

Abstract
Childhood neurotransmitter disorders are increasingly recognised as an expanding group of inherited neurometabolic syndromes. They are caused by disturbance in synthesis, metabolism, and homeostasis of the monoamine neurotransmitters, including the catecholamines (dopamine, norepinephrine, and epinephrine) and serotonin. Disturbances in monoamine neurotransmission will lead to neurological symptoms that often overlap with clinical features of other childhood neurological disorders (such as hypoxic ischaemic encephalopathy, cerebral palsy, other movement disorders, and paroxysmal conditions); consequently, neurotransmitter disorders are frequently misdiagnosed. The diagnosis of neurotransmitter disorders is made through detailed clinical assessment, analysis of cerebrospinal fluid neurotransmitters, and further supportive diagnostic investigations. Early and accurate diagnosis of neurotransmitter disorders is important, as many are amenable to therapeutic intervention. The principles of treatment for monoamine neurotransmitter disorders are mainly directly derived from understanding these metabolic pathways. In disorders characterized by enzyme deficiency, we aim to increase monoamine substrate availability, boost enzyme co-factor levels, reduce monoamine breakdown, and replace depleted levels of monoamines with pharmacological analogs as clinically indicated. Most monoamine neurotransmitter disorders lead to reduced levels of central dopamine and/or serotonin. Complete amelioration of motor symptoms is achievable in some disorders, such as Segawa’s syndrome, and, in other conditions, significant improvement in quality of life can be attained with pharmacotherapy. In this review, we provide an overview of the clinical features and current treatment strategies for childhood monoamine neurotransmitter disorders.
Key Points
| Monoamine neurotransmitter disorders associated with abnormal dopamine metabolism are associated with predominantly neurological phenotypes |
| Mononeurotransmitter disorders are underrecognised despite many being treatable and pharmacoresponsive and should be considered in those presenting with movement disorder and unexplained cerebral palsy |
| Treatment strategies should be tailored to the specific neurotransmitter defect with input from the expert neurologist |
Background
The monoamine neurotransmitters include the catecholamines dopamine, norepinephrine and epinephrine, and serotonin. These biogenic amines are critically involved in signaling pathways in the central and peripheral nervous system. They play a direct role in the regulation of movement, activity levels, mood, attention, and sleep, as well as maintenance of vascular tone, blood flow, thermoregulation, and modulation of pain mechanisms [1, 2].
Disruption of monoamine neurotransmitter synthesis, metabolism, and homeostasis leads to diverse neurological manifestations in childhood and include developmental delay, motor disorders, epilepsy, autonomic dysfunction, and neuropsychiatric features. More specific clinical features suggestive of dopamine disorders include dystonia, with fluctuation of symptoms (e.g. diurnal variation), tremor, brisk deep tendon reflexes, oculogyric crises, palpebral ptosis, axial hypotonia, hypersalivation, developmental delay, feeding difficulties, excessive sweating, and temperature instability [3]. These features overlap with clinical symptoms described in other neurological syndromes such as hypoxic ischemic encephalopathy, cerebral palsy, paroxysmal disorders, and the epileptic encephalopathies, which can make the diagnosis of neurotransmitter disorders challenging. An understanding of the biochemical pathways of monoamine neurotransmitter synthesis, metabolism, and transport is valuable in accurate diagnosis and also underpins the available therapeutic options for this spectrum of disorders.
Dopamine and Serotonin Biosynthesis: Rationale for Treatment Strategies
The process of neurotransmission requires (1) synthesis of the neurotransmitter in the presynaptic nerve terminal, (2) storage in presynaptic vesicles, (3) regulated release into the synaptic cleft, (4) action at specific receptors at the postsynaptic interface, and (5) a means of termination of action of the released neurotransmitter (either degradation or reuptake). The monoamine biosynthesis pathway in its entirety is illustrated in Fig. 1.
Fig. 1.
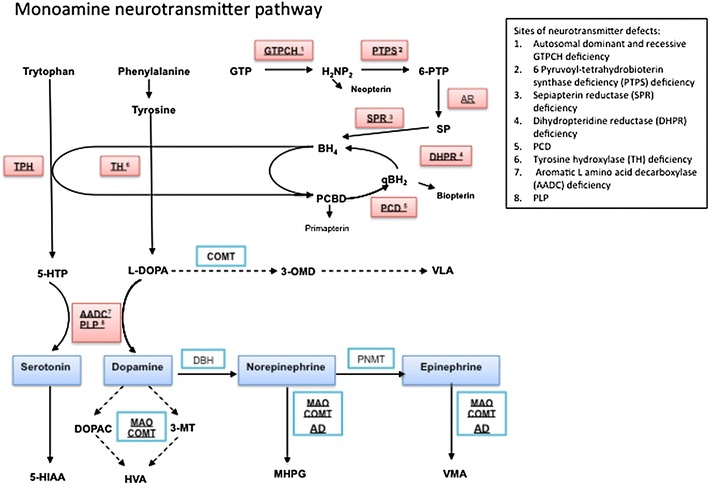
Flow diagram of the monoamine neurotransmitter biosynthesis pathway. The initial substrates for dopamine and serotonin synthesis are aromatic amino acids tyrosine and tryptophan that enter the brain via the large neutral amino acid transporter. They are hydroxylated by tyrosine hydroxylase (TH) and tryptophan hydroxylase (TPH) to levodopa (l-dopa) and 5-hydroxytryptophan (5-HTP), respectively, and are both subsequently decarboxylated by aromatic l-amino acid decarboxylase (AADC) to yield the active neurotransmitters dopamine and serotonin. AADC activity is dependent on its cofactor pyridoxal 5 phosphate. Dopamine and serotonin are further catabolized by monoamine oxidase (MAO) and catechol-O-methyl transferase (COMT) to form 3-methyl 4-hydroxyphenylglycol (MHPG) and homovanillic acid (HVA) from dopamine and 5-hydroxyindoleacetic acid (5-HIAA) from serotonin. These are the stable metabolites measured in the cerebrospinal fluid for neurotransmitter analysis, and are often the key to diagnosis of a childhood monoamine neurotransmitter disorder. l-dopa is metabolised by COMT to 3-OMethyldopa (3-OMD) and then Vanillactic acid (VLA). Both tyrosine hydroxylase and tryptophan hydroxylase activity require the co-factor tetrahydrobiopterin (BH4). Therefore enzymatic deficiencies in the pterin synthesis pathway that affect the levels of this essential cofactor will lead to reduced levels of the monoamine neurotransmitters. BH4 is synthesized in four steps from guanosine triphosphate (GTP) that are dependent on the enzyme activity of guanine triphosphate cyclohydrolase 1 (GTPCH 1), 6-pyruvoyl-tetrahydrobiopterin synthase (PTPS), aldose reductase (AR), and sepiapterin reductase (SPR). The major site of regulation of BH4 biosynthesis is at the level of GTP cyclohydrolase. After coupling as an active co-factor to the aromatic amino hydroxylases (tyrosine and tryptophan hydroxylase), BH4 is regenerated through oxidation by tetrahydrobiopterin-4α-carbinolamine to form quinoid dihydrobiopterin (qBH2) and is subsequently converted back to the active co-factor by dihydrobiopteridine reductase (DHPR). The biogenic amines are illustrated in blue boxes, with the sites of enzyme or cofactor deficiency leading to neurotransmitter disorder highlighted in red boxes with corresponding key for abbreviations used. AD aldehyde dehydrogenase, DOPAC 3,4-dihydroxyphenylacetic acid, DBH dopamine β hydroxylase, GTPCH guanosine triphosphate cyclohydrolase, H 2 NP 2 dihydroneopterin triphosphate, 3-MT 3-methoxytyramine, PCD pterin-4α-carbinolamine dehydratase, PLP pyridoxal phosphate, PNMT phenylethanolamine N-methyltransferase, TH tyrosine hydroxylase, VMA vanillylmandelic acid
The principles of treatment for monoamine neurotransmitter disorders are directly derived from understanding these metabolic pathways. The sites of common dopaminergic-specific pharmacotherapy are summarized in Fig. 2. In disorders characterized by enzyme deficiency, we aim to increase monoamine substrate availability, boost enzyme co-factor levels, reduce monoamine breakdown, and replace depleted levels of monoamines with pharmacological analogs as clinically indicated. The treatments and doses discussed in this review are collated from the expertise and experience cited in landmark papers reporting these disorders. As the majority are rare conditions, robust evidence for efficacy remains limited, and doses and drug strategies are more reliant on expert opinion.
Fig. 2.
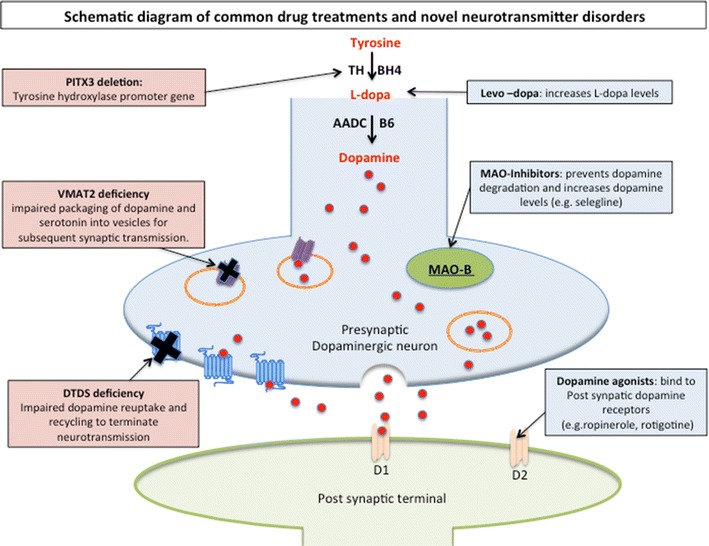
Schematic diagram dopaminergic neurotransmission and sites of common drug treatments and novel neurotransmitter disorders. The presynaptic neuron is illustrated in blue, with dopamine is represented as red circles and postsynaptic neuron in green. Following dopamine synthesis, it is packaged into synaptic secretory vesicles (orange circles) through VMAT (purple symbol). Presynaptic uptake of dopamine is-facilitated by dopamine transporter (blue transmembrane protein symbol) and deficiencies in VMAT and DAT are the most recent neurotransmitter transport disorders to be described. Sites for pharmaco-treatment are replacement of L-dopa, and alternative stimulation of the postsynaptic dopamine receptors (light orange symbols). MAO-B is represented as a green oval and acts to metabolize dopamine into HVA. Inhibition of MAO-B is a pharmaco-target to increase intrinsic dopamine. AADC aromatic amino acid decarboxylase, B6 pyridoxal phosphate, BH4 tetrahydrobiopterin, D 1 postsynaptic dopamine receptor type 1, D 2 postsynaptic dopamine receptor type 2, DAT dopamine transporter, DTDS dopamine transport deficiency syndrome, MAO monoamine oxidase, MAO-B monoamine oxidase type B, PITX3 PITX3 gene, TH tyrosine hydroxylase, VMAT2 vesicular monoamine transporter 2
Most neurotransmitter disorders lead to reduced levels of central dopamine and/or serotonin. Treatment of neurotransmitter disorders can therefore include the following strategies.
Replacement of Precursors of Dopamine and Serotonin
Levodopa (l-dopa) and 5-hydroxytryptophan (5-HTP) can be used to replenish central dopamine and serotonin. l-dopa therapy has been used for decades in dystonia, with fluctuation and Parkinson’s disease, where dopamine deficiency is attributed in part to presynaptic dopaminergic neuronal degeneration [4]. l-dopa is also used to treat several childhood-onset monoamine neurotransmitter disorders where dopamine deficiency results from either impaired dopamine or pterin synthesis, with the aim of increasing central dopamine levels. Knowledge and understanding of the specific neurotransmitter disorder being treated is paramount, as l-dopa is not indicated in all monoamine neurotransmitter disorders. For example, it is considered inappropriate for the vast majority of aromatic l-amino acid decarboxylase (AADC) deficiency patients and not used in dopamine transporter deficiency syndrome (DTDS). Knowledge of the specific defect also guides the appropriate dose ranges typically used, as these also differ from one neurotransmitter disorder to another (see Table 1 for example, where the l-dopa dosage range for autosomal dominant guanosine triphosphate [GTP] cyclohydrolase [AD GCH] deficiency is different to that for treatment of tyrosine hydroxylase [TH] deficiency). For all dopamine deficiency syndromes, a slow and steady titration regimen is recommended to assess efficacy and minimize risk of dose-related side effects.
Table 1.
List of childhood monoamine neurotransmitter disorders with clinical features and summary of drug treatments used
| Disorder | Clinical features | CSF neurotransmitter profile and other investigations | Drug treatment and cited dose ranges used | |
|---|---|---|---|---|
| AD GCH deficiency (Segawa’s syndrome) | Dopa-responsive dystonia, classically ‘evening equinus’, diurnal fluctuation | Low CSF HVA (± HIAA), low pterins, normal plasma phenylalanine | l-dopa 1–5 mg/kg/day (often <3 mg/kg/day is sufficient for clinical response) [17, 18] | |
| AR GCH deficiency | Truncal hypotonia, dystonia, developmental delay, seizures | Low CSF HVA, HIAA, BH4 and neopterin, raised plasma phenylalanine in most cases | BH4 1–10 mg/kg/day [33, 35] | |
| l-dopa 1–10 mg/kg/day [33, 35] | ||||
| 5-HTP 1–8 mg/kg/day [33, 35] | ||||
| PTPS deficiency | Hypotonia, hypokinesia, rigidity, chorea, dystonia, oculogyric crisis | Low HVA and HIAA, biopterin, raised plasma phenylalanine | BH4 1–12 mg/kg/day [33, 35] | |
| l-dopa 4–18 mg/kg/day [33, 35] | ||||
| 5-HTP 1–10 mg/kg/day [33, 35] | ||||
| Other treatments: Dopamine agonists and MAOI to avoid dopamine-related off-on phenomena [35] | ||||
| SRD deficiency | Axial hypotonia, dystonia, oculogyric crisis, diurnal fluctuation, CP-like presentation | Low CSF HVA, HIAA with raised biopterin, BH2 and SP, normal plasma phenylalanine | l-dopa 0.5–2 mg/kg/day [5, 39, 40] | |
| 5-HTP 1–6 mg/kg/day [5, 39, 40] | ||||
| Other treatments: MAOI (selegiline 0.03–2mg/kg/day) [5] | ||||
| DHPR deficiency | Bulbar dysfunction, dyskinesia, tremor, dystonia, choreathetosis | Low CSF HVA, HIAA, folate, raised CSF BH2 level, normal or raised biopterin levels, raised plasma phenylalanine | l-dopa 5–13 mg/kg/day [33, 36] | |
| 5-HTP 3–11 mg/kg/day [33, 36] | ||||
| Folinic acid (calcium folinate) 15 mg/day [BNFC 2014] | ||||
| Other treatments: Dopamine agonists and MAOI to avoid dopamine-related off-on phenomena [12] | ||||
| TH deficiency type A | Parkinsonism–dystonia, hypokinesia or bradykinesia, rigidity, diurnal variation | Low CSF HVA with normal 5HIAA and normal pterin profile, low ratio HVA:HIAA (usually <1.0), raised serum prolactin | l-dopa (low dose) 3–10 mg/kg/day (titrated slowly according to response) [42] | |
| TH deficiency type B | Focal or generalized dystonia with crises; severe parkinsonism, hypotonia, oculogyric crises, tremor, ptosis, hypersalivation, autonomic disturbance | Low CSF HVA with normal 5-HIAA, low ratio HVA:HIAA (usually <1.0), raised prolactin | l-dopa <0.5–2 mg/kg/day (titrated slowly according to response) [42] | |
| AADC deficiency | Hypotonia, oculogyric crisis, hypokinesia, chorea, dystonia, bulbar dysfunction | Low HVA, 5-HIAA, 3-methoxy-4-hydroxyphenylglycol with raised 5- HTP, l-dopa, and 3-ortho-methyldopa; AADC enzyme activity in plasma is very low or absent; urinary catecholamines raised (vanillactic acid, 3-orthomethyldopa) | Pyridoxine 20–160 mg/kg/day [8] | |
| Folinic acid (calcium folinate) 15 mg/day [BNFC 2014] | ||||
|
Other treatments: Dopamine agonists: [48] Rotigotine, patch dose as per BNFC; 2,4,6,8 mg dose patches available and applied once a day (doses 0.17–0.25 mg/kg/day used) (other dopamine agonists used; pergolide, bromocriptine, pramipexole, ropinirole) [45, 46] MAOI selegiline 0.03–2 mg/kg/day [45, 46] Trihexyphenydyl 1–12 mg/day titrated (often higher doses are tolerated but titrated slowly) Benztropine 1–4 mg/day Clonidine 0.1–3 total mg/day [BNFC initial test dose is recommended with higher doses used with caution due to antihypertensive action] Benzodiazepines | ||||
| PNPO deficiency | Severe pharmaco-resistant neonatal epileptic encephalopathy, history of in utero seizure onset, prematurity | Low CSF HVA and HIAA, raised l-dopa, 5-HTP + 3 ortho-methyldopa, raised CSF (glycine, taurine, histidine, and threonine) | Pyridoxal phosphate 30–50 mg/kg/day [58] | |
| PITX3 deletion | Mild learning difficulties, hyperactivity, sleep disturbance, distinctive facial features, hypoplastic middle 5th phalanges | Low CSF HVA and HIAA, absent CSF l-dopa, low CSF biopterin | l-dopa 1–2.5 mg/kg/day [63] | |
| Brain dopamine-–serotonin vesicular transport disease | Axial hypotonia, oculogyric crises, parkinsonism, tremor, facial dyskinesia, ptosis, bulbar dysfunction, sleep disturbance | Normal CSF neurotransmitter profile, urine raised HVA and HIAA |
Dr R Alkhater personal communication for treatment regimen. Initial treatment: Dopamine agonist [66] Pramipexole 0.01–0.02mg/kg/day divided to twice per day, if the patient tolerates this without side effects (usually older patients) I double the dose depending on the gait dystonia. Other treatments: Trihexyphenidyl starting dose of 0.2mg/kg/day divided to twice per day dose. Thus far the highest dose used is 4 mg/day l-dopa (sinemet): 3.2 years into treatment l-dopa was introduced at 0.05–0.08 mg/kg/day and gait improved even more. The doses are titrated according to the clinical situation, guided by the side effects, the gait and the fine motor coordination |
|
| Dopamine transporter deficiency syndrome | Feeding difficulties, irritability, axial hypotonia and dyskinetic movement disorder, progressive dystonic and dyskinetic movement disorder with eye involvement | Raised HVA, normal HIAA, HVA:HIAA ratio >5 |
Dopamine agonist Pramipexole: [66] (Kurian, personal communication) for dosage titration: 5 mcg/kg/day in 3 divided doses and increase to 35 mcg/kg/day (in 3 divided doses) Ropinirole: [66] as per BNFC (titration 0.25 mg once daily starting dose and increasing by 0.25 mg every 3 days) (ranges 0.5–4 mg per day are used) |
|
Drug treatments listed are cited from expertise and experience reported in significant published series on the childhood monoamine neurotransmitter disorders. The majority are rare disorders and, therefore, robust evidence for efficacy remains limited. Where drug treatment use and response is detailed but doses are not detailed within a cited source, a dose range is quoted from the recommendations of the BNFC, 2014 edition
AADC aromatic l-amino acid decarboxylase, AD GCH autosomal dominant, BH2 dihydrobiopterin, BH4 tetrahydrobiopterin, BNFC British National formulary for children, CP cerebral palsy, CSF cerebrospinal fluid, DHPR dihydropteridine reductase, GCH guanosine triphosphate cyclohydrolase, HIAA hydroxyindoleacetic acid, HTP hydroxytryptophan, HVA homovanillic acid, l -dopa levodopa, MAOI monoamine oxidase inhibitor, PNPO pyridoxamine 5′-phosphate oxidase, PTPS 6 pyruvoyl-tetrahydrobiopterin synthase, SP sepiapterin, SRD sepiapterin reductase deficiency, TH tyrosine hydroxylase
l-dopa given in preparations in combination with carbidopa
Peripheral AADC inhibitors such as carbidopa or benserazide are frequent concomitant treatments with l-dopa to prevent peripheral decarboxylation, thereby enabling more l-dopa to reach the central nervous system. Unlike l-dopa, carbidopa and benserazide cannot cross the blood–brain barrier. Within the central nervous system, l-dopa is further metabolized to dopamine by the enzyme AADC (Fig. 1). l-dopa administration is associated with a range of side effects such as nausea, vomiting, dose-related dyskinesia, hallucinations, and mood disorders.
5-HTP is used as an adjunct treatment for pterin defects where there are low central serotonin levels. 5-HTP is the biosynthetic precursor to serotonin that can be administered orally and then crosses the blood–brain barrier to reach the central nervous system. The aim of 5-HTP treatment is to increase serotonin precursor levels, with dosage often being limited by side effects, predominantly nausea, vomiting, and diarrhea. The clinical treatment response is more difficult to assess with 5-HTP than with l-dopa, but improvement in sleep, motor and cognitive aspects in the pterin disorder sepiapterin reductase (SPR) deficiency have been attributed to 5-HTP therapy [5].
Supplementation of Enzyme Co-Factors
Further strategies to increase central neurotransmitter availability include co-factor supplementation to boost enzyme activity and thereby increase bioavailability of monoamines. There are two key enzyme co-factors in the monoamine neurotransmitter synthesis pathway, as previously discussed (Fig. 1), namely tetrahydrobiopterin (BH4) and pyridoxal phosphate (B6).
Primary synthesis of dopamine and serotonin through TH and tryptophan hydroxylase is dependent on BH4, and thus BH4 deficiency leads to reduced levels of both neurotransmitters. Three known BH4 deficiency disorders (autosomal recessive GCH, dihydropteridine reductase [DHPR], and 6-pyruvoyl-tetrahydropterin synthase [PTPS] deficiency) are associated with hyperphenylalaninemia and, in these, BH4 co-factor supplementation may be utilized and is more commonly given as combination treatment with l-dopa. BH4 may help to reduce phenylalanine levels, as significantly raised phenylalanine is associated with acute neurological presentation with encephalopathy and longer-term neurological sequelae. BH4 supplementation has been used infrequently with pterin defects without hyperphenylalaninemia (sometimes in SPR deficiency and very rarely in AD GTP cyclohydrolase [GCH] deficiency). Again, the indication for BH4 supplementation is dependent on knowledge of the specific pterin defect identified.
B6 is another essential co-factor for reactions catalyzed by AADC, required for the conversion of 5-HTP and l-dopa to serotonin and dopamine, respectively, and thus an essential co-factor supplement in AADC deficiency to potentiate residual enzyme activity.
Use of Monoamine Analogs
Dopamine agonists are used to directly stimulate the postsynaptic dopamine receptors in disorders of dopamine deficiency (Fig. 2). Agonists are subdivided into D1-like (D1, D5) and D2 (D2, D3, D4) receptors [6]. Several dopamine agonists are available, including those with an ergoline structure (bromocriptine, pergolide, cabergoline) as well as non-ergoline structured compounds (rotigotine, pramipexole, ropinirole, aripiprazole). Each has an individual dopamine receptor affinity profile, although, to date, the clinical application of differing properties is unclear. The dopamine agonists used in movement disorders have higher binding affinity towards D2 receptors and are commonly tried as a second-line agent in children who have either not tolerated or not responded to l-dopa therapy in our clinical practice. The main side effects of dopamine agonists are nausea and vomiting, which can be managed well with concomitant use of domperidone, and clinicians should be vigilant for the occurrence of excessive sleepiness and emotional lability. In general, if a patient has failed to gain significant benefit from one dopamine agonist, it is unlikely another will be successful, although it may be helpful to consider alternative preparations such as rotigotine patches that allow transdermal controlled drug release over 24 h if dose-related fluctuations have been experienced with orally administered agents.
Use of Agents That Modify Metabolism of Dopamine/Serotonin
Monoamine oxidase type A (MAO-A) primarily metabolizes norepinephrine and serotonin, whilst MAO type B (MAO-B) primarily metabolizes dopamine into homovanillic acid (HVA) (Fig. 1). MAO-B inhibitors are therefore used to prevent the metabolism of dopamine at the synaptic level. Selegiline is commonly used in children (Fig. 2). MAO-B inhibitors can be used as an alternative/adjunct therapy in conditions unresponsive to l-dopa or to enable reduction of l-dopa dose. Selegiline is the l-isomer of N-propynyl-methamphetamine and an irreversible inhibitor of MAO-B. It is metabolized to an amphetamine derivative that can lead to stimulant effects [7]. This may be beneficial but can have intolerable side effects, including sleep disturbance and hallucinations, as well as dry mouth and hypotension [7]. At higher doses of more than 10 mg, selegiline loses its selectivity and can inhibit both MAO-A and MAO-B enzymes. MAO-A is also involved in the metabolism of tyramine, which is contained in foods such as cheese, and thus blockade of MAO-A is associated with raised tyramine levels and subsequently can lead to hypertensive crisis, especially when l-dopa is also given [7].
Selective serotonin-reuptake inhibitors (SSRIs) are also used in childhood neurotransmitter disorders associated with low cerebral serotonin. Their mode of action is to increase the extracellular level of serotonin by inhibiting its presynaptic reuptake, thereby boosting endogenous serotonin levels. They are most commonly used in psychiatric disorders but have been used in neurotransmitter movement disorders. Fluoxetine has been used in AADC deficiency [8] because it exhibits high-affinity binding to the serotonin transporter, inhibiting reuptake inhibition of synaptic serotonin and enhancing neurotransmission with minimal effects on norepinephrine and dopamine uptake, and has minimal binding to postsynaptic receptors [9]. Other drugs targeting the serotonergic system have been used with variable response, including zolmitriptan and buspirone in AADC deficiency [10]. The use of MAO-B inhibitors (selegiline) in conjunction with SSRIs can lead to serotonergic crisis with clinical features of dyskinesia, confusion, and hypertension and is managed with combined withdrawal of both medications.
The Role of Folate
Cerebral folate deficiency (CFD) is characterized by decreased levels of cerebrospinal fluid (CSF) 5-methyltetrahydrofolate in the presence of normal peripheral serum folate. This causes a broad clinical spectrum of neurological disorders, including epilepsy, learning difficulties, and motor disorders. Primary CFD is attributed to pathogenic mutations in FOLR1 [11], but CFD can also be secondary to another neurological disorder (such as a mitochondrial cytopathy), drug-induced or idiopathic. CFD is specifically associated with DHPR and AADC deficiency. Treatment with folinic acid has been successful in restoring cerebral folate levels and thus it is important to supplement with folinic acid to prevent further neurological impairment. DHPR is thought to play an important role in maintaining folate in its active form [12]. It is recommended that folate replacement with oral calcium folinate/folinic acid is instigated routinely in DHPR deficiency. Furthermore, the catabolism of excess l-dopa in AADC deficiency by catechol-O-methyltransferase (COMT) to 3-OMD is dependent on the methyl donor S-adenosylmethionine (SAM), which in turn can deplete 5-methyltetrahydrofolate [13, 14]. There is therefore a risk of CFD in both AADC deficiency and other neurotransmitter disorders requiring chronic l-dopa supplementation that may also necessitate supplementation with folinic acid.
In this review, we delineate the clinical features of the dopamine pathway neurotransmitter disorders, and describe disorder-specific treatment rationales and strategies, ancillary pharmacotreatments, and novel therapeutic advances.
Disorders of Tetrahydrobiopterin (BH4) Synthesis
BH4 deficiency represents a heterogeneous group of metabolic disorders caused by defects in the pterin synthesis or regenerating pathway (Fig. 1). Disorders of BH4 deficiency may result in hyperphenylalaninemia in some conditions, as well as decreased monoamine levels and folate levels in the CSF. Autosomal recessive GCH-I, DHPR, and PTPS deficiencies are classically recognized as BH4 biosynthesis disorders leading to hyperphenylalaninemia. In contrast, AD GCH deficiency, SPR deficiency, and pterin-4α-carbinolamine dehydratase (PCD) deficiency present without hyperphenylalaninemia.
BH4 deficiencies are treatable disorders and thus thorough diagnostic investigations are mandatory in all patients with (1) clinical symptoms of dopamine/serotonin deficiency, (2) elevated bloodspot phenylalanine levels detected in newborn screening, and (3) unexplained neurological presentations and raised phenylalanine detected on plasma amino acid analysis during neurological investigations [15]. The diagnostic work-up of suspected BH4 deficiencies includes (1) the analysis of pterins (neopterin, biopterin, and primapterin) in urine or dried blood spots (DBSs), (2) the measurement of DHPR activity in the DBSs, (3) CSF HVA and 5-hydroxyindoleacetic acid (5-HIAA) (are usually decreased, except in some mild forms of PTPS), and (4) CSF pterin analysis (characteristic CSF pterin profiles for each BH4 disorder).
Autosomal Dominant Guanosine Triphosphate (GTP) Cyclohydrolase Deficiency
The rate-limiting step of BH4 biosynthesis is the GCH-catalyzed conversion of GTP to dihydroneopterin triphosphate. Both AD and recessive forms of the disease are recognized. AD GCH deficiency is also commonly known as Dopa-responsive dystonia (DRD), Segawa disease, and DYT5a [16], and is the most commonly recognized form of inherited dystonia in childhood, with an incidence of 0.5 per 100,000 [17] (Table 1). It has a female predominance, with a reported female-to-male ratio from 2:1 to 6:1 [17] and a classical clinical presentation of childhood-onset postural dystonia at modal age 6 years (range 1–11 years) [16]. The postural dystonia most commonly presents in a lower limb, most classically with unilateral in-turning of the foot (pes equinovarus) [16]. The most distinctive features of AD GCH deficiency include the marked diurnal fluctuation (often manifesting as ‘evening equinus’) [18] as well as the obvious and sustained response to l-dopa, leading to resolution of motor symptoms in the majority of cases [18, 19]. Segawa [18] has subsequently further delineated the spectrum of clinical phenotypes categorized by age at onset and postulated movement presentation, relating basal ganglia maturation to pathophysiological mechanism. Childhood onset at a slightly older age (8 years) commonly presents with a different phenotype of action dystonia of the neck and shoulder causing retrocollis and may also be associated with oculogyric crisis. Onset after 10 years of age commonly presents with an asymmetric upper limb postural tremor, whilst adult-onset presentation includes upper limb tremor with some features of parkinsonism (such as rigidity) affecting gait [19]. Other movement phenotypes have been described in adults, such as writer’s cramp and spasmodic dysphonia [20]. Clinicians should be aware of atypical presentations such as apparent ‘diplegic cerebral palsy’, ‘hereditary spastic paraparesis’, and paroxysmal exercise-induced dyskinesia, as the response to l-dopa can be profoundly life altering, with multiple reports of restoration of ambulation and full resolution of motor symptoms in the literature [21–23].
The diagnosis of GCH deficiency is achieved through a combination of biochemical, genetic, and therapeutic approaches. The biochemical hallmark identified on CSF neurotransmitter analysis reveals low levels of HVA (± HIAA) and decrease in biopterin and neopterin levels, with normal plasma phenylalanine levels. CSF findings in AD GCH deficiency are not always classical and other biochemical and genetic investigations are often required to make this diagnosis. As well as its role in the monoamine synthesis pathway, BH4 is also a cofactor in the action of phenylalanine hydroxylase in the liver for phenylalanine clearance. The oral phenylalanine load can therefore be used to determine a pterin defect in patients with DRDs, particularly those without hyperphenylalaninemia. A 100 mg/kg load of phenylalanine is administered, and phenylalanine levels are measured at 1, 2, 4, and 6 h. A raised phenylalanine:tyrosine ratio would suggest a BH4 pathway defect, especially if there was associated decrease in biopterin concentration [24]. Phenylalanine loads need to be interpreted with caution, as false-positive and -negative results are sometimes seen; the test is neither 100 % sensitive nor specific for pterin defects [25]. Genetic confirmation of GCH deficiency is reached through testing for heterozygous mutations of the GCH1 gene on chromosome 14q22.1-q22.2, of which over 100 mutations have been identified to date. However, variable penetrance has been observed [20].
Treatment of Autosomal Dominant (AD) GTP Cyclohydrolase Deficiency
In AD GCH deficiency, the cardinal clinical feature of dopa responsiveness is elicited by a therapeutic trial of l-dopa. A trial of combined l-dopa/carbidopa is initiated at low dose (0.5–1 mg/kg/day) and gradually titrated to a standard treatment do l-dopa it is unlikely that the diagnosis is AD GCH deficiency [18] (Table 1). l-dopa is effective in almost all patients without relation to age of onset or longevity of clinical course [18, 19]. Gradual titration is recommended, as rapid increment of dose can lead to aggravation of active dystonia symptoms and intolerable dyskinesia. These side effects are ameliorated with dose reduction [18, 26]. We recommend the use of l-dopa in preparations containing adjunct carbidopa (in ratios of l-dopa:carbidopa of 4:1–10:1) to limit peripheral l-dopa side effects, as previously discussed. Response to l-dopa is sustained, and the motor complications secondary to tolerance encountered in long-term l-dopa therapy (such as those seen in Parkinson’s disease) are not observed in AD GCH deficiency. The natural course of AD GCH deficiency is an initial progressive dystonia that stabilizes, and there is usually no requirement to increment the l-dopa dosage with age or length of treatment [19]. In contrast to many other conditions of dopamine deficiency, it has been observed that there is a decrease in l-dopa dosage requirement to control symptoms over time, as reported in 20 % of adult patients who developed l-dopa-related dyskinesia at long-term follow-up [27].
There have been few reports of other pharmacotherapies used in AD GCH deficiency. Additional BH4 combined with l-dopa has been used infrequently and reported to achieve complete resolution of symptoms, but BH4 monotherapy has not been successful [18]. This is recapitulated in animal models, where it has been shown that administration of BH4 has failed to stimulate dopamine synthesis despite brain BH4 levels being elevated sevenfold [28].
Autosomal Recessive GTP Cyclohydrolase Deficiency
Autosomal recessive GCH deficiency is characterized by a complex neurological syndrome of truncal hypotonia, dystonia, other movement disorders, autonomic dysfunction, seizures, and developmental delay [29] (Fig. 1). Affected neonates are often identified on newborn screening (raised phenylalanine level). CSF HVA and 5-HIAA, BH4, neopterin, and urine pterins are all reduced. The diagnosis is made through genetic testing for homozygous or compound heterozygous mutations in the GCH1 gene, and enzymatic activity can be assayed in skin fibroblasts [29]. More recently, reports have suggested a continuous spectrum between AD and autosomal recessive GCH deficiency, and the division between dominant and recessive disease forms appears less distinct than previously reported. The distinguishing biochemical feature of hyperphenylalaninemia in recessive disease is no longer a key feature, with several reports of autosomal recessive GCH deficiency presenting without hyperphenylalaninemia and infantile-onset neurological symptoms [20, 30, 31].
Treatment of Autosomal Recessive GTP Cyclohydrolase Deficiency
Supplementation of BH4 (titrated dosage 1–10 mg/kg/day) is used to augment the activity of phenylalanine hydroxylase [33] (Table 1). The use of BH4 monotherapy does not appear to sufficiently increase the synthesis of monoamine neurotransmitters, and l-dopa and 5-HTP supplementation are often also required. In autosomal recessive GCH deficiency, higher doses of l-dopa are required than in AD GCH, titrated up to 10 mg/kg/day. 5-HTP is also titrated up to doses of 8 mg/kg/day [33].
Pyruvoyl-tetrahydropterin Synthase (PTPS) Deficiency
PTPS is the most common pterin defect associated with hyperphenylalaninemia [32, 33] (Fig. 1). There is a wide phenotypic spectrum, ranging from mildly affected cases (children may be relatively asymptomatic on medication) to quite severe forms of disease. Neonates with PTPS are frequently small for gestational age with poor suck, hypotonia, and microcephaly [34]. The range of movement disorders observed include hypotonia, hypokinesia, rigidity, chorea, dystonia, and oculogyric crisis and, in the severe form, can be associated with learning disabilities, epilepsy, and psychiatric features [25].
In the majority, hyperphenylalaninemia is detected on neonatal blood spot screening with specific features of reduced biopterin and raised neopterin concentrations in urine. In the majority, both CSF HVA and 5-HIAA are decreased but may also be normal in some patients [34]. PTPS deficiency is caused by mutations in the PTS gene, and the disease is more frequently observed in Asian populations. There is good genotype–phenotype correlation, with mild phenotypes associated with mutations preserving high residual enzyme activity reported [35].
Treatment of PTPS Deficiency
Early diagnosis and treatment is possible, with detection through neonatal hyperphenylalaninemia screening on blood spots (Table 1). The pharmacological aim is to replace BH4 (1–12 mg/kg/day). l-dopa/carbidopa (with l-dopa at 4–18 mg/kg/day) and 5-HTP (3–10 mg/kg/day) are also given, with administration of these three drugs initiated at low dose with slow increments to avoid side effects [36]. Medication is usually divided into three to four doses per day to avoid off-on phenomena. Other agents used as adjunctive or alternatives to l-dopa include selegiline, entacapone, and pramipexole [36, 37].
Sepiapterin Reductase (SR) Deficiency
SPR deficiency is a rarer autosomal recessive pterin disorder with normal phenylalanine levels caused by mutations in the SPR gene (Fig. 1). The triad of upward gaze, paroxysmal stiffening, and hypotonia tends to occur early in infancy as one of the earliest symptoms [38]. The core clinical features are axial hypotonia, motor and language delay, oculogyric crisis, weakness, and dystonia, with diurnal fluctuation of symptoms [5]. Parkinsonian features, sleep disturbance, and behavioral and psychiatric abnormalities are also frequently associated. Only 8 % have normal cognitive abilities, with the majority having mild to moderate learning disability [5, 39]. The diagnosis is made by CSF analysis, which shows low HVA and HIAA with raised total biopterin, dihydrobiopterin (BH2) and sepiapterin levels. Urine pterins and plasma phenylalanine levels are normal, but the phenylalanine load test is frequently positive.
Treatment of SR Deficiency
The enzyme activity of SPR is reduced and therapeutic approaches involve dopamine and serotonin precursor supplementation (Table 1). Most patients display a dramatic response to l-dopa/carbidopa, with improvement in motor and sleep symptoms [40]. Most patients seem to respond to combination therapy of l-dopa and 5-HTP. Some experience side effects such as dose-related dyskinesia; therefore, a very low starting dosage (~0.5–2 mg/kg/day in three to four doses) with slow increment to the target dose is recommended. The independent benefits of 5-HTP have been difficult to assess, but modest improvement in sleep, motor, and cognitive aspects have been reported, with 5-HTP doses ranging from 1 to 6 mg/kg/day. The majority of patients continue to have mild motor and cognitive problems. The side effects of vomiting, diarrhea, and abdominal pain are reported with 5-HTP use in this condition but are resolved by reduction in dose [5].
Disorder of BH4 Regeneration
Dihydropteridine Reductase (DHPR) Deficiency
DHPR deficiency results in a defect of BH4 regeneration, and is usually detected on newborn screening (hyperphenylalaninemia) and subsequently confirmed on direct enzyme activity measurement on DBS (Fig. 1). The clinical features are recognized to be more severe than in the other pterin defects, with disease onset in early infancy or even earlier in the neonatal period, presenting with feeding difficulties, bulbar dysfunction, hypersalivation, microcephaly, and developmental delay. Associated movement disorders include limb hypertonia with truncal hypotonia, dyskinesia, tremor, dystonia, and choreoathetosis [33]. Learning disability and seizures are observed frequently in this group. Early recognition and prompt treatment of DHPR deficiency is vital, as 40 % of DHPR-deficient neonates are asymptomatic [33]. CSF neurotransmitter profile shows low concentrations of HVA, HIAA, and folate, raised BH2, and raised/normal biopterin levels. The disorder is caused by mutations in the QDPR gene.
Treatment of DHPR Deficiency
The therapeutic aims are to replace CSF dopamine and serotonin as previously described in other pterin disorders, with l-dopa/carbidopa (5–13 mg/kg/day of l-dopa) and 5-HTP (3–11 mg/kg/day) [33] (Table 1). The use of monoamine oxidase inhibitors (MAOIs) and dopamine agonists, such as selegiline, entacapone, and pramipexole, has been reported in a recent international survey on patients with BH4 deficiency [33]. The use of BH4 supplementation remains controversial, as DHPR deficiency patients accumulate 7,8 BH2, which is considered to be neurotoxic with additional inhibitory effects on aromatic amino acid hydroxylases. BH2 has also been reported to uncouple neuronal nitric oxide synthase (NOS) [41]. In addition, DHPR is thought to play an important role in maintaining folate in its active form [40]. The risk of associated CFD (that may cause further neurological impairment) has led to the recommendation that replacement with oral calcium folinate/folinic acid is commenced routinely in DHPR deficiency.
Pterin 4-α Carbinolamine Dehydratase Deficiency
Pterin 4-α carbinolamine dehydratase is required for the regeneration of BH4 after phenylalanine hydroxylation, and the enzyme deficiency leads to a mild form of hyperphenylalaninemia associated with raised levels of urinary 7-biopterin. PCD deficiency is caused by mutations in the PCDB gene; affected patients may present with transient hypertonia but the majority are asymptomatic with normalization of the phenylalanine with normal diet. The neurotransmitters are normal, and this condition is considered benign [32] (Fig. 1).
Disorder of Dopamine Synthesis
Tyrosine Hydroxylase (TH) Deficiency
TH deficiency is an autosomal recessive condition caused by mutations in the TH gene (Fig. 1). TH catalyses the conversion of tyrosine to l-dopa, which is the rate-limiting step in catecholamine biosynthesis. This leads to reduced CSF HVA with normal HIAA and reduced ratio HVA:HIAA (usually <1.0). The clinical phenotypic spectrum is expanding, with two classical phenotypes recognized: (1) type A, with progressive extrapyramidal movement disorder with hypokinetic rigid syndrome and dystonia (onset in infancy or childhood), (2) and type B, with complex encephalopathy (onset in the neonatal period or early infancy) [42]. The majority of patients are identified with type A phenotype, and diurnal variation of symptoms is frequently observed.
Children with type B TH deficiency present within the first 3 months of life with severe Parkinsonism and hypotonia, with cognitive impairment affecting the majority. Focal or generalized dystonia with dystonic crises, oculogyric crises, tremor, ptosis, hypersalivation, and autonomic disturbance are also associated [42]. Up to 50 % of TH patients have hyperprolactinemia, which may rarely manifest as galactorrhea [42].
More recently, an intermediate clinical phenotype has been reported [43]. Additionally, within the type A subgroup, there are also some children who present later in childhood with gait instability and walking difficulties [42], suggesting an expanding phenotypic spectrum of disease.
Treatment of TH Deficiency
The dosing regimen for l-dopa treatment in TH deficiency needs to be tailored specifically to minimize l-dopa hypersensitivity, otherwise intolerable side effects of dyskinesia are observed, even at lower dose ranges [44] (Table 1). In type B TH deficiency, the initiating dose commences at <0.5 mg/kg/day (in four to six divided doses per day) with slow titration (over weeks to months) to upper dose ranges of 2 mg/kg/day according to response [42]. In type A disease, slow titration of l-dopa doses to treatment ranges between 3 and 10 mg/kg/day are used with reported response [42]. In those who tolerate l-dopa well, there is a good or moderate response, with improvement of motor symptoms and movement disorder. Cognitive abilities are often impaired but are stable in the majority [42]. The response to l-dopa can be variable and often ancillary pharmacological agents are used to managed dystonia (discussed in ancillary pharmacotherapies).
Aromatic l-Amino Acid Decarboxylase (AADC) Deficiency
The clinical presentation and phenotype of AADC deficiency is extremely variable, with onset ranging from 4 months to adulthood, although the majority of patients present in childhood. Hypotonia and oculogyric crisis are cardinal features of AADC deficiency; 50 % of patients present with other movement disorders, including hypokinesia, chorea, dystonia, and bulbar dysfunction [45] (Fig. 1). Associated features of monoamine neurotransmitter deficiency, such as sleep disturbance, autonomic dysfunction (primarily ptosis, excessive sweating, temperature instability, nasal congestion, hypersalivation, and hypotension), insufficient stress response, and irritability are also observed with cognitive impairment in the majority [45, 46]. The characteristic CSF neurotransmitter profile shows low HVA, 5-HIAA, and 3-methoxy-4-hydroxyphenylglycol with raised 5-HTP, l-dopa, and 3-ortho-methyldopa. AADC enzyme activity in plasma is very low or absent and urinary catecholamines are raised (vanillactic acid, 3-orthomethyldopa) [45]. There are no specific neuroimaging features, but a spectrum of non-specific changes are reported, including cerebral atrophy, white matter abnormalities, and thinning of the corpus callosum. Just under 100 patients have been identified with AADC deficiency caused by mutations in the DDC gene, with a founder mutation identified in Taiwanese Chinese patients (IVS6+4A>T) and one-third of known patients being of Southern Chinese origin [47].
Treatment of AADC Deficiency
The pharmacological treatment of AADC usually includes a combination of dopamine agonists, MAOIs, pyridoxine and folinic acid [10] (Table 1). l-Dopa is not currently used in the majority of AADC deficiency patients, as l-dopa accumulation leads to depletion of SAM that is potentiated by the AADC deficiency [48]. However, three siblings with an homozygous point mutation (c.387 G>A) in exon 3 showed significant response to l-dopa and pyridoxal-5-phosphate, with improvement in dystonia but persistence of behavioral problems. This specific mutation leads to a distinct decreased affinity between AADC enzyme and l-dopa, affecting the catalytic site or binding site for the pyridoxal phosphate (PLP) cofactor and thus l-dopa is therapeutic [13].
Supplementation with the AADC enzyme cofactor pyridoxine to a maximum dose of 200 mg/day is standard treatment for AADC deficiency. AADC deficiency is treated with vitamin B6 in the form of pyridoxine with the aim of boosting residual AADC activity. Pyridoxine is first phosphorylated to pyridoxine 5′-phosphates by pyridoxine kinase and subsequently converted to PLP by pyridox(am)ine 5′-phosphate oxidase (PNPO) [49]. There does not appear to be a clinical response to pyridoxine treatment [46], but biochemical response has been observed with increased CSF HVA levels (vitamin B6 at 100 mg/kg per day) [50] and increased plasma serotonin levels (vitamin B6 at 200 mg/day) [51]. An efficacious dose of pyridoxine in AADC deficiency has not been established. Often vitamin B6 dose reduction is necessary due to gastrointestinal side effects [13, 14]. There is growing in vitro evidence that an optimal level of PLP is important for AADC maintenance and stability. Indeed, cell line models show reduced AADC enzyme levels and activity in PLP-depleted PC12 cells and, in the presence of PLP and an l-dopa analog, the AADC was protected from proteolysis [52]. Catabolism of excess l-dopa in this condition is initiated by COMT that converts l-dopa to 3-OMD. The process is dependent on the methyl donor SAM, which in turn can deplete 5-methyltetrahydrofolate [13, 14]. The risk of CFD in this condition therefore necessitates supplementation with folinic acid/calcium folinate.
Adjunct treatments are frequently used in AADC deficiency and include dopamine receptor agonists such as pergolide, bromocriptine, pramipexole, and ropinirole. More recently, rotigotine patches have been used with some success [48]. However, the response to dopamine agonists is highly variable, with some reported improvements in voluntary movements and absence of response in others [48, 53].
MAOIs such as selegiline and tranylcypromine are also used frequently in AADC deficiency. The action of MAO-A inhibitors is to prevent the breakdown of dopamine and leads to accumulated dopamine and stimulated receptors that can have an upstream feedback effect of ultimately decreasing AADC activity [54]. MAO-B inhibitors act on increasing AADC messenger RNA (mRNA) and protein potentiating the neuronal response to dopamine without increasing dopamine or metabolite concentrations. This effect was observed to be dose dependent in mouse models receiving selegiline 1 mg/kg/day, with a selective effect on MAO-B inhibition and increase in AADC mRNA and protein, whilst high-dose 10 mg/kg/day selegiline treatment inhibits both MAO-A and -B that leads to overall AADC inhibition [55]. This effect of high-dose MAOIs and inhibition of AADC is also observed in high non-selective MAOI phenelzine [56].
Patients with AADC deficiency experience autonomic dysfunction due to reduced production of noradrenaline and adrenaline, and current autonomic interventions in AADC deficiency are usually targeted to specific symptoms such as oxymetazoline for nasal congestion. This is a sympathomimetic nasal spray that has been successfully used to reduce nasal congestion in one case [46]. In addition, serotonin is the precursor of melatonin; consequently, in AADC deficiency, melatonin is likely to be deficient and may be contributory to abnormal sleep patterns [10]. Melatonin can be given directly as a treatment and has been reported to be beneficial for the control of sleep disturbances in at least two cases [46].
Symptoms of dystonia in AADC deficiency have been treated with adjunct agents that are further discussed in the ancillary drug treatments section. Overall, the response to treatment in children with AADC deficiency is variable and often disappointing, which has resulted in the investigation of novel therapies such as gene therapy for this condition [57].
Pyridoxamine 5′-Phosphate Oxidase Deficiency
B6 is an essential cofactor for reactions catalyzed by AADC. Autosomal recessive pyridoxamine 5′phosphate oxidase (PNPO) deficiency leads to reduction in synthesis of B6 from pyridoxine and pyridoxamine [49]. This reduces recycling of B6 from pyridamine phosphate, which consequently impairs dopamine and serotonin. The neurotransmitter profile is sometimes (but not always) similar to that seen in AADC deficiency, with low CSF HVA and HIAA and high levels of l-dopa, 5-HTP, and 3OMD, with raised CSF glycine, taurine, histidine, and threonine). If this pattern of CSF neurotransmitters and amino acids is seen, AADC deficiency should be excluded by measurement of blood AADC activity and also exclusion of pyridoxine-dependent epilepsy (due to antiquitin gene mutations) through measurement of urinary aminoadipic semialdehyde. Diagnosis of PNPO deficiency is suspected if a low CSF B6 level is detected, and is confirmed through testing for pathogenic mutations in the PNPO gene on chromosome 17q21.32 [58]. Children present with a severe pharmaco-resistant neonatal epileptic encephalopathy, and often there is a history of in utero seizure onset and prematurity [59]. They respond to treatment with B6 (but not pyridoxine), which can lead to seizure reduction.
In suspected cases of PNPO deficiency or intractable neonatal/early infantile seizures, B6 should be trialed at 30 mg/kg/day in three to four divided doses for 3–5 days. There is risk of prolonged apnea during a trial of B6, and close cardiorespiratory monitoring and precautions for respiratory support should be in place as a safeguard. In confirmed cases, 30–50 mg/kg/day B6 daily should be used in the long term.
Disorders of Dopamine Synthesis Regulation
The PITX3 gene (Fig. 2) activates the TH promoter and thus regulates dopamine production [60]. Pitx3 (−/−) mice have a selective loss of dopaminergic neurons in the substantia nigra and ventral tegmental area, leading to the significantly reduced dopamine levels in the nigrostriatal pathway and in the dorsal striatum. The murine model manifests anomalous striatum-dependent cognitive impairment and neurobehavioral activity [61, 62]. Treatment with l-dopa, dopamine, or dopamine receptor agonists in these mice leads to reversal of several of their sensorimotor impairments. A deletion fragment at chromosome 10q24.32 leading to PITX3 gene deletion was identified in an adolescent with mild learning difficulties, psychiatric symptoms, hyperactivity, behavior problems, and sleep disturbance. He had distinctive facial features, including high forehead, open mouth, synophrys, short broad nose, and hypoplastic middle phalanges of fifth fingers. Neuroimaging was normal, but CSF neurotransmitter analysis shows low CSF HVA and HIAA, absent l-dopa, and low biopterin. l-dopa treatment was initiated at 1 mg/kg/day and increased to 2.5 mg/kg/day with improvement in behavior, attention, and sleep [63].
Disorders of Dopamine Transport
Brain Dopamine—Serotonin Vesicular Transport Disease
Brain dopamine–serotonin vesicular transport disease is a novel neurotransmitter ‘transportopathy’ that has been recently identified (Fig. 2). It is caused by mutations in the SLC18A2 that encodes for the vesicular monoamine transporter 2 (VMAT2) (Fig. 2). This protein is responsible for dopamine–serotonin loading into the synaptic vesicles for subsequent monoamine neurotransmission [64]. A loss-of-function mutation in SLC18A2 was identified in a highly consanguineous family with eight members affected. The clinical features at presentation in childhood included developmental delay and axial hypotonia in infancy, with an eye movement disorder and oculogyric crises. In adolescence, clinical features of bulbar dysfunction, sleep disturbance, ptosis, hypomimia, and facial dyskinesia, tremor, ataxia, and parkinsonian shuffling gait were reported. Neuroimaging studies were normal and CSF neurotransmitter profile (available in one case) was normal. The urinary neurotransmitters were abnormal, showing raised levels of HIAA, HVA, and low urinary epinephrine and dopamine. There was an adverse reaction to l-dopa, with deterioration in the movement disorder (worsening chorea and dystonia). However, a trial of the dopamine receptor agonist pramipexole resulted in complete and sustained amelioration of the parkinsonism–dystonia motor symptoms, leading to restoration of ambulation in some, with tolerable side effects of hyperactivity and weight loss observed [65].
Dopamine Transporter Deficiency Syndrome
Dopamine transporter deficiency is an autosomal recessive condition caused by pathogenic mutations of SLC6A3 that encodes for the dopamine transporter [64] (Fig. 2). Loss-of-function mutations lead to defective presynaptic uptake of dopamine, with accumulation of synaptic dopamine, which is catabolized and results in the characteristic raised HVA levels in CSF. The dopamine transporter does not affect the serotonin pathway and thus CSF HIAA levels are normal. The CSF neurotransmitter profile of raised CSF HVA:HIAA ratio >5 is described in all reported cases of this disorder [66]. DTDS clinically presents in infancy with feeding difficulties and irritability with axial hypotonia. Over time, children develop a progressive dystonic and dyskinetic movement disorder with eye involvement [66]. This condition is under-recognized and frequently misdiagnosed as dyskinetic cerebral palsy. During childhood, there is evolution of prominent parkinsonian features, with bradykinesia, hypomimia, rigidity, and resting tremor, with death in some patients in adolescence [66]. The phenotypic spectrum of this condition is expanding, with the first adults with DTDS now recognized, and indeed increasingly considered as a differential for monogenic causes of juvenile and early onset parkinsonism [67, 68]. The majority of patients are unfortunately unresponsive to nearly all currently available therapeutic agents, including l-dopa, anticholinergics, benzodiazepines, and deep brain stimulation. l-dopa and dopamine agonists such as ropinirole/pramipexole have been used with limited improvement in motor symptoms in a minority of patients [65].
Secondary Neurotransmitter Abnormalities
Abnormal neurotransmitter profiles are recognized in other neurological conditions and therefore are not an exclusive feature of disorders of dopamine synthesis or metabolism. These so-called ‘secondary’ neurotransmitter abnormalities are increasingly recognized and reported in several neurological disorders [69] (such as pontocerebellar hypoplasia, epilepsy, hypoxic-ischemic encephalopathy, mitochondrial disorders, infection, and primary genetic syndromes), and may be indicative of secondary dopaminergic derangement due to dopaminergic tract degeneration/dysfunction or effects on dopamine metabolism at a cellular level.
The clinical interpretation of HVA levels and therapeutic strategies can be challenging, and results will often require discussion with both the neurometabolic laboratory and a neurologist with neurotransmitter expertise [70]. In patients with low HVA of undetermined or secondary origin, the role of l-dopa remains unclear. Certainly, in some patients with obvious symptoms and signs of dopaminergic deficiency (such as Parkinsonism, dystonia, oculogyric crises), there may be a role for the slow introduction and gradual increase of l-dopa to tolerated doses. A proportion of these patients show improvement in motor function [71]. The possibility of treating such patients with l-dopa and 5-HTP for central dopamine and serotonin deficiency to improve clinical symptoms and neurological outcome should therefore be considered [70]. The use of neurotransmitter-replacement therapy in patients with CSF neurotransmitter abnormality in the absence of classical features of dopaminergic deficiency remains controversial.
Ancillary Pharmacotherapies
The disorders of monoamine neurotransmitter synthesis most commonly manifest with hyperkinetic symptoms such as dystonia, tremor, and chorea. Often, such symptoms do not fully respond to standard pharmacotreatments aimed at restoration of the defect in the monoamine pathway. Other adjunctive treatments are also often required. Dystonia, especially, can be difficult to manage in a number of disorders [72]. Symptomatic management aims to relieve abnormal postures, reduce discomfort, and improve function and range of movement, and thus requires the combined approach of pharmacotherapies with physical therapy and orthotic management to prevent contractures.
Anticholinergic medications are most beneficial in the treatment of generalized and segmental dystonia and overall, trihexyphenydyl is generally well tolerated, starting at a low dose of around 1 mg/day and gradually incrementing to a maximum of 12 mg/day. It is well recognized that children tolerate higher dosages than adults, and often doses are increased much beyond these recommended doses. The mechanism of action is likely to be a central antimuscarinic effect [73]. It is frequently used in AADC deficiency and also sometimes in the management of GCH deficiency, especially for those who have experienced l-dopa-related dyskinesia [17, 26, 74, 75]. The side effects are dose related and include drowsiness, confusion, hallucinations, urinary retention, constipation, dry secretions, anorexia, confusion, and psychosis [75]. These are best avoided through slow titration and can be ameliorated with dose reduction. Benztropine is another anticholinergic drug used for dystonia management; it has both anticholinergic and antimuscarinic effects, and blocks dopamine uptake by inhibiting presynaptic uptake. Doses of 1–4 mg/day are recommended [75].
GABA-ergic agents are also used in the management of dystonia. Benzodiazepines can also be used in intractable dystonia and act by binding to the GABA2 receptors-ion channel complex, thereby increasing chloride entry into the cell and enhancing GABA-mediated inhibition. The main side effect is sedation that can be minimized through careful titration of dose. Oral baclofen is a GABA agonist that stimulates the metabotropic GABA-B autoreceptor, but the mechanism of benefit for dystonia is unknown. The starting dose is 0.25 mg/kg three times a day (tid), increasing every 3 days to maximum age-related dose (recommendations 5 mg tid in children aged >10 years). However, higher doses (with doses being gradually increased to avoid side effects) are often well tolerated in older children/adolescents. From personal experience, many older children tolerate the adult dosing recommendations of 10 mg tid. The main side effects are lethargy, dry mouth, and dizziness; rapid withdrawal may produce seizures, and tapered withdrawal is advised.
Clonidine, an α adrenergic agonist, is widely used for tics in Tourette syndrome and in paroxysmal autonomic instability with dystonia (PAID) but is now increasingly used in generalized dystonia. The main side effect is hypotension; sedation and an in-hospital test dose of 0.1 mg/kg is advised at the start of a treatment trial. The initial dose is 0.1 mg/kg/day in three divided doses, increasing gradually to 3 mg/kg/day.
Tetrabenazine is also used in hyperkinetic disorders and is often a first-/second-line agent for chorea. It is a benzoquinolizine that acts by inhibiting VMAT2 and prevents repackaging of dopamine and serotonin into vesicles for neurotransmission; it leads to monoamine neurotransmitter depletion and in particular dopamine depletion [76]. This reduces dopaminergic neurotransmission in hyperkinetic movement disorders; clinicians should be aware of the main side effects of sedation, Parkinsonism, and low mood.
In AADC deficiency and DTDS, episodes of dystonic crisis are frequently experienced and require emergency management to prevent complications such as extreme pain, severely disrupted sleep pattern, abnormal posture preventing seating, rhabdomyolysis, myoglobinuria, and dystonia-triggered diaphragmatic splinting, and respiratory failure. This often requires hospitalization for aggressive muscle relaxation and sedation and to ablate status dystonicus, with treatments such as intravenous midazolam infusion, morphine, and, in some cases, general anesthesia.
Gabapentin has been used for neuropathic pain and adult movement disorders such as restless leg syndrome. A recent study reported improvement in dystonia in 49 children with associated improvement in transfers, sleep, sitting tolerance, mood, tone, involuntary contractions, and pain. No unexpected side effects were reported at higher doses [76]. This is an encouraging novel indication for gabapentin, but further evaluation is required to assess dosage requirement and response.
Future Treatments
A proportion of dopamine metabolism disorders respond satisfactorily to monoamine replacement and enhancement pharmacotreatments, whilst others respond poorly, such as patients with AADC deficiency and DTDS. Thus, novel treatments are being developed.
Alternative novel treatment strategies are in the early stages of in vitro development, and include studies on pseudoexom-exclusion therapy with antisense morphilino oligonucleotides. Using this approach, the PTPS enzyme activity in fibroblast cell lines from three PTPS-deficient patients with intronic mutations resulting in splicing defects was restored [77].
In addition the role of COMT inhibitors for the treatment of neurotransmitter disorders may need further exploration, especially as they have been trialed in Parkinson’s disease. Early pharmacological agents were highly toxic and short acting with poor bioavailability; however, there have been several approaches to develop novel COMT inhibitors that are safe and efficacious as future therapeutic agents [78].
Furthermore, gene transfer technologies have been developed to deliver an adeno associated virus (AAV) encoding hAADC for stable expression of AADC enzyme in the striatum. This experimental technology has previously been utilized in clinical trials for adult Parkinson’s disease. AAV hAADC gene therapy was reported to show stable expression, with improvement in parkinsonian rating scores and reduction in dopaminergic therapy dosage and clinical safety [79]. Such studies have paved the way for clinical trials in gene therapy for AADC deficiency. Recently, four children underwent AAV-mediated transfer of human AADC gene through stereotactic injection bilaterally to the putamen. This has shown promising results, with improvement in motor symptoms in all the patients, clinical safety, and increased dopamine and serotonin levels following gene transfer [57]. Larger clinical trials will be more informative as to the relative benefit of gene therapy in this group of patients.
Research into dopamine metabolism and dopaminergic neurons was previously limited by the availability of in vivo and in vitro models of dopaminergic neuronal disease. With the advent of pathogenic dopaminergic neuronal modeling through induced pluripotent stem cells, the potential for future research to inform disease mechanisms and develop novel treatments such as enzyme-replacement therapy and gene therapy appear highly promising.
Conclusion
Childhood neurological disorders of dopamine dysregulation are increasingly recognized as an expanding group of inherited neurometabolic syndromes. They remain challenging conditions to diagnose, often mimicking other neurological syndromes. The principles of treatment are to replace central monoamine neurotransmitters, prevent monoamine breakdown, enhance enzyme activity, and increase dopamine receptor activity. For some disorders, therapeutic interventions can ablate all symptoms but this is far from universal in this group of disorders. Patients will often need adjunct therapy for symptomatic control of features such as dystonia, sleep disturbance, and gastrointestinal symptoms. Treatment is also governed by clinical response and tolerability of side effects. Treatment strategies are often specific to the neurotransmitter disorder subtype (Table 1); therefore, knowledge of the biochemical pathway of monoamine neurotransmitter synthesis metabolism underpins accurate diagnosis and appropriate therapeutic options in this spectrum of disorders. Future research will no doubt elucidate novel therapeutic strategies to ameliorate the more pharmaco-resistant disorders of dopamine metabolism.
Acknowledgments
We thank Dr Reema Alkhater for her personal communication on the drug treatment of Brain dopamine-serotonin vesicular deficiency. MAK is funded by a Wellcome Intermediate Clinical Fellowship. JN is funded by a Medical Research Council (MRC) Clinical Research Training Fellowship. Both MAK and JN are funded in addition by Great Ormond Street Hospital Children’s Charities (GOSHCC). The authors did not receive specific funding for preparation of this manuscript and declare no conflicts of interest.
References
- 1.Goldstein DS. Catecholamines in the periphery. Overview Adv Pharmacol. 1998;42:629–639. doi: 10.1016/s1054-3589(08)60806-6. [DOI] [PubMed] [Google Scholar]
- 2.Yildiz O, Smith JR, Purdy RE. Serotonin and vasoconstrictor synergism. Life Sci. 1998;62:1723–1732. doi: 10.1016/S0024-3205(97)01166-1. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez-Alvarez E. Movement disorders in children: recent advances in management. Indian J Pediatr. 2009;76:531–536. doi: 10.1007/s12098-009-0138-6. [DOI] [PubMed] [Google Scholar]
- 4.Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351:2498–2508. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- 5.Friedman J, Roze E, Abdenau JE, et al. Sepiapterin reductase deficiency: a treatable mimic of cerebral palsy. Ann Neurol. 2012;71:520–530. doi: 10.1002/ana.22685. [DOI] [PubMed] [Google Scholar]
- 6.Seeman P, Van Tol HHM. Dopamine receptor pharmacology. Trends Pharmcol Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 7.Westlund KN. The distribution of monoamine oxidases A and B in normal human brain. In: Leiberman A, Olanow CW, Youdim MBH, Tipton K, editors. Monoamine oxidase inhibitors in neurological disease. New York: Marcel Dekker; 1994. pp. 1–20. [Google Scholar]
- 8.Manegold C, Hoffmann GF, Degen I, et al. Aromatic l-amino acid decarboxylase deficiency: clinical features, drug therapy and follow up. J Inherit Metab Dis. 2009;32:371–380. doi: 10.1007/s10545-009-1076-1. [DOI] [PubMed] [Google Scholar]
- 9.Bobo WV, Shelton RC. Efficacy, safety and tolerability of SymbyaxAR for acute-phase management of treatment-resistant depression. Expert Rev Neurother. 2010;10:651–670. doi: 10.1586/ern.10.44. [DOI] [PubMed] [Google Scholar]
- 10.Allen GF, Land JM, Heales SJR. A new perspective on the treatment of aromatic l-amino acid decarboxylase deficiency. Mol Genet Metab. 2009;97:6–14. doi: 10.1016/j.ymgme.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Grapp M, Just IA, Linnankivi T, et al. Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain. 2012;135(Pt 7):2022–2031. doi: 10.1093/brain/aws122. [DOI] [PubMed] [Google Scholar]
- 12.Porta F, Mussa A, Concolino D, et al. Dopamine agonists in dihydropteridine reductase deficiency. Mol Genet Metab. 2012;105:582–584. doi: 10.1016/j.ymgme.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Chang YT, Sharma R, Marsh JL, et al. Levodopa responsive aromatic l-amino acid decarboxylase deficiency. Ann Neurol. 2004;55:435–438. doi: 10.1002/ana.20055. [DOI] [PubMed] [Google Scholar]
- 14.Surtees R, Hyland K. l-3,4-dihydroxyphenylalanine (Levodopa) lowers central nervous system S-adenosylmethionine concentrations in humans. J Neurol Neurosurg Psychiatry. 1990;53:569–572. doi: 10.1136/jnnp.53.7.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet Neurol. 2010;376:1417–1427. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- 16.Segawa M. Autosomal dominant GTP cyclohydrolase 1 (AD GCH1) deficiency (Segawa disease, dystonia 5, DYT 5) Chang Gung Med J. 2009;32:1–11. [PubMed] [Google Scholar]
- 17.Furukawa Y, Pagon RA, Adam MP, et al. GTP cyclohydrolase 1-deficient dopa-responsive dystonia. Gene reviews. Seattle: University of Washington; 1993–2013.
- 18.Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain Dev. 2011;22:195–201. doi: 10.1016/j.braindev.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol. 1976;14:215–233. [PubMed] [Google Scholar]
- 20.Trender-Gerhard I, Sweeney MG, et al. Autosomal-dominant GTPCH1-deficiency DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry. 2009;80:839–845. doi: 10.1136/jnnp.2008.155861. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Ki CS, Kim DS, et al. Dopa-responsive dystonia with a novel initiation mutation in the GCH1 gene misdiagnoses as cerebral palsy. J Korean Med Sci. 2011;26:1244–1246. doi: 10.3346/jkms.2011.26.9.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jan MMS. Misdiagnosis in children with Dopa responsive dystonia. J Pediatr Neurol. 2004;31:298–303. doi: 10.1016/j.pediatrneurol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Dale RC, Melchers A, Fung VS, et al. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev Med Child Neurol. 2010;52:583–586. doi: 10.1111/j.1469-8749.2010.03619.x. [DOI] [PubMed] [Google Scholar]
- 24.Saunders-Pullman R, Blau N, Hyland K, et al. Phenylalanine loading as a diagnostic test for DRD interpreting the utility of the test. Mol Genet Metab. 2004;83:207–212. doi: 10.1016/j.ymgme.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Kurian MA, Gissen P, Smith M, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol. 2011;10:721–733. doi: 10.1016/S1474-4422(11)70141-7. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Laso E, Beyer K, Opladen T, et al. Dyskinesias as a limiting factor in the treatment of Segawa disease. Pediatr Neurol. 2012;46:404–406. doi: 10.1016/j.pediatrneurol.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang WJ, Calne JKC, Tsui R, et al. The long-term response to levodopa in dopa-responsive dystonia. Parkinson Relat Disord. 2008;8:1–5. doi: 10.1016/S1353-8020(00)00084-5. [DOI] [PubMed] [Google Scholar]
- 28.Sumi-Ichinose C, Urano F, Kuroda R, et al. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. A study from 6-pyruvolytetrahydrobiopterin synthase knockout mice. J Biol Chem. 2001;276:41150–41160. doi: 10.1074/jbc.M102237200. [DOI] [PubMed] [Google Scholar]
- 29.Bonafe L, Thony B, Leimbacjer W, et al. Diagnosis of dopa-responsive dystonia and other tetrahydrobiopterin disorders by the study of biopterin metabolism in fibroblasts. Clin Chem. 2001;47:477–485. [PubMed] [Google Scholar]
- 30.Horvath GA, Stockler-Ipsiroglu SG, Salvarinova-Zivkovi R, et al. Autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia: Evident of a phenotypic continuum between dominant and recessive forms. Mol Genet Metab. 2008;94:127–131. doi: 10.1016/j.ymgme.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Opladen T, Hoffmann G, Horster F, et al. Clinical and biochemical characterisation of patients with early infantile onset of autosomal recessive GTP cyclohydrolase 1 deficiency without hyperphenylalaninemia. Mov Disord. 2011;25:157–161. doi: 10.1002/mds.23329. [DOI] [PubMed] [Google Scholar]
- 32.Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis. 2009;32:333–342. doi: 10.1007/s10545-009-1067-2. [DOI] [PubMed] [Google Scholar]
- 33.Opladen T, Hoffmann GF, Blau N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J Inherit Metab Dis. 2012;35:963–973. doi: 10.1007/s10545-012-9506-x. [DOI] [PubMed] [Google Scholar]
- 34.Smith I, Dhondt JL. Birthweight in patients with defective biopterin metabolism. Lancet. 1983;33:2417–2424. doi: 10.1016/s0140-6736(85)91474-6. [DOI] [PubMed] [Google Scholar]
- 35.Leuzzi V, Carducci CA, Carducci CL, et al. Phenotypic variability, neurological outcome and genetic background of 6-pyruvoyl-tetrahydrobiopterin synthase deficiency. Clin Genet. 2010;26:157–161. doi: 10.1111/j.1399-0004.2009.01306.x. [DOI] [PubMed] [Google Scholar]
- 36.Jaggi L, Zurfluh MR, Schuler A, et al. Outcome and long term follow up of 36 patients with tetrahydrobiopterin deficiency. Mol Genet Metab. 2008;93:295–305. doi: 10.1016/j.ymgme.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 37.Porta F, Mussa A, Concolino D, et al. Dopamine agonists in 6-pyruvoyl tetrahydrobiopterin synthase deficiency. Neurology. 2009;73:633–636. doi: 10.1212/WNL.0b013e3181b38983. [DOI] [PubMed] [Google Scholar]
- 38.Dill P, Wagner M, Somerville A, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012;31(78):29–32. doi: 10.1212/WNL.0b013e3182452849. [DOI] [PubMed] [Google Scholar]
- 39.Neville BG, Parascandalo R, Farrugia R, et al. Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain. 2005;128:2291–2296. doi: 10.1093/brain/awh603. [DOI] [PubMed] [Google Scholar]
- 40.Friedman J, Hyland K, Blau N, et al. Dopa-responsive hypersomnia and mixed movement disorder due to sepiapterin reductase deficiency. Neurology. 2006;67:2032–2035. doi: 10.1212/01.wnl.0000247274.21261.b4. [DOI] [PubMed] [Google Scholar]
- 41.Crabtree MJ, Tatham Al, Al Wakeel Y, et al. Quantitative regulation of intracellular endothelila nitric-oxide synthase (eNOS) coupling by tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP-cyclohydroxylase I expression. J Biol Chem. 2009;294:1136–1144. doi: 10.1074/jbc.M805403200. [DOI] [PubMed] [Google Scholar]
- 42.Willemsen MA, Verbeek MM, Kamsteeg EJ, et al. Tyrosine hydroxylase deficiency; a treatable disorder of brain catecholamine biosynthesis. Brain. 2010;133:1810–1822. doi: 10.1093/brain/awq087. [DOI] [PubMed] [Google Scholar]
- 43.Pons R, Syrengelas D, Syrengelas A, et al. Tyrosine hydroxylase: the Greek experience. Eur J Paediatr Neurol. 2013;15(Suppl 1):S19–S20. doi: 10.1016/S1090-3798(13)70063-1. [DOI] [Google Scholar]
- 44.Pons R, Syrengelas D, Youroukos S, et al. Levodopa-induced dyskinesias in tyrosine hydroxylase deficiency. Mov Disord. 2013;28:1058–1063. doi: 10.1002/mds.25382. [DOI] [PubMed] [Google Scholar]
- 45.Brun L, Ngu LH, Chang GS, et al. Clinical and biochemical features of aromatic l-amino acid decarboxylase deficiency. Neurology. 2010;75:64–71. doi: 10.1212/WNL.0b013e3181e620ae. [DOI] [PubMed] [Google Scholar]
- 46.Pons R, Ford B, Chiriboga CA, et al. Aromatic amino acid decarboxylase deficiency: clinical features, treatment, prognosis. Neurology. 2004;62:1058–1065. doi: 10.1212/WNL.62.7.1058. [DOI] [PubMed] [Google Scholar]
- 47.Lee H, Tsai CR, Chi CS, et al. Aromatic l-amino acid decarboxylase deficiency in Taiwan. Eur J Paediatr Neurol. 2009;13:135–140. doi: 10.1016/j.ejpn.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 48.Mastrangelo M, Caputi C, Galosi S, et al. Transdermal rotigotine in the treatment of aromatic l-amino acid decarboxylase deficiency. Mov Disord. 2012;28:556–557. doi: 10.1002/mds.25303. [DOI] [PubMed] [Google Scholar]
- 49.Allen GF, Neergheen V, Oppenheim M, et al. Pyridoxal 5′phosphate deficiency causes a loss of aromatic l-amino acid decarboxylase in patient and human neuroblastoma cells, implications for aromatic l-amino acid decarboxylase and vitamin B6 deficiency states. J Neurochem. 2010;114:87–96. doi: 10.1111/j.1471-4159.2010.06742.x. [DOI] [PubMed] [Google Scholar]
- 50.Hyland K, Surtees RAH, Rodeck C, et al. Aromatic l-amino acid decarboxylase deficiency: clinical features, diagnosis and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology. 1992;42:1980–1988. doi: 10.1212/WNL.42.10.1980. [DOI] [PubMed] [Google Scholar]
- 51.Maller A, Hyland K, Milstein S, et al. Aromatic l-amino acid decarboxylase deficiency: clinical feature. J Child Neurol. 1997;12:349–354. doi: 10.1177/088307389701200602. [DOI] [PubMed] [Google Scholar]
- 52.Matsuda N, Hayaashi H, Miyatake T, et al. Instability of the apo form of aromatic a l-amino acid decarboxylase activity in vivo and in vitro: implications for the involvement of the flexible loop that covers the active site. J Biochem. 2004;135:33–42. doi: 10.1093/jb/mvh004. [DOI] [PubMed] [Google Scholar]
- 53.Swodoba KJ, Saul JP, McKenna CE, et al. Aromatic l-amino acid decarboxylase deficiency overview of clinical features and outcomes. Ann Neurol. 2003;54:264–270. doi: 10.1002/ana.10631. [DOI] [PubMed] [Google Scholar]
- 54.Paterson IA, Juorio AV, Berry MY, et al. Inhibition of monoamine oxidase-B by deprenyl potentials neuronal response to dopamine agonistics but does not inhibit dopamine catabolism in the rat striatum. J Pharmacol Exp Ther. 1991;258:1019–1026. [PubMed] [Google Scholar]
- 55.Cho AM, Duchemin NH, Neff M, et al. Modulation of tyrosine hydroxylase and aromatic l-amino acid decarboxylase after inhibiting monoamine oxidase-A. Eur J Pharmacol. 1996;310:51–59. doi: 10.1016/S0014-2999(96)00530-4. [DOI] [PubMed] [Google Scholar]
- 56.Dyck LE, Dewar KM. Inhibition of aromatic l-amino decarboxylase and tyrosine aminotransferase by the monoamine oxidase inhibitor phenelzine. J Neurochem. 1986;46:1899–1903. doi: 10.1111/j.1471-4159.1986.tb08511.x. [DOI] [PubMed] [Google Scholar]
- 57.Hwu WL, Muramatsu S, Tseng SH, et al. Gene therapy for aromatic l-amino acid decarboxylase deficiency. Sci Transl Med. 2012;4:134ra61. doi: 10.1126/scitranslmed.3003640. [DOI] [PubMed] [Google Scholar]
- 58.Mills PB, Footitt EJ, Mills KA, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency) Brain. 2010;133:218–259. doi: 10.1093/brain/awq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mills PB, Surtees RA, Champion MP, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′ phosphate oxidase. Hum Mol Genet. 2005;14:1077–1086. doi: 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- 60.Reddy SD, Rayala SK, Ohshiro K, et al. Multiple coregulatory control of tyrosine hydroxylase gene transcription. Proc Natl Acad Sci USA. 2011;108:4200–4205. doi: 10.1073/pnas.1101193108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang DY. 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson’s disease. J Neurosci. 2005;25:2132–2137. doi: 10.1523/JNEUROSCI.3718-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hwang D-Y, Ardayfio P, Kang UJ, Semina EV, Kim KS. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res Mol Brain Res. 2003;114:123–131. doi: 10.1016/S0169-328X(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 63.Katarzyna Derwinska K, Mierzewska H, Goszczanska A, et al. Clinical improvement of the aggressive neurobehavioral phenotype in a patient with a deletion of PITX3 and the absence of l-DOPA in the cerebrospinal fluid. Am J Med Genet B Neuropsychiatr Genet. 2012;159:236–242. doi: 10.1002/ajmg.b.32020. [DOI] [PubMed] [Google Scholar]
- 64.Rilstone JJ, Alkhater RA, Minassian BA. Brain dopamine–serotonin vesicular transport disease and its treatment. NEJM. 2013;368:543–550. doi: 10.1056/NEJMoa1207281. [DOI] [PubMed] [Google Scholar]
- 65.Kurian MA, Zhen J, Cheng SY, et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism dystonia. J Clin Invest. 2009;119:1595–1603. doi: 10.1172/JCI39060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kurian MA, Li Y, Zhen J, et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 2011;10:54–62. doi: 10.1016/S1474-4422(10)70269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng J, Zhen J, Meyer E, et al. Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain. 2014;137:1107–1119. doi: 10.1093/brain/awu022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henriksen FH, Yasmeen S, Skjørringe T, et al. Atypical dopamine transporter deficiency syndrome in an adult male: molecular characterisation of new transporter variants. Program No.10.11. 2013 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience; 2013. http://www.sfn.org/~/media/SfN/Documents/AnnualMeeting/FinalProgram/FullAbstractPDFs/AbstractPDFs_Nano.ashx (Online).
- 69.García-Cazorla A, Serrano M, Pérez-Dueñas B, et al. Secondary abnormalities of neurotransmitters in infants with neurological disorders. Dev Med Child Neurol. 2007;49:740–744. doi: 10.1111/j.1469-8749.2007.00740.x. [DOI] [PubMed] [Google Scholar]
- 70.Kurian MA. What is the role of dopamine in childhood neurological disorders? Dev Med Child Neurol. 2013;55(6):493–494. doi: 10.1111/dmcn.12130. [DOI] [PubMed] [Google Scholar]
- 71.Ng J, Tuschl K, Kinali M, et al. TH gene-negative infantile onset severe dopamine deficiency syndrome: a novel neurotransmitter disorder? Dev Med Child Neurol. 2013;5(S1):15. [Google Scholar]
- 72.Breakfield XO, Blood A, Li Y, et al. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–243. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- 73.Bhidayasiri R, Tarsay D. Treatment of dystonia. Expert Rev Neurother. 2006;6:863–886. doi: 10.1586/14737175.6.6.863. [DOI] [PubMed] [Google Scholar]
- 74.Lopez-Laso E, Ochoa-Sepulveda JJ, Ochoa-Amor JJ, et al. Segawa syndrome due to mutation Q89X in the GCH1 gene: a possible founder effect in Cordoba (southern Spain) J Neurol. 2009;256:1816–1824. doi: 10.1007/s00415-009-5198-z. [DOI] [PubMed] [Google Scholar]
- 75.Pettibone DJ, Totaro JA, Pflueger AB. Tetrabenazine-induced depletion of brain monoamines: characterization and interaction with selected antidepressants. Eur J Pharmacol. 1984;20:425–430. doi: 10.1016/0014-2999(84)90562-4. [DOI] [PubMed] [Google Scholar]
- 76.Liow N, Marianczak J, Kirk E. et al. Gabapentin can improve dystonia severity, transfers, sitting, sleep, mood and pain in children EJPN. 2013;S18:O58–O2017. [Google Scholar]
- 77.Brasil S, Viecelli HM, Meili D, et al. Pseudoexom exclusion by antisense therapy in 6-pyruvoyl-tetrahydrobiopterin synthase deficiency. Hum Mutat. 2011;32:1019–1027. doi: 10.1002/humu.21529. [DOI] [PubMed] [Google Scholar]
- 78.Jatana N, Apoorva N, Malik S, et al. Inhibitors of catechol-O-methyl transferase in the treatment of neurological disorders. Central Nerv Syst Agents Med Chem. 2014;2013(13):166–194. doi: 10.2174/1871524913666140109113341. [DOI] [PubMed] [Google Scholar]
- 79.Eberling JL, Jaqust WJ, Christine CW, et al. Results from phase 1 safety trial of hAADC gene therapy for Parkinson disease. Neurology. 2008;80:1980–1983. doi: 10.1212/01.wnl.0000312381.29287.ff. [DOI] [PubMed] [Google Scholar]