of MHC class I peptide binding prediction servers: applications for vaccine research. BMC Immunol. 2008;9:8. doi: 10.1186/1471-2172-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang GL, Ansari HR, Bradley P, et al. Machine learning competition in immunology - Prediction of HLA class I binding peptides. J Immunol Methods. 2011;374(1-2):1–4. doi: 10.1016/j.jim.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Dores GM, Anderson WF, Curtis RE, et al. Chronic lymphocytic leukaemia and small lymphocytic lymphoma: overview of the descriptive epidemiology. Br J Haematol. 2007;139(5):809–819. doi: 10.1111/j.1365-2141.2007.06856.x. [DOI] [PubMed] [Google Scholar]
- 23.Zenz T, Mertens D, Küppers R, Döhner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10(1):37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 24.Del Giudice I, Chiaretti S, Tavolaro S, et al. Spontaneous regression of chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood. 2009;114(3):638–646. doi: 10.1182/blood-2008-12-196568. [DOI] [PubMed] [Google Scholar]
- 25.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkhardt UE, Hainz U, Stevenson K, et al. Autologous CLL cell vaccination early after transplant induces leukemia-specific T cells. J Clin Invest. 2013;123(9):3756–3765. doi: 10.1172/JCI69098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidney J, Southwood S, Oseroff C, del Guercio MF, Sette A, Grey HM. Measurement of MHC/peptide interactions by gel filtration. Curr Protoc Immunol; 2001. Chapter 18:Unit 18-13. [DOI] [PubMed] [Google Scholar]
- 29.Cai A, Keskin DB, DeLuca DS, et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin Cancer Res. 2012;18(20):5761–5772. doi: 10.1158/1078-0432.CCR-12-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jäger E, Gnjatic S, Nagata Y, et al. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci U S A. 2000;97(22):12198–12203. doi: 10.1073/pnas.220413497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardwick N, Buchan S, Ingram W, et al. An analogue peptide from the Cancer/Testis antigen PASD1 induces CD8+ T cell responses against naturally processed peptide. Cancer Immun. 2013;13:16. [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundegaard C, Lund O, Nielsen M. Prediction of epitopes using neural network based methods. J Immunol Methods. 2011;374(1-2):26–34. doi: 10.1016/j.jim.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sette A, Vitiello A, Reherman B, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol. 1994;153(12):5586–5592. [PubMed] [Google Scholar]
- 36.Fritsch ED, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans [published online ahead of print March 3, 2014]. Cancer Immunol Res. [DOI] [PMC free article] [PubMed]
- 37.Collin GB, Cyr E, Bronson R, et al. Alms1-disrupted mice recapitulate human Alström syndrome. Hum Mol Genet. 2005;14(16):2323–2333. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collin GB, Marshall JD, Ikeda A, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat Genet. 2002;31(1):74–78. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 39.Liu HJ, Tan YR, Li ML, Liu C, Xiang Y, Qin XQ. Cloning of a novel protein interacting with BRS-3 and its effects in wound repair of bronchial epithelial cells. PLoS ONE. 2011;6(8):e23072. doi: 10.1371/journal.pone.0023072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forbes SA, Tang G, Bindal N, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38(Database issue):D652–D657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai C, Rajaram M, Zhou X, et al. Activation of multiple cancer pathways and tumor maintenance function of the 3q amplified oncogene FNDC3B. Cell Cycle. 2012;11(9):1773–1781. doi: 10.4161/cc.20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla SA, Rajasagi M, Dixon PM, et al. Sensitive detection of somatic class I HLA mutations using exome capture sequencing. (Submitted)
- 43.Ene CI, Fine HA. Many tumors in one: a daunting therapeutic prospect. Cancer Cell. 2011;20(6):695–697. doi: 10.1016/j.ccr.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 44.Longo DL. Tumor heterogeneity and personalized medicine. N Engl J Med. 2012;366(10):956–957. doi: 10.1056/NEJMe1200656. [DOI] [PubMed] [Google Scholar]
- 45.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu YC, Yao X, Li YF, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. 2013;190(12):6034–6042. doi: 10.4049/jimmunol.1202830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown SD, Warren RL, Gibb EA, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743–750. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–1091. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 52.Dubey P, Hendrickson RC, Meredith SC, et al. The immunodominant antigen of an ultraviolet-induced regressor tumor is generated by a somatic point mutation in the DEAD box helicase p68. J Exp Med. 1997;185(4):695–705. doi: 10.1084/jem.185.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer. 2004;4(5):371–380. doi: 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- 55.Nishida T, Hudecek M, Kostic A, et al. Development of tumor-reactive T cells after nonmyeloablative allogeneic hematopoietic stem cell transplant for chronic lymphocytic leukemia. Clin Cancer Res. 2009;15(14):4759–4768. doi: 10.1158/1078-0432.CCR-09-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang W, Choi J, Zeng W, et al. Graft-versus-leukemia antigen CML66 elicits coordinated B-cell and T-cell immunity after donor lymphocyte infusion. Clin Cancer Res. 2010;16(10):2729–2739. doi: 10.1158/1078-0432.CCR-10-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rammensee HG, Singh-Jasuja H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev Vaccines. 2013;12(10):1211–1217. doi: 10.1586/14760584.2013.836911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stickel JS, Weinzierl AO, Hillen N, et al. HLA ligand profiles of primary renal cell carcinoma maintained in metastases. Cancer Immunol Immunother. 2009;58(9):1407–1417. doi: 10.1007/s00262-008-0655-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neumann A, Hörzer H, Hillen N, et al. Identification of HLA ligands and T-cell epitopes for immunotherapy of lung cancer. Cancer Immunol Immunother. 2013;62(9):1485–1497. doi: 10.1007/s00262-013-1454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity. 1996;4(6):565–571. doi: 10.1016/s1074-7613(00)80483-5. [DOI] [PubMed] [Google Scholar]
- 61.Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482(7385):405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]