

Abstract
The global burden of advanced stage cervical cancer remains significant, particular in resource poor countries where effective screening programs are absent. Unfortunately, a proportion of patients will be diagnosed with advanced stage disease, and may suffer from persistent or recurrent disease despite treatment with combination chemotherapy and radiation. Patients with recurrent disease have a poor salvage rate, with an expected 5-year survival of less than 10%. Recently, significant gains have been made in the antiangiogenic arena; nonetheless the need to develop effective alternate targeted strategies is implicit. As such, a review of molecular targeted therapy in the treatment of this disease is warranted. In an era of biologics, combined therapy with cytotoxic drugs and molecular targeted agents, represents an exciting arena yet to be fully explored.
Keywords: Angiogenesis, Recurrent cervical cancer, Targeted therapy
INTRODUCTION
In 2011 an estimated 529,800 cases of cervical cancer were diagnosed worldwide, with 275,100 deaths [1]. This global burden is attributable to the disproportionately high incidence of cervical cancer in developing, resource poor countries lacking adequate health care infrastructure and screening programs. Regionally, in the United States, an estimated 12,360 cases will be diagnosed in 2014, with 4,020 deaths; it is anticipated that this number will continue to decline as human papilloma virus (HPV) vaccination rates rise, and the focus shifts to primary prevention [2]. Despite advances in screening, vaccination and treatment of early stage disease, a proportion of patients will be diagnosed with advanced stage (stage IVB), recurrent or persistent cervical cancer. Specifically, patients with International Federation of Obstetrics and Gynecology (FIGO) stage IB-IIA disease have a recurrence risk ranging from 10% to 20% despite primary chemo-radiation, while those patients diagnosed FIGO stage IIB-IVA have a 50% to 70% chance of disease recurrence [3]. For this subset of patients, systemic chemotherapy remains the standard treatment.
In the setting of advanced stage or recurrent disease cure is exceedingly rare, and goals of care are centered on palliation of symptoms, and control of disease burden [4]. Since the publication of the initial studies exploring single agent cisplatin in the treatment of cervical cancer, a number of effective drugs have been identified, including paclitaxel, ifosfamide and topotecan, although none have exhibited significant gains with respect to overall survival (OS) [5,6,7,8,9,10,11,12,13,14,15,16]. Ultimately, various combination-based regimens were investigated, with modest gains in response rate, which analogously failed to translate into a meaningful OS advantage [17,18,19,20,21]. Importantly, the combination regimen of cisplatin and paclitaxel has been established as the backbone for future chemotherapy trials, although OS approaches only 13 months [5]. Additionally, responses to this regimen are uniformly temporary, and effective second and third line therapeutic regimens are lacking.
The poor oncologic outcome in this patient population represents and unmet clinical need, and catalyzed the exploration of novel treatment paradigms. Most recently, the results of Gynecologic Oncology Group (GOG) protocol 240 were presented and published, illustrating a 3.7 months improvement in OS with the incorporation of the antiangiogenic agent bevacizumab to a chemotherapy backbone [22]. In an era of molecular medicine, the development of additional biologic therapies, to be used solely or in conjunction with cytotoxic chemotherapy, is implicit. Currently, a number of biologic agents targeting various molecular pathways including epidermal growth factor receptor (EGFR), histone deacetylase, matrix metalloproteinase, check point inhibitors, Notch pathway, and mammalian target of rapamycin (mTOR) are under clinical development (Table 1). This article will explore molecularly targeted drugs developed for the treatment of cervical cancer.
Table 1.
Molecular pathways targeted in cervical cancer
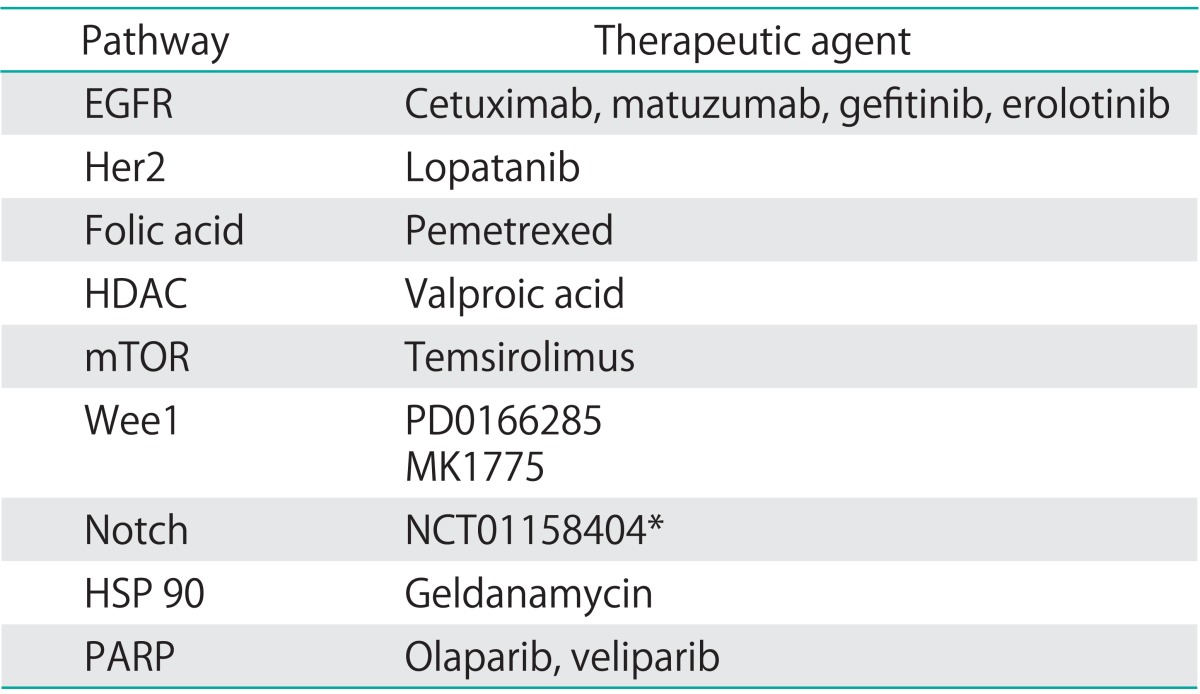
EGFR, epidermal growth factor receptor; HDAC, histone deacetylase; Her2, human epidermal growth factor receptor 2; HSP, heat shock protein; mTOR, mammalian target or rapamycin; PARP, poly-ADP ribose polymerase; Wee1, nuclear kinase belonging to serine/threonine family of protein kinases.
*Clinical trial reference number for HSP inhibitor in cervical cancer.
NOVEL NONANGIOGENIC TARGETED THERAPIES IN CERVICAL CANCER
1. Targeting EGFR and Her2
The EGFR is a trans membrane protein involved in signaling pathways critical for cell survival (Fig. 1) [23]. EGFR overexpression has been shown to correlate with resistance to cytotoxic therapy and radiation in squamous cell cancers [24,25,26,27]. Additionally, the majority of cervical cancer patients (54% to 71%) exhibit EGFR expression, with correlative studies associating increased expression with prognosis and tumor aggressiveness [28]. More importantly, the addition of cetuximab to radiotherapy in the treatment of head and neck squamous cancers resulted in a statistically significant prolongation in OS and locoregional control at 2 years, providing the biologic rational for study in cervical cancer [29].
Fig. 1.

Cytoplasmic/traditional and nuclear modes of the epidermal growth factor receptor (EGFR) signalling pathway. The EGFR signalling pathway exerts its biological effects via two major modes of actions, namely, cytoplasmic/traditional (A) and nuclear (B) modes. (A) The cytoplasmic EGFR pathway is consisted of four major modules: PLC-γ-CaMK/PKC, Ras-Raf-MAPK, PI-3K-Akt-GSK and signal transducer and activator of transcriptions (STATs). Activation of these signalling modules often leads to tumorigenesis, tumour proliferation, metastasis, chemoresistance and radioresistance. (B) The nuclear EGFR pathway can be initiated by ligand binding and exposure to vitamin D, radiation, cisplatin, heat and H2O2. Following nuclear translocalization, nuclear EGFR interacts with DNA-binding transcription factors, E2F1 and STAT3, and activates expression of B-Myb and inducible nitric oxide synthase (iNOS), respectively. Nuclear EGFR also upregulates cyclin D1 gene expression. Increased expression of cyclin D1 and B-Myb contributes to accelerated G1/S cell cycle progression and, on the other hand, elevated iNOS is associated with tumour proliferation and metastasis. Upon DNA damage and oxidative/heat stress, EGFR enters the cell nucleus and interacts with DNA-PK, leading to DNA repair and radioresistance. Reprinted from Lo and Hung [23].
To date 2 anti-EGFR antibodies have been developed and studied in cervical cancer: cetuximab and matuzumab [28]. Cetuximab, is a chimeric immunoglobulin G2 monoclonal antibody that targets the extracellular domain of the EGFR. Alternatively, matuzumab is a humanized immunoglobulin G1 monoclonal anti-EGFR antibody.
A total of four studies were completed, reporting on the safety an efficacy of cetuximab in the treatment of cervical cancer. Two studies used single agent cetuximab [30,31], while one combined the monoclonal antibody with cisplatin [32], and the final study combined cetuximab with cisplatin and topotecan [33].
Unfortunately, response rates were less than anticipated during trial design. When used as a single agent, cetuximab failed to result in a clinical response [30]. The median progression free survival (PFS) and OS were 1.9 and 6.7 months, respectively. Combining cetuximab with cisplatin did not translate into improved outcomes. Farley et al. [32] investigated cetuximab at a loading dose of 400 mg/m2 followed by 250 mg/m2 on days 1, 8, and 15 every 21 days in combination with cisplatin 30 mg/m2 on day 1 and 8. Sixty-nine patients with advanced, persistent or recurrent cervical cancer were eligible and evaluable, and the clinical objective response rate was 11%. The authors concluded that there was little or no evidence that cetuximab was beneficial in this patient population beyond single agent cisplatin.
Lastly, a 3-drug combination of cetuximab, cisplatin, and topotecan was studied in patients with advanced cervical cancer not amenable to curative treatment [33]. This study was terminated early, after only 19 subjects enrolled, due to unacceptable toxicity, with three treatment related deaths.
Matuzumab has also been studied in patients with cervical cancer progressing after treatment with platinum-based chemotherapy, with data presented in abstract form at the 2005 ASCO Annual Meeting. Amongst 38 evaluable patients, there were two partial responses and nine patients with stable disease. Reported grade 3/4 AEs included hepatotoxicity, diarrhea, fainting, anorexia, fatigue, abdominal pain, and pancreatitis (1 of each) [28].
Analogous to receptor tyrosine kinase inhibition in the angiogenic cascade, investigators explored anti-EGFR tyrosine-kinase inhibitors (TKIs) in the treatment of cervical cancer (Table 2) [30,32,33,34,35,36]. EGFR signal transduction is dependent on receptor auto-phosphorylation followed by signal transduction. Inhibition via small molecule TKIs results in inhibition of phosphorylation and interruption of signal transduction.
Table 2.
EGFR inhibition in the treatment of cervical cancer
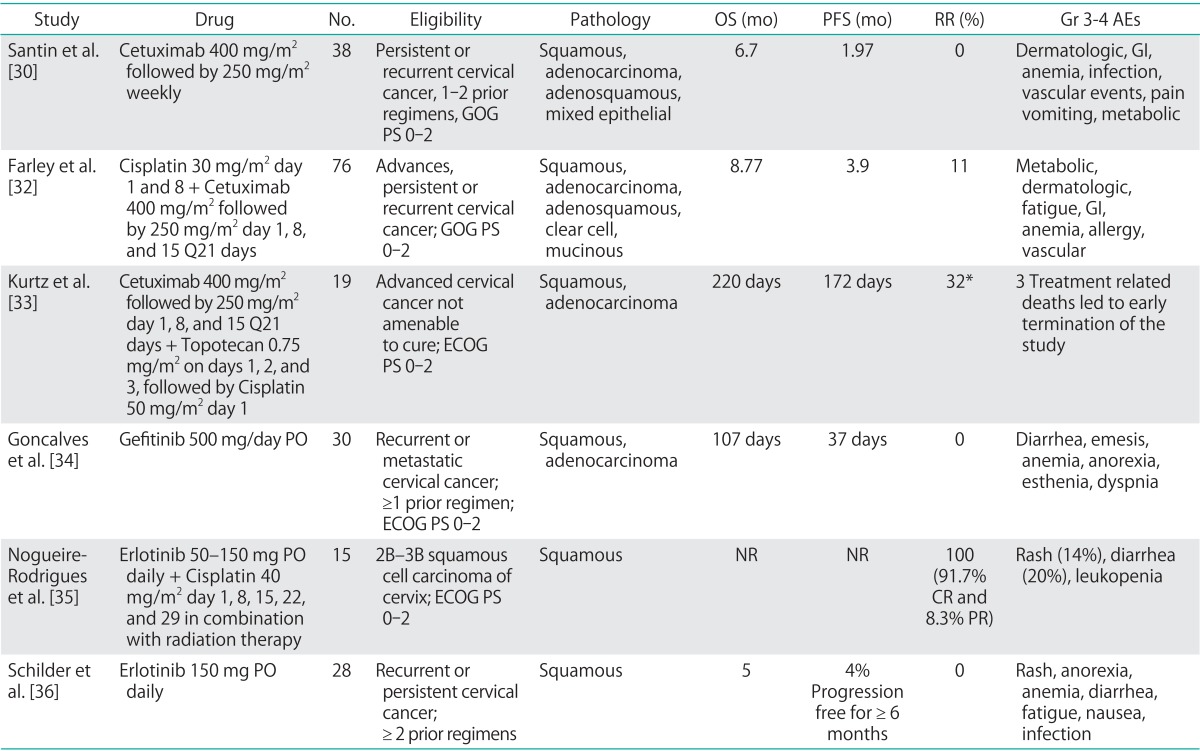
AE, adverse events; CR, complete response; ECOG, European College of Obstetrics and Gynecology; EGFR, epidermal growth factor receptor; GI, gastrointestinal; GOG, Gynecologic Oncology Group; Gr, grade; NR, not reported; OS, overall survival; PFS, progression free survival; PO, per oral; PR, partial response; PS, performance status; RR, response rate.
*Intent to treat analysis in the 19 subjects enrolled on study.
Three trials have reported outcomes in patients with cervical cancer treated using anti-EGFR TKI's. Two, phase 2 trials investigated the use of oral, single agent gefitinib and erlotinib in patients with recurrent, persistent or metastatic cervical cancer, receiving at least one prior cytotoxic chemotherapy regimen [34,36]. In both studies, there was a lack of an observed objective response, signaling that these small molecule TKIs were unlikely to exhibit clinical activity as single agents in the advanced/recurrent setting. An additional phase 1 study, explored the combination of erlotinib orally with concurrent cisplatin+external beam radiation and brachytherapy. A total of 15 patients with clinical stage 2B-3B squamous cell carcinoma of the cervix were enrolled on trial. The authors reported a 91.7% complete response rate and an 8.3% partial response rate. No data on long-term outcome was provided. The most commonly reported grade 3/4 AEs associated with this class of targeted agents include: rash, nausea, anorexia, diarrhea, anemia, fatigue, and infection.
The only published study to date exploring Her2/neu inhibition in the treatment of cervical cancer was previously reviewed [37] (Table 2). Amongst the human EGFR (HER) family, HER2 is unique in that it exists in a constitutively active form. Dimerization results in receptor activation, phosphorylation and down stream signaling with oncogenic gene transcription. Prior studies indicated an association between increased Her2 expression and improved prognosis in cervical cancer patients [38]. Unfortunately, lapatinib was found to have only modest activity, with a 5% objective response rate, and was associated with an up to 13% rate of grade 3/4 AEs.
2. Antifolate agents
More recently, a novel chemotherapeutic combination regimen of pemetrexed (an anti-folate, which disrupts folate-dependent metabolic processes) and cisplatin was investigated in patients with cervical carcinoma. Initial phase I trials of single agent pemetrexed completed in South Africa indicated a 21% response rate in chemotherapy naïve cervical cancer patients. Recently, GOG 127T, a phase II trial evaluating pemetrexed in the treatment of recurrent cervical carcinoma in patients who failed prior chemotherapy, completed accrual. It was well tolerated and demonstrated a 15% response rate.
Given the above, protocol 77GG was developed, exploring the combination regimen of cisplatin 50 mg/m2+pemetrexed 500 mg/m2 in patients with recurrent, metastatic cervical cancer. The results were presented at the ASCO Annual Meeting in June 2013. Patients had received no prior therapeutic chemotherapy aside from concurrent with primary radiation therapy. From September 2008 to November 2011, five GOG member institutions enrolled 55 patients. Fourteen of the enrolled subjects (29%) received greater than nine cycles. The most common grade two toxicities included neutropenia (35%), leukopenia (28%), and metabolic (28%). There were one complete and 16 partial responses (RR 31%). Median response duration was 7 months and survival 12 months.
3. Histone deacetylase inhibitors in cervical cancer
Over the last several years significant interest in epigenetic modifications resulting in alteration of tumor suppressor gene expression has catalyzed the investigation of novel treatment strategies using histone deacetylase inhibitors (HDACIs). In its transcriptionally silent state, chromatin is composed of nucleosomes in which the histones have low level acetylation [39]. Acetylation of histone proteins subsequently results in relative charge neutrality, allowing for DNA unfolding and transcriptional access. HDACs remove the acetyl groups, returning an overall positive charge, which is thought to inhibit transcription of tumor suppressor genes in various malignancies [39,40,41].
The most extensively studied HDACI in cervical carcinoma is a drug commonly used in the treatment of epilepsy, bipolar disorder, and major depression, valproic acid. Valproic acid acts as a specific inhibitor of class I HDACs and induces proteosomal degradation of HDAC2, leading to cell differentiation, growth arrest and death in vitro and in vivo [42]. To date, four studies have reported on the effects of HDACI on oncologic outcome in patients with cervical cancer.
In the primary setting, Chavez-Blanco et al. [43] conducted a phase I study exploring the impact of magnesium valproate use on histone acetylation in 12 patients with stage 2B to 4B cervical carcinoma. All subjects were treated with magnesium valproate after a baseline tumor biopsy and blood sampling at the following dose levels (four patients each): 20, 30, or 40 mg/kg for 5 days via oral route. At day 6, tumor and blood sampling were repeated and the study protocol ended. Tumor acetylation of H3 and H4 histones and HDAC activity were evaluated by Western blot and colorimetric HDAC assay respectively. Blood levels of valproic acid were determined at day 6 once the steady state was reached.
Ten patients were evaluated for H3 and H4 acetylation and HDAC activity. After treatment, investigators observed hyper-acetylation of H3 and H4 in the tumors of nine and seven patients, respectively, whereas 6 patients demonstrated hyperacetylation of both histones. Serum levels of valproic acid ranged from 73.6 to 170.49 mg/mL. Tumor deacetylase activity decreased in eight patients (80%), whereas two had either no change or a mild increase. There was a statistically significant difference between pre- and posttreatment values of HDAC activity (mean, 0.36 vs. 0.21; two-tailed t-test p<0.0264). There was no correlation between H3 and H4 tumor hyperacetylation with serum levels of valproic acid. The authors concluded that magnesium valproate at a dose between 20 and 40 mg/kg inhibited deacetylase activity and hyperacetylated histones in tumor tissues.
The combined use of hydralazine, a DNA methyltransferase inhibitor, and valproic acid has also been studied in a double-blind randomized phase 3 trial [44]. DNA demethylation results in reactivation and expression of tumor suppressor genes, which was hypothesized to synergize with HDAC inhibition. Patients received hydralazine at 182 mg for rapid, or 83 mg for slow acetylators, and valproate at 30 mg/kg, beginning a week before chemotherapy and continuing until disease progression. A total of 36 patients were enrolled, 17 treated with hydralazine and valproic acid (HV) and 19 with placebo (PLA), both groups receiving combination topotecan and cisplatin. The median number of cycles was 6. There were four partial responses in the HV arm, and one in the PLA arm. At a median follow-up time of 7 months, the median PFS was 6 months for the PLA arm and 10 months for the HV arm (p=0.0384, two tailed). Molecular correlates with response and survival from this trial are yet to be analyzed.
The same combination was assessed in the up front setting in patients with stage 3B squamous and adenosquamous cervical cancer [45]. A total of 22 patients received weekly cisplatin 40 mg/m2 + pelvic radiation, in combination with hydralazine 30 mg/kg administered three times daily until completion of intracavitary radiation therapy. The reported response rate was 100%, although delay in brachytherapy administration precluded assessment of the impact of epigenetic therapy.
4. mTOR in cervical cancer
mTOR plays an integral role in angiogenesis, cell growth, proliferation, and survival. Activation of the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway begins with growth factor receptor tyrosine kinase ligand binding, resulting in activation of PI3K. The primary role of activated PI3K is to convert phosphatidylinositol-4,5-bis-phosphate to phosphatidylinositol-3,4,5-triphosphate (PIP3) [46]. Accumulation of PIP3 at the cell surface then results in phosphorylation and activation of Akt, a protein serine-threonine kinase. In the absence of PTEN inhibition, Akt phosphorylates and inhibits the tuberous sclerosis complex (TSC), leading to mTOR activation. Activated mTOR subsequently forms 2 different multiprotein complexes, mTOR complex 1 and mTOR complex 2, associated with the regulatory associated protein of mTOR (raptor) [47]. Ultimately, phosphorylation and activation of 2 separate down stream signaling molecules, eukaryotic translation initiation factor 4E binding protein and ribosomal protein S6 kinase 1, promotes the translation of proteins involved in cell growth, angiogenesis, proliferation and survival [46,48,49,50].
High risk HPV related E6 has been shown to cause the rapid degradation of TSC2, resulting in TORC1 activation and downstream mTOR signaling. Additionally, HeLa cells are defective in the tumor suppressor LKB1, which inhibits mTOR via TSC2 stimulation.
The sole mTOR inhibitor studied in the treatment of cervical cancer is temsirolimus. In a combined phase 1 study, two patients with advanced stage and recurrent squamous cell cervical cancer were included amongst 15 total subjects. One patient had stable disease, and the median time to progression was 3 months. Unfortunately, the combination regimen of topotecan+temsirolimus was not tolerated in patients with a history of pelvic radiation therapy.
5. WEE1 inhibition and mitotic catastrophe
In normal, nonmalignant cells, cell cycle progression is a carefully orchestrated process, with emphasis on mutation prevention. Normally, several checkpoints ensure genomic integrity, including G1-S transition, S-phase, and the G2-M transition [51]. Normal cells commonly repair DNA damage during the G1-arrest, with cancer cells commonly exhibiting a deficient G1-arrest, relying more on G2-arrest to facilitate DNA repair. This process is carefully regulated by a series of cyclin dependent kinases, including the protein kinase WEE1.
In normal cells, WEE1 phosphorylates, and inactivates cyclin dependent kinase CDK1 preventing premature entry into mitosis while DNA integrity is ensured. Thus, WEE1 functions as a mitotic inhibitor, regulating the G2-M transition. WEE1 has been shown to be overexpressed in various cancer types, including cervical cancer. The scientific rational for WEE1 targeting is based on data indicated a deficient G1-S checkpoint, resulting in increased DNA damage at the G2-M transition in cancer cells. Thus, abrogation of the G2-M arrest releases cells with unrecognized and unrepaired DNA damage into premature mitosis, resulting in mitotic catastrophe and apoptotic cell death [52]. Combination treatment with DNA damaging cytotixic agents and WEE1 inhibition, is therefore hypothesized to prime these cells for apoptosis.
Preclinical studies using cancer cell lines and animal models identified WEE1 as a potential therapeutic targerget using RNA interference screening. Preclinical in vitro studies illustrated inhibition of cervical cancer cell outgrowth, using the WEE1 inhibitor, PD0166285 [52]. Additionally, knockdown of WEE1 in cervical cancer cells, but not in normal human epithelial cells, in combination with adriamycin, induced apoptosis [53]. These findings were supported by in vivo experiments, in which combination carboplatin and MK1775 (WEE1 inhibitor) resulted in tumor growth reduction for HeLa-luc xenograft animal models.
The above findings led to the development of a phase 1/2 clinical trial (NCT01076400) evaluating MK1775 in combination with topotecan and cisplatin in patients with advanced stage, metastatic and recurrent cervical cancer.
6. Notch signaling, cell fate, and cervical cancer
Notch genes encode transmembrane receptors that are highly conserved and are involved in cell fate decision (Fig. 2) [54,55]. Signaling via this pathway is dependent on direct contact between adjacent cells expressing the Notch receptors and ligands. Ultimately, down stream signaling results in regulation of cell fate specification, differentiation, proliferation and survival [56,57,58,59]. Notch receptors and their ligands have been shown to be overexpressed in various malignancies, including cervical cancer. The earliest studies using Notch specific antibodies, detected greater Notch gene product expression in cervical cancer specimens relative to high-grade dysplasia [60,61].
Fig. 2.

The Notch signaling pathway and its roles in cancer metastasis. The Notch receptors are activated by the Delta-like and Jagged families of ligands expressed on adjacent cells. Upon γ-secretase-mediated proteolysis, NICD proteins translocate to the nucleus and bind to the DNA binding protein CSL, taking the place of the corepressors (CoRs). NICD forms a complex with the DNA binding protein CSL and coactivators (CoAs), leading to the transcriptional activation of Notch target genes. The activation of Notch signaling in tumor microenvironment could promote epithelial-mesenchymal transition (EMT), the anoikis-resistance of tumor cells and maintain the homeostasis of angiogenesis, the morphology of vasculatures and the self-renewal of cancer stem cells (CSCs). Reprinted from Hu et al. [54] with permission from Springer.
Importantly, the basal epithelial cells normally found at the ectocervical-endocervical junction are prone to metaplastic transformation, and strongly stain for Notch1, Notch2 and their ligand jagged. Specifically, squamous and columnar epithelium normally cover the ectocervix and endocervical canal, with a reserve cell population at the squamocolumnar junction. This population of reserve cells (also known as basal cells) can differentiate into squamous or columnar epithelium. HPV infection of this cell population (at the transformation zone) is thought to result in premalignant and malignant transformation, and may be related to aberrations in Notch signaling and cell fate decisions.
The relationship between cervical cancer and Notch, however, is complex, with in vitro assays indicating a requirement for both E6 and E7 oncogene expression prior to soft agar colony formation in an immortalized keratinocyte cell line [60,61]. Ultimately, regulated Notch signaling was found to be associated with cervical cancer progression, although later experimental studies indicated that Notch overexpression, beyond an undetermined threshold, results in apoptotic cell death and counteracts HPV-induced cellular transformation. A resolution of these apparent contradictory observations may emerge as we gain insight into the cancer stem cell evolutionary paradigm [62].
There are currently several clinical trials in various stages of accrual investigating the clinical utility of Notch inhibition in patients with metastatic caricnoma. An industry sponsored prospective phase 1, dose escalation, study was conducted in patients with metastatic recurrent carcinoma, using a Notch inhibitor (NCT01158404). The study is closed to accrual, with results pending.
7. Heat shock protein 90
Antiangiogenic agents have shown promise in the treatment of cervical cancer, heralded with the presentation of GOG 240 [22]. Unfortunately however, acquired resistance continues to be a clinical dilemma, as a cancer cells utilize redundant pathways to allow for continued growth and proliferation.
In an effort to target several proangiogenic pathways simultaneously, researchers have explored interruption of ubiquitous heat shock protein 90 (HSP90) activity. Mechanistically, HSP90 is required for proper protein folding (for its substrate molecules), and simultaneously functions as a scaffold protein, facilitating interactions between receptor tyrosine kinases and their downstream substrates. Thus, inhibition of HSP90 allows for interruption of several 'cancer signaling pathways' simultaneously, although in a nonspecific manner (Fig. 3) [63,64,65]. One of the most prominent pro-angiogenic pathways up-regulated in patients with cancer is the hypoxia inducible factor (HIF)/vascular endothelial growth factor (VEGF) cascade. In a hypoxic environment, there is up-regulation of HIF, with subsequent activity, resulting in VEGF activation, and angiogenic signaling. Importantly, several key mediators of this pathway including HIF, VEGF receptor, tumor growth factor alpha (TGF-α), and EGFR are dependent on HSP90 function. Given this interdependence, inhibition of HSP90 is hypothesized to result in impaired signaling along these parallel pathways, suppressing tumor angiogenesis [66].
Fig. 3.
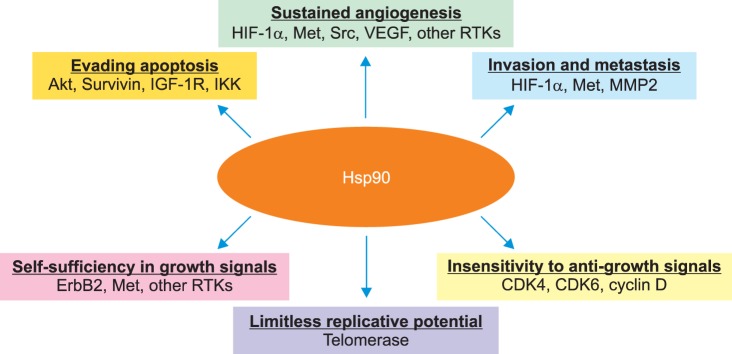
The cancer chaperone Hsp90. Hsp90 plays a central role in supporting all of the six hallmark traits of cancer, suggested by Hanahan and Weinberg, by chaperoning a subset of client proteins as shown. CDK4, 6, cyclin-dependent kinase 4, 6; HIF-1α, hypoxia-inducible factor-1α; IGF-1R, insulin-like growth factor-1 receptor; IKK, inhibitor of kappa B kinase; MMP2, matrix metalloproteinase 2; RTK, receptor tyrosine kinase; VEGF, vascular endothelial growth factor. Reprinted from Koga et al. [63] with permission from International Institute of Anticancer Research.
In patients with cervical cancer, correlative clinical studies conducted as early as 1986, indicated an association between increased HSP expression and carcinogenesis, tumor size and cellular proliferation [64]. Additionally, HSP70 expression was associated with poor outcomes in patients with cervical cancer [67].
Bisht et al. [68] investigated the impact of the HSP90 inhibitor, Geldanamycin, on two cervical cancer cell lines (HeLa and SiHa) in conjunction with radiation. HSP90 inhibition resulted in significant tumor cytotoxicity and radiosensitization, suggesting a potential therapeutic utility. Additionally, VEGF expression was down regulated in cells treated with the geldanamycin analog, 17-allylamino-17-demethoxy (17-AAG). There are no current active clinical trials investigating the use of HSP90 inhibition in patients with cervical cancer.
8. PARP inhibition in the treatment of cervical cancer
Poly ADP ribose polymerase (PARP) catalyzes the poly ADP-ribosylation of proteins involved in DNA repair [69]. Currently, this class of antineoplastic therapy is being extensively studied in patients with epithelial ovarian cancer, with an emphasis on patients with germline breast cancer susceptibility gene (BRCA) mutations. Deficiency in homologous recombination (HR) results in enhanced cell death with exposure to PARP inhibitors. Biologically, PARP inhibitors result in the creation of large numbers of single stand breaks in DNA, that when left unrepaired, lead to double strand breaks (DSBs) at replication forks (Fig. 4) [69,70]. In vitro assays indicated that cells deficient in HR (BRCA mutants) are unable to maintain genomic integrity in the contexts of large numbers of DNA DSBs, and are sensitive to PARP inhibition.
Fig. 4.
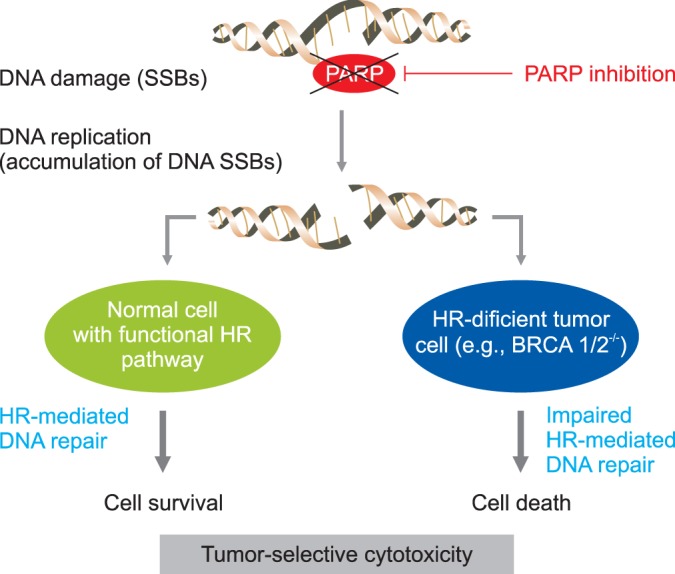
Mechanism for synthetic lethality in breast cancer susceptibility gene (BRCA) 1/2 deficient cancer. Poly ADR-ribose polymerase (PARP) inhibition produces tumor-selective synthetic lethaity. When PARP action is inhibited, SS are converted to double strand break (DSB) at replication. In cells with functional HR pathway, the DSB will be repaired. In cells with a dysfunctional HR pathway, as is the case with BRCA 1 and 2, the lesions go unrepaired and cell death ensues. HR, homologous recombination; SSB, single-strand break. Reprinted from Saijo [70].
Importantly, it was discovered that the activity of PARP inhibitors is not limited to BRCA mutation carriers, indicating that PARP inhibition may result in synthetic lethality when alternate DNA repair genes are deficient. This finding subsequently opened the door to the investigation of PARP inhibitors in cancers with deficiencies in HR, aside from known BRCA mutants.
The use of PARP inhibitors in cervical cancer is in its infancy. In a study of HeLa cell lines resistant to cisplatin with constitutively hyperactivated PARP1, Michels et al. [71] were able to illustrate the cytotoxic effects of PARP inhibition. Interestingly, these investigators were also able to show that elevated levels of poly ADP-ribose (PAR) predicted response to PARP inhibition in vitro and in vivo more than PARP1 expression itself [69]. A phase 1 trial is presently enrolling patients with cervical cancer along with other gynecologic malignancies to investigate the combination of olaparib with carboplatin in refractory or recurrent disease (NCT01237067). Another phase 1/2 trial is investigating the use of veliparib with cisplatin and paclitaxel in advanced, persistent, or recurrent cervical cancer (NCT01281852). The results of the above studies are anxiously anticipated as we try and identify alternate therapeutic options in a population of patients where few effective therapies exist.
CONCLUSIONS
In summary, several nonangiogenic molecular pathways have been identified as potential therapeutic targets in the treatment of cervical cancer. To date, none has been studied in a prospective phase 3 trial, although promising phase 2 data is emerging. Despite the above, several questions remain including long-term toxicity with use of the above agents, identifying the appropriate regimen (i.e., in combination with cytotoxic agents, as single agents or in the maintenance setting), using translational end points to help predict response, and ultimately understanding acquired resistance. It is evident that the field of molecular therapeutics is in its infancy within the cervical cancer domain, and we anxiously await the growth of this field and the identification of effective therapeutic agents.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Diaz-Padilla I, Monk BJ, Mackay HJ, Oaknin A. Treatment of metastatic cervical cancer: future directions involving targeted agents. Crit Rev Oncol Hematol. 2013;85:303–314. doi: 10.1016/j.critrevonc.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Greer BE, Koh WJ, Abu-Rustum NR, Apte SM, Campos SM, Chan J, et al. Cervical cancer. J Natl Compr Canc Netw. 2010;8:1388–1416. doi: 10.6004/jnccn.2010.0104. [DOI] [PubMed] [Google Scholar]
- 5.Thigpen T, Shingleton H, Homesley H, Lagasse L, Blessing J. Cis-platinum in treatment of advanced or recurrent squamous cell carcinoma of the cervix: a phase II study of the Gynecologic Oncology Group. Cancer. 1981;48:899–903. doi: 10.1002/1097-0142(19810815)48:4<899::aid-cncr2820480406>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 6.McGuire WP, 3rd, Arseneau J, Blessing JA, DiSaia PJ, Hatch KD, Given FT, Jr, et al. A randomized comparative trial of carboplatin and iproplatin in advanced squamous carcinoma of the uterine cervix: a Gynecologic Oncology Group study. J Clin Oncol. 1989;7:1462–1468. doi: 10.1200/JCO.1989.7.10.1462. [DOI] [PubMed] [Google Scholar]
- 7.Fracasso PM, Blessing JA, Wolf J, Rocereto TF, Berek JS, Waggoner S. Phase II evaluation of oxaliplatin in previously treated squamous cell carcinoma of the cervix: a gynecologic oncology group study. Gynecol Oncol. 2003;90:177–180. doi: 10.1016/s0090-8258(03)00253-1. [DOI] [PubMed] [Google Scholar]
- 8.Thigpen T, Blessing JA, Gallup DG, Maiman M, Soper JT. Phase II trial of mitomycin-C in squamous cell carcinoma of the uterine cervix: a Gynecologic Oncology Group study. Gynecol Oncol. 1995;57:376–379. doi: 10.1006/gyno.1995.1157. [DOI] [PubMed] [Google Scholar]
- 9.Sutton GP, Blessing JA, Adcock L, Webster KD, DeEulis T. Phase II study of ifosfamide and mesna in patients with previously-treated carcinoma of the cervix: a Gynecologic Oncology Group study. Invest New Drugs. 1989;7:341–343. doi: 10.1007/BF00173765. [DOI] [PubMed] [Google Scholar]
- 10.Look KY, Blessing JA, Levenback C, Kohler M, Chafe W, Roman LD. A phase II trial of CPT-11 in recurrent squamous carcinoma of the cervix: a gynecologic oncology group study. Gynecol Oncol. 1998;70:334–338. doi: 10.1006/gyno.1998.5129. [DOI] [PubMed] [Google Scholar]
- 11.Schilder RJ, Blessing JA, Morgan M, Mangan CE, Rader JS. Evaluation of gemcitabine in patients with squamous cell carcinoma of the cervix: a Phase II study of the gynecologic oncology group. Gynecol Oncol. 2000;76:204–207. doi: 10.1006/gyno.1999.5671. [DOI] [PubMed] [Google Scholar]
- 12.Bookman MA, Blessing JA, Hanjani P, Herzog TJ, Andersen WA. Topotecan in squamous cell carcinoma of the cervix: a phase II study of the Gynecologic Oncology Group. Gynecol Oncol. 2000;77:446–449. doi: 10.1006/gyno.2000.5807. [DOI] [PubMed] [Google Scholar]
- 13.Curtin JP, Blessing JA, Webster KD, Rose PG, Mayer AR, Fowler WC, Jr, et al. Paclitaxel, an active agent in nonsquamous carcinomas of the uterine cervix: a Gynecologic Oncology Group Study. J Clin Oncol. 2001;19:1275–1278. doi: 10.1200/JCO.2001.19.5.1275. [DOI] [PubMed] [Google Scholar]
- 14.McGuire WP, Blessing JA, Moore D, Lentz SS, Photopulos G. Paclitaxel has moderate activity in squamous cervix cancer: a Gynecologic Oncology Group study. J Clin Oncol. 1996;14:792–795. doi: 10.1200/JCO.1996.14.3.792. [DOI] [PubMed] [Google Scholar]
- 15.Garcia AA, Blessing JA, Vaccarello L, Roman LD Gynecologic Oncology Group Study. Phase II clinical trial of docetaxel in refractory squamous cell carcinoma of the cervix: a Gynecologic Oncology Group Study. Am J Clin Oncol. 2007;30:428–431. doi: 10.1097/COC.0b013e31803377c8. [DOI] [PubMed] [Google Scholar]
- 16.Muggia FM, Blessing JA, Waggoner S, Berek JS, Monk BJ, Sorosky J, et al. Evaluation of vinorelbine in persistent or recurrent nonsquamous carcinoma of the cervix: a Gynecologic Oncology Group Study. Gynecol Oncol. 2005;96:108–111. doi: 10.1016/j.ygyno.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 17.Bloss JD, Blessing JA, Behrens BC, Mannel RS, Rader JS, Sood AK, et al. Randomized trial of cisplatin and ifosfamide with or without bleomycin in squamous carcinoma of the cervix: a gynecologic oncology group study. J Clin Oncol. 2002;20:1832–1837. doi: 10.1200/JCO.2002.07.045. [DOI] [PubMed] [Google Scholar]
- 18.Long HJ, 3rd, Bundy BN, Grendys EC, Jr, Benda JA, McMeekin DS, Sorosky J, et al. Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: a Gynecologic Oncology Group Study. J Clin Oncol. 2005;23:4626–4633. doi: 10.1200/JCO.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 19.Monk BJ, Sill MW, McMeekin DS, Cohn DE, Ramondetta LM, Boardman CH, et al. Phase III trial of four cisplatin-containing doublet combinations in stage IVB, recurrent, or persistent cervical carcinoma: a Gynecologic Oncology Group study. J Clin Oncol. 2009;27:4649–4655. doi: 10.1200/JCO.2009.21.8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore DH, Blessing JA, McQuellon RP, Thaler HT, Cella D, Benda J, et al. Phase III study of cisplatin with or without paclitaxel in stage IVB, recurrent, or persistent squamous cell carcinoma of the cervix: a gynecologic oncology group study. J Clin Oncol. 2004;22:3113–3119. doi: 10.1200/JCO.2004.04.170. [DOI] [PubMed] [Google Scholar]
- 21.Omura GA, Blessing JA, Vaccarello L, Berman ML, Clarke-Pearson DL, Mutch DG, et al. Randomized trial of cisplatin versus cisplatin plus mitolactol versus cisplatin plus ifosfamide in advanced squamous carcinoma of the cervix: a Gynecologic Oncology Group study. J Clin Oncol. 1997;15:165–171. doi: 10.1200/JCO.1997.15.1.165. [DOI] [PubMed] [Google Scholar]
- 22.Tewari KS, Sill MW, Long HJ, 3rd, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370:734–743. doi: 10.1056/NEJMoa1309748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milas L, Mason K, Hunter N, Petersen S, Yamakawa M, Ang K, et al. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin Cancer Res. 2000;6:701–708. [PubMed] [Google Scholar]
- 25.Huang SM, Harari PM. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin Cancer Res. 2000;6:2166–2174. [PubMed] [Google Scholar]
- 26.Bianco C, Bianco R, Tortora G, Damiano V, Guerrieri P, Montemaggi P, et al. Antitumor activity of combined treatment of human cancer cells with ionizing radiation and anti-epidermal growth factor receptor monoclonal antibody C225 plus type I protein kinase A antisense oligonucleotide. Clin Cancer Res. 2000;6:4343–4350. [PubMed] [Google Scholar]
- 27.Akimoto T, Hunter NR, Buchmiller L, Mason K, Ang KK, Milas L. Inverse relationship between epidermal growth factor receptor expression and radiocurability of murine carcinomas. Clin Cancer Res. 1999;5:2884–2890. [PubMed] [Google Scholar]
- 28.Zagouri F, Sergentanis TN, Chrysikos D, Filipits M, Bartsch R. Molecularly targeted therapies in cervical cancer: a systematic review. Gynecol Oncol. 2012;126:291–303. doi: 10.1016/j.ygyno.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 29.del Campo JM, Prat A, Gil-Moreno A, Perez J, Parera M. Update on novel therapeutic agents for cervical cancer. Gynecol Oncol. 2008;110(3) Suppl 2:S72–S76. doi: 10.1016/j.ygyno.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 30.Santin AD, Sill MW, McMeekin DS, Leitao MM, Jr, Brown J, Sutton GP, et al. Phase II trial of cetuximab in the treatment of persistent or recurrent squamous or non-squamous cell carcinoma of the cervix: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;122:495–500. doi: 10.1016/j.ygyno.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hertlein L, Lenhard M, Kirschenhofer A, Kahlert S, Mayr D, Burges A, et al. Cetuximab monotherapy in advanced cervical cancer: a retrospective study with five patients. Arch Gynecol Obstet. 2011;283:109–113. doi: 10.1007/s00404-010-1389-1. [DOI] [PubMed] [Google Scholar]
- 32.Farley J, Sill MW, Birrer M, Walker J, Schilder RJ, Thigpen JT, et al. Phase II study of cisplatin plus cetuximab in advanced, recurrent, and previously treated cancers of the cervix and evaluation of epidermal growth factor receptor immunohistochemical expression: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;121:303–308. doi: 10.1016/j.ygyno.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurtz JE, Hardy-Bessard AC, Deslandres M, Lavau-Denes S, Largillier R, Roemer-Becuwe C, et al. Cetuximab, topotecan and cisplatin for the treatment of advanced cervical cancer: a phase II GINECO trial. Gynecol Oncol. 2009;113:16–20. doi: 10.1016/j.ygyno.2008.12.040. [DOI] [PubMed] [Google Scholar]
- 34.Goncalves A, Fabbro M, Lhomme C, Gladieff L, Extra JM, Floquet A, et al. A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol Oncol. 2008;108:42–46. doi: 10.1016/j.ygyno.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 35.Nogueira-Rodrigues A, do Carmo CC, Viegas C, Erlich F, Camisao C, Fontao K, et al. Phase I trial of erlotinib combined with cisplatin and radiotherapy for patients with locally advanced cervical squamous cell cancer. Clin Cancer Res. 2008;14:6324–6329. doi: 10.1158/1078-0432.CCR-07-5112. [DOI] [PubMed] [Google Scholar]
- 36.Schilder RJ, Sill MW, Lee YC, Mannel R. A phase II trial of erlotinib in recurrent squamous cell carcinoma of the cervix: a Gynecologic Oncology Group Study. Int J Gynecol Cancer. 2009;19:929–933. doi: 10.1111/IGC.0b013e3181a83467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monk BJ, Mas Lopez L, Zarba JJ, Oaknin A, Tarpin C, Termrungruanglert W, et al. Phase II, open-label study of pazopanib or lapatinib monotherapy compared with pazopanib plus lapatinib combination therapy in patients with advanced and recurrent cervical cancer. J Clin Oncol. 2010;28:3562–3569. doi: 10.1200/JCO.2009.26.9571. [DOI] [PubMed] [Google Scholar]
- 38.Lee CM, Shrieve DC, Zempolich KA, Lee RJ, Hammond E, Handrahan DL, et al. Correlation between human epidermal growth factor receptor family (EGFR, HER2, HER3, HER4), phosphorylated Akt (P-Akt), and clinical outcomes after radiation therapy in carcinoma of the cervix. Gynecol Oncol. 2005;99:415–421. doi: 10.1016/j.ygyno.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 39.Takai N, Kira N, Ishii T, Nishida M, Nasu K, Narahara H. Novel chemotherapy using histone deacetylase inhibitors in cervical cancer. Asian Pac J Cancer Prev. 2011;12:575–580. [PubMed] [Google Scholar]
- 40.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 41.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 42.Atmaca A, Al-Batran SE, Maurer A, Neumann A, Heinzel T, Hentsch B, et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer. 2007;97:177–182. doi: 10.1038/sj.bjc.6603851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chavez-Blanco A, Segura-Pacheco B, Perez-Cardenas E, Taja-Chayeb L, Cetina L, Candelaria M, et al. Histone acetylation and histone deacetylase activity of magnesium valproate in tumor and peripheral blood of patients with cervical cancer: a phase I study. Mol Cancer. 2005;4:22. doi: 10.1186/1476-4598-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, Gonzalez-Fierro A, de la Cruz-Hernandez E, et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer: preliminary results. Med Oncol. 2011;28(Suppl 1):S540–S546. doi: 10.1007/s12032-010-9700-3. [DOI] [PubMed] [Google Scholar]
- 45.Candelaria M, Cetina L, Perez-Cardenas E, de la Cruz-Hernandez E, Gonzalez-Fierro A, Trejo-Becerril C, et al. Epigenetic therapy and cisplatin chemoradiation in FIGO stage IIIB cervical cancer. Eur J Gynaecol Oncol. 2010;31:386–391. [PubMed] [Google Scholar]
- 46.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 47.Diaz-Padilla I, Duran I, Clarke BA, Oza AM. Biologic rationale and clinical activity of mTOR inhibitors in gynecological cancer. Cancer Treat Rev. 2012;38:767–775. doi: 10.1016/j.ctrv.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Lee DF, Hung MC. All roads lead to mTOR: integrating inflammation and tumor angiogenesis. Cell Cycle. 2007;6:3011–3014. doi: 10.4161/cc.6.24.5085. [DOI] [PubMed] [Google Scholar]
- 49.Liu D, Hou P, Liu Z, Wu G, Xing M. Genetic alterations in the phosphoinositide 3-kinase/Akt signaling pathway confer sensitivity of thyroid cancer cells to therapeutic targeting of Akt and mammalian target of rapamycin. Cancer Res. 2009;69:7311–7319. doi: 10.1158/0008-5472.CAN-09-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–522. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 52.Dixon H, Norbury CJ. Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle. 2002;1:362–368. doi: 10.4161/cc.1.6.257. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther. 2004;3:305–313. doi: 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- 54.Hu YY, Zheng MH, Zhang R, Liang YM, Han H. Notch signaling pathway and cancer metastasis. Adv Exp Med Biol. 2012;727:186–198. doi: 10.1007/978-1-4614-0899-4_14. [DOI] [PubMed] [Google Scholar]
- 55.Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011;1815:197–213. doi: 10.1016/j.bbcan.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66:1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis J. Notch signalling and the control of cell fate choices in vertebrates. Semin Cell Dev Biol. 1998;9:583–589. doi: 10.1006/scdb.1998.0266. [DOI] [PubMed] [Google Scholar]
- 58.Lewis J. Notch signalling: a short cut to the nucleus. Nature. 1998;393:304–305. doi: 10.1038/30597. [DOI] [PubMed] [Google Scholar]
- 59.Simpson P. Developmental genetics: the Notch connection. Nature. 1995;375:736–737. doi: 10.1038/375736a0. [DOI] [PubMed] [Google Scholar]
- 60.Daniel B, Rangarajan A, Mukherjee G, Vallikad E, Krishna S. The link between integration and expression of human papillomavirus type 16 genomes and cellular changes in the evolution of cervical intraepithelial neoplastic lesions. J Gen Virol. 1997;78:1095–1101. doi: 10.1099/0022-1317-78-5-1095. [DOI] [PubMed] [Google Scholar]
- 61.Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci U S A. 1995;92:6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maliekal TT, Bajaj J, Giri V, Subramanyam D, Krishna S. The role of Notch signaling in human cervical cancer: implications for solid tumors. Oncogene. 2008;27:5110–5114. doi: 10.1038/onc.2008.224. [DOI] [PubMed] [Google Scholar]
- 63.Koga F, Kihara K, Neckers L. Inhibition of cancer invasion and metastasis by targeting the molecular chaperone heat-shock protein 90. Anticancer Res. 2009;29:797–807. [PubMed] [Google Scholar]
- 64.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hwang YJ, Lee SP, Kim SY, Choi YH, Kim MJ, Lee CH, et al. Expression of heat shock protein 60 kDa is upregulated in cervical cancer. Yonsei Med J. 2009;50:399–406. doi: 10.3349/ymj.2009.50.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bohonowych JE, Gopal U, Isaacs JS. Hsp90 as a gatekeeper of tumor angiogenesis: clinical promise and potential pitfalls. J Oncol. 2010;2010:412985. doi: 10.1155/2010/412985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abd el All H, Rey A, Duvillard P. Expression of heat shock protein 70 and c-myc in cervical carcinoma. Anticancer Res. 1998;18(3A):1533–1536. [PubMed] [Google Scholar]
- 68.Bisht KS, Bradbury CM, Mattson D, Kaushal A, Sowers A, Markovina S, et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 2003;63:8984–8995. [PubMed] [Google Scholar]
- 69.Reinbolt RE, Hays JL. The role of PARP inhibitors in the treatment of gynecologic malignancies. Front Oncol. 2013;3:237. doi: 10.3389/fonc.2013.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saijo N. Present status and problems on molecular targeted therapy of cancer. Cancer Res Treat. 2012;44:1–10. doi: 10.4143/crt.2012.44.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Michels J, Vitale I, Galluzzi L, Adam J, Olaussen KA, Kepp O, et al. Cisplatin resistance associated with PARP hyperactivation. Cancer Res. 2013;73:2271–2280. doi: 10.1158/0008-5472.CAN-12-3000. [DOI] [PubMed] [Google Scholar]