

Abstract
Osteosarcoma (OS) is the most common non-hematologic primary tumor of bone in children and adults. High-dose cytotoxic chemotherapy and surgical resection have improved prognosis, with long-term survival for non-metastatic disease approaching 70%. However, most OS tumors are high grade and tend to rapidly develop pulmonary metastases. Despite clinical advances, patients with metastatic disease or relapse have a poor prognosis. Toward a better understanding of the molecular pathogenesis of human OS, several genetically modified OS mouse models have been developed and will be reviewed here. However, better animal models that more accurately recapitulate the natural progression of the disease are needed for the development of improved prognostic and diagnostic markers as well as targeted therapies for both primary and metastatic OS.
Keywords: osteosarcoma, conditional mouse models, germ-line mouse models, animal models, p53, RB
Introduction
Osteosarcoma (OS) is a highly malignant form of bone cancer characterized by osteoid production. Although OS comprises <1% of cancers diagnosed in the United States, it is the most common primary malignancy of the bone (1, 2). It occurs predominantly after the first decade of life during periods of skeletal growth, with a second peak incidence in the geriatric patient population (1, 3). The vast majority of OS in children, adolescents, and young adults is high grade and begins in the intramedullary space of metaphyseal locations in long bones of the lower extremity. This suggests a relationship with active growth plates. After a low incidence in individuals between 25 and 59 years of age, the incidence of OS rises again in individuals over 60 years of age, and is most often associated with Paget’s disease or radiation exposure (1, 2). This may suggest that the underlying pathogenesis is not identical in young and older patients. Conventional OS presents in three major subtypes based on histological classification: osteoblastic, fibroblastic, and chondroblastic. Osteoblastic is the most common (around 60%) with fibroblastic and chondroblastic being equally represented (4).
Osteosarcoma is characterized by a local invasion of bone and soft tissue, loss of the function of the affected extremity, and distant metastasis, most often to the lung (90%). Metastases are also found in bone (8–10%) and rarely in lymph nodes (5). Treatment involves aggressive removal of the primary tumor to afford local control via limb sparing surgery or amputation. Systemic chemotherapy (both prior to and after tumor removal) is used to suppress development of metastasis and effect cure. The most common chemotherapy regimens comprise the drugs, cisplatin, doxorubicin, and high-dose methotrexate in combination (6–8). Although chemotherapy slows tumor growth, it can induce cardiomyopathy, hearing loss, and risk of secondary malignancy (8, 9). In patients without metastases at the time of diagnosis (80–90%), surgical treatment in combination with chemotherapy has resulted in long-term survival rates that approach 70%. In contrast, for patients with established metastases there is currently no reliable therapeutic option to provide long-term tumor control. Despite intensive efforts to improve both chemotherapeutics and surgical management, 40% of all OS patients succumb to the disease. Specifically, the clinical outcome for metastatic OS remains poor; fewer than 30% of patients who present metastases survive 5 years after initial diagnosis. Therefore, there is an urgent need for the development of novel therapeutics for OS agents with increased capacity to eliminate systemic tumor burden as well as reduced toxicity in healthy tissues.
Etiology of OS
Osteosarcoma is characterized by a complex karyotype and a lack of recurrent translocations. Genetic approaches have identified several genes of potential importance in the development and progression of the disease (10–12). However, the widespread chromosomal alterations of the OS genome have limited the interpretation of these findings. Genetic alterations of OS are usually sporadic though genetic predisposition has been documented in patients with Li-Fraumeni and retinoblastoma syndrome. Somatic deletions and point mutations in P53 occur in approximately 50% of human OS (13–16) and half of those mutations are associated with loss of the remaining allele (14). Additionally, almost 70% of OS have at least one RB allele alteration (17, 18). Homozygous deletions of RB are seen in 23% of tumors, while point mutations appear in 6% (18, 19). In addition, numerous alterations that disrupt the RB pathway have also been reported; for example, the loss of function at the INK4a/ARF locus and the amplification of CDK4 have been found to occur (one or the other) in 22% of OS (20–22). The prevalence of these alterations would suggest that the deregulation of both G1/S and G2/M checkpoint in the cell cycle are a common event in OS.
For this, a tumor of unknown origin, chaotic genetics, early onset, and aggressive behavior, there is a need for more representative models to learn more about the biology of OS.
Animal Models in OS
Animal models hold significant promise in increasing our understanding of the genetic basis of OS and more importantly, in advancing preclinical studies aimed to the rational development of new therapeutic approaches as well as their validation prior to clinical trials.
In order for any animal model of human disease to be useful and informative, it is preferable to accurately recapitulate the natural course of the disease. Unfortunately, the etiology and pathogenesis of OS are not completely understood; therefore, the establishment and induction of representative experimental models are challenging and incomplete. Currently, there is not a robust animal model of OS that fully represents its biological and clinical features. The ideal would be one in which there was a naturally occurring primary bone lesion and spontaneous pulmonary metastases. To date, the major species used to generate OS models are mouse and rat; however, OS arising in dogs is also of note as a validated model of spontaneous OS.
Many aspects of the biology of the disease have been determined from a variety of animal model approaches. Genetically modified mouse models of OS have given the field much insight. However, spontaneous OS, secondary OS as a consequence of animals receiving radiation, human and murine OS cell lines, and xenotransplantation studies are also important to understand the biology of this malignancy.
Canine Models
Spontaneous OS is much more common in large dogs than in humans, making the dog an attractive candidate model to study human disease (23). Canine OS is indistinguishable from human tumors at the histological and gene expression levels (24–27). The primary differences between the two are the age of development and the prevalence of the disease. In dogs, OS is a disease of older, large breed dogs (6–12 years of age), and it is estimated that over 10,000 cases occur annually in the United States. The median disease-free interval following surgery alone is 4 months, and after surgery with chemotherapy, 13 months. This high prevalence and the relatively rapid rate of disease progression provide the opportunity to model metastasis development and progression and evaluate novel treatment options in a relatively short period of time (28–32). Many of the genes involved in human OS pathogenesis appear to participate in canine OS, including P53, RB, and PTEN (33–36).
Although canine OS serves as an excellent comparative tumor model for human OS, there are some limitations to be considered. First, OS affects skeletally mature, geriatric dogs, which is different from humans where the peak of incidence occurs during adolescence. Second, some breeds have specific heritable germ-line mutations in certain genes that may influence OS biology, progression, and response to treatment without driving the initiation of the disease (37).
Secondary OS after Radiation
The development of rodent OS models began with the exposure of rats and mice to chemical and radioactive carcinogens (38–40). Of note, among those was the development of OS in rats treated with P32-orthophosphate, which resulted in a high incidence (41). These models yielded tumors that histologically resembled the human cancer and produced cell lines that complement human OS studies (42). Despite the high penetrance of the models, their relevance remains unclear since the majority of OS in humans is sporadic, while the carcinogen-induced murine model is more representative of a therapy induced disease.
Xenotransplantation Studies
There is a significant amount of literature related to the development and use of xenograft and allograft models of human and murine OS cells injected into immunocompromised mice. The injected cells form a solid tumor locally grown within days or weeks after implantation (42, 43). The use of these systems has become a prominent tool in current oncological research due to the quick onset, its affordable cost, and ease of handling and maintenance. In addition, OS donor-derived cells may metastasize to the lungs, providing an opportunity to investigate primary and secondary tumor growth. The principal limitation is that the approach uses fully developed OS cells and therefore does not provide information about the initiation of the tumor and its etiology. Furthermore, since the tumor microenvironment can contribute significantly to the tumor behavior, such interactions may be lost when establishing the disease by direct introduction into a recipient animal (44–46). In certain circumstances, the injected cell line may not be metastatic in the rodent context, making it impossible to study the dissemination of the disease. Despite these limitations, many groups have successfully used this model to identify factors involved in OS migration (47, 48) and more importantly for screening drugs with tumoricidal potential (49). Distinct advantages of the subcutaneous cell suspension injection model are high rate of incidence and reproducibility that allows for accurate titration of cell numbers in the inoculum to quantify tumorigenic potential of the injected cells.
A variation of injecting cell suspensions into recipient animals is to transplant pieces of tumor directly harvested from the patient. The advantage is that the human malignant cells can grow in its native environment maintaining the heterogeneity that may be required for their proliferation, which in some reports has been shown to enhance tumor growth and metastasis. With the use of cell suspension and transplants, murine host cells can infiltrate the tumor, possibly influencing the activities of the tumor cells, and in some cases, cells of the rodent host can overgrow the human cell population (50). Alternatively orthotopic, intratibial implantation of OS cells has been shown to induce OS at local and metastatic sites (proximal tibia and lung) (43, 51–53). This approach allows the study of primary tumor formation within a more native context as well as the early stages of metastatic progression of OS, thereby reconstituting the entire metastatic process. Its use, however, is limited by a lack of reproducibility due in part to the technical skill required to perform the implantation and the associated lack of quantifiable inoculum.
Genetically Engineered Mouse Models
Of the sarcomas with complex karyotypes, OS is one of the most well-studied as exemplified by the development of numerous mouse models available for this disease. The ability to alter specifically the expression of individual genes (by loss or gain of function) became available in the mouse with the evolution of gene targeting technologies (54, 55).
Many murine OS models have been developed to recapitulate the P53 and RB mutations in hereditary and sporadic human OS. Germ-line deletion of P53 resulted in an OS incidence of 4% in homozygous P53 null mice (56) and 25% in heterozygous P53 mice (57), underlying the importance of altered P53 in driving OS. This unexpected ratio of tumor formation, though, is likely due to the early lethality seen in the homozygous null population. Further, the rapid development, the higher incidence of other tumors (mostly lymphomas), and the long latency of OS (58) necessitate the sacrifice of the mice before OS onset, hampering in many cases the utility of these models. The role of P53 was further highlighted by tumor analysis of P53 knock-in mice containing a mutant copy of P53R172H (corresponding to the R175H hot-spot mutation in humans) that not only develop primary tumors but also metastasize to the lungs as well as other organs (59, 60). Conversely, mice with germ-line deletions of Rb did not develop OS: homologous deletion of Rb is embryonic lethal and the heterozygotes are not predisposed to OS (61, 62).
The application of conditional gene regulation and the availability of tissue specific Cre expressing mouse lines (63) have greatly enhanced our ability to generate specific models of mesenchymal osteogenic lineage that more faithfully resemble human OS (55, 64). The majority of these models have used the loss of P53 with or without the disruption of the Rb pathway to generate penetrant OS models (54). They use conditional gene deletion approaches restricted to multipotent mesenchymal progenitors, early committed osteoblasts (pre-osteoblasts) and the osteoblast population (Figure 1) (Table 1).
Figure 1.
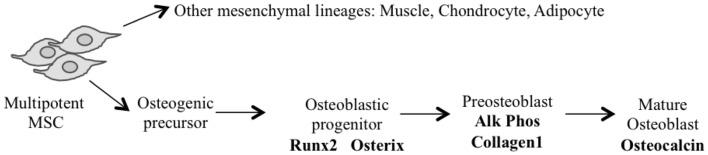
Model of osteoblast differentiation and putative stage of Cre expression is shown.
Table 1.
Summary of genetically modified OS murine models.
| Cell | Cre | Gene | OS penetrance (%) | Other tumors | Metastatic disease |
|---|---|---|---|---|---|
| MSC/skeletal | Prx-1 | p53fl/+ | 22 (65) | ||
| Progenitors | p53fl/fl | 61 (65); 62(66) | PDS (32%), LY (3%), LPS (3%); RMS (15%), PDS (12%) | Yes (24%) | |
| p53fl/fl-Rbfl/+ | 92 (66) | RMS (9%), PDS (18%), HIB (4%) | |||
| p53fl/fl-Rbfl/fl | 18 (65); 29 (66) | PDS (57%), LY (14%); RMS (12%), PDS (3%), HIB (91%) | |||
| Pre-osteoblasts | Osx | p53fl/fl | 100 (67); 100 (68) | Yes (32%); yes (40%) | |
| p53fl/fl-Rbfl/+ | 53 (67); 100 (68) | ||||
| p53fl/fl-Rbfl/fl | 72 (67); 100 (68) | Multiple tumors per animal; concurrent HIB (20–25%) | Yes (37%) | ||
| shp53 | 100 (69) | 0% | Yes (83.33%) | ||
| shp53-Rbfl/+ | 100 (69) | 0% | Yes (58.82%) | ||
| shp53-Rbfl/fl | 100 (69) | 0% | Yes (85.72%) | ||
| Col1α1–3.6 | p53fl/fl | 60 (70) | |||
| Osteoblasts | Col1α1–2.3 | p53fl/fl | 85 (65) | ||
| Og2 | SV40 Tag | 100 (71) | Yes (90%) |
LPS, liposarcoma; LY, lymphoma; RMS, rhabdomyosarcoma; PDS, poorly differentiated sarcoma; HIB, hibernomas.
Using Cre recombinase activated by the gene promoter of Paired related homebox 1 (Prx1-Cre) (72) that deletes LoxP flanked alleles in the early limb mesenchyme (multipotential cells), 22% of mice with P53-mediated heterozygosity developed OS. Not surprisingly, homozygous deletion of P53 had a threefold increase in OS incidence over the heterozygous animals. In contrast, the deletion of Rb in the mesenchymal Prx expressing progenitors did not produce any OS tumors (65, 66). Interestingly, the highest incidence (92%) of OS occurred with the combined deletion of one allele of Rb with homozygous P53 deletion (66). Homozygous deletion of both genes resulted in more non-specific tumor formation with only 18% OS tumors and the remainder being poorly differentiated soft tissue sarcomas (PD-STS) and lymphoma (65, 66).
For a more restricted deletion of genes in the osteoblast lineage, promoters of genes ranging from those expressed early in the commitment of progenitors as Osterix 1 and Collagen1 α1–3.6 to those expressed in more lineage-restricted osteoblast precursors such as Collagen1 α1–2.3 and osteocalcin (Og2) have been used. Development of OS with a penetrance of 100% (67, 68) has been observed following osteoblast specific deletion of P53 using Osterix-mediated Cre expression (Osx-Cre) (73). As with mesenchymal progenitors, Rb deletions have no effect and combined deletion of Rb and P53 in osteoblasts once again generated fibroblastic or undifferentiated OS with high penetrance (100%) (67, 68). Potential translational utility is the existence of short-latency spontaneous metastatic OS similar to human tumors in which cells are arrested in their differentiation (67, 68). Although the greatest proportion of tumors was OS when P53 was conditionally deleted, neuroendocrine tumors and hibernomas were also reported to be generated in several mice (67, 68). However, Walkley et al. enriched the C57BL/6 background of the mouse strain and the percentage of hibernomas was reduced, suggesting a possible impact of mouse strains in the phenotype observed (69). A recent study in mice that expressed SV40 T/t antigen (Tag) in mature osteoblasts under the Og2 (74) showed OS with complete penetrance (71) and 90% incidence of lung metastases. Further analysis of the tumors derived from this model revealed a recurrent genomic deletion of the Prkar1a gene in a specific subset also in human OS. Transgenic shRNA has been used to specifically knock down P53 (rather than delete) using the Osx-Cre transgene (69). These mice develop osteoblastic OS with a 100% penetrance, and although they have a longer latency to tumor onset, they more often develop in long bones and are highly metastatic (lung and liver), features similar to human OS. This model has not developed any non-OS tumors.
Independent of the stage of development in which Cre becomes active, the latency of OS is essentially the same when comparing either P53 alone or in combination with Rb. The use of Cre in more primitive cells (Prx), however, leads to the development of tumors of other mesenchymal lineages at higher frequency.
Possibly providing insight into the initiating events of OS (70), a prominent cellular feature of conditional inactivation of P53 in osteoblastic progenitors is the hyperproliferation of osteoblasts prior to tumor formation. Rb has been proposed to have a role in influencing late osteoblast differentiation by interacting with Runx2 (75). However, a number of independent studies have shown that the removal of Rb alone is not sufficient to induce OS. The different experimental approaches strongly suggest that mutation in the p53 pathway can serve as an initiating event in OS, with a subsequent mutation in the Rb pathway strongly accelerating tumor development.
These engineered mouse models of OS reproduce many features of human OS including similar gene-transcription signatures (76) and cytogenetic complexity. However, the sites of primary tumor formation in Cre–loxP mice do not recapitulate the spontaneous human disease. The majority of lesions (85%) arise in axial skeletal sites (mandibule, maxilla, rib/vertebra, skull, sternum) while on 13.6% of tumors developed from the appendicular skeleton (hind leg, front leg) (68). This contrasts with the anatomic distribution of OS diagnosed in humans, with the distal femur, proximal tibia, and proximal humerus being the most common sites involved and only 10% develop in the axial skeleton, most commonly the pelvis (5). Only in one study (69) did the tumor arise primarily in long bones. In addition, the observed frequency of distant metastases was comparatively low when compared to human disease except for the P53 knockdown model (69). As opposed to a complete deletion of P53, the primary tumor cells proliferated slower and the animals did not have to be sacrificed for local tumor size prior to completion of the metastatic process. Furthermore, the primary site of metastases in human OS is predominantly the lung parenchyma while in Cre–loxP mice, sites of metastases were more diverse with both the lung and liver being affected in almost equal proportions.
Other genes such as C-FOS (77, 78), TWIST (79), p14ARF (80), p16INK4a (81), PRKAR1A (71), and p21CIP (82) have also been implicated in OS pathogenesis based on studies of human OS samples. Their mutation appears to complement the defects in the P53 and RB pathways, and their involvement in osteosarcomagenesis is also demonstrated from genetically engineered mouse models. They provide important information regarding the genetics of OS, but the long latency combined with low penetrance makes utilization of these models less practical.
Targeted Therapies in OS
Osteosarcoma is very resistant to therapy and therefore there is an urgent need to effectively treat affected patients. The emergence of new anti-cancer drugs and the small number of patients eligible for early-phase clinical trials present another challenge in the clinical testing of novel compounds for OS treatment. As discussed earlier, xenotransplantation models have provided the greatest utility for preclinical screening of drugs with tumoricidal potential. To this end, the National Cancer Institute (NCI) has implemented the Pediatric Preclinical Testing Program (PPTP), a consortium of institutions across the United States and in Australia. Its objective is to identify agents with significant activity in panels of mouse xenograft models representing the most common pediatric cancers including OS (83). The program has been successful, leading to Phase I and II clinical trials for cixutumumab, sorafenib, and rapamycin for OS treatment. (84–86). In each case, these agents demonstrated high levels of response in the PPTP and were well-tolerated with promising anti-tumor activity in some adult and pediatric patients.
The use of spontaneous and transgenic OS models for high throughput screening of anti-OS drugs is hampered due to practical considerations associated with the cost and time of generating sufficient numbers of animals for statistically meaningful data. This is due to variations in disease onset as well as tumor heterogeneity, incidence, and progression. However, the recent generation of transgenic animals expressing shRNAs to knock down P53 (69) represents a potential breakthrough with respect to preclinical screening. Unlike conventional Cre-mediated gene deletion approaches, P53 knock down mice exhibited 100% penetrance for osteoblastic OS (the most common form of the disease). Moreover, the tumors were most frequently present in long bones and preferentially disseminated to the lungs, consistent with human OS.
Another consideration for preclinical testing in in vivo models is the accurate measurement of the disease burden at non-accessible sites. The use of in vivo imaging offers the opportunity to detect and monitor the development and progression of the disease. However, imaging systems are costly and not always widely accessible for many researchers. OS has the advantage that the primary tumor in genetically engineered mouse models appears in long bones and is therefore more accessible than abdominal tumors. The monitoring/visualization of micrometastases represents a greater challenge due to their small size. Inaccurate evaluation of metastatic spread in preclinical studies potentially leads to disappointing results in clinical trials. Consequently, there is great interest in refining the methods to enable reproducible and ultrasensitive detection of metastases at the single cell level. The main focus therefore is on techniques, which allow the detection of tumor cells in vivo, such as microcomputer tomography (micro-CT), positron emission tomography (PET), bioluminescence, or fluorescence imaging.
Conclusion
Our understanding of human OS biology is hindered by its rapid onset, low prevalence, and absence of predisposing conditions or precursor lesions. With limited human tissue available for study, animal models provide a valuable tool to investigate the underlying mechanisms driving tumor initiation, progression, metastatic events, and therapeutic interventions. While these models have yet to faithfully recapitulate all aspects of OS, there is no doubt that the study of OS animal models has enabled insight into the genetics of tumor initiation as well as the cellular and molecular profiles of tumor growth and metastasis. In particular, gene knockout studies have been instrumental in identifying genetic mutations that promote OS tumor initiation (P53), as well as co-operative mutations that increase disease incidence (RB, c-FOS).
With the use of cell lineage specific markers, it is now possible to introduce genetic mutations by sequential targeting from early precursor (multipotent mesenchymal cell) to more mature osteoblastic cells (osteoblast to osteocyte) to investigate OS incidence and tumor pathology. With this strategy, Prx1 and Osx have been used to identify mesenchymal and osteoprogenitor cells, respectively, following conditional mutation of P53. It remains to be seen, however, whether these populations are truly distinct, as Prx1 could be coexpressed with Osx in a certain subpopulation of cells. Another consideration particularly relevant in OS is its tumor heterogeneity among patients, which suggests that multiple cell types could act as cell of origin. Additionally, this concept of heterogeneity calls into question the utility of models exploiting single gene manipulation. Its consideration may permit a more systematic analysis of the genetic lesions involved in OS initiation and progression and could serve as a platform for the identification of early disease biomarkers. Cell of origin identification may also have important implications in the prevention of relapse and elucidate key molecular pathways and driver mutations that could lead to new therapeutic approaches to prevent the disease.
Thus, although for now, conventional orthotopic and subcutaneous transplantation models will remain indispensable to continue the study of OS in vivo, new models of spontaneous OS need to be developed to further our understanding of OS biology. Models that accurately reproduce the establishment of spontaneous micrometastases are necessary to investigate novel antimetastatic agents, as this clinical scenario is most often the lethal event for patients with this form of cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer (2009) 125(1):229–34 10.1002/ijc.24320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer (2009) 115(7):1531–43 10.1002/cncr.24121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKenna RJ, Schwinn CP, Higinbotham NL. Osteogenic sarcoma in children. CA Cancer J Clin (1966) 16(1):26–8 [DOI] [PubMed] [Google Scholar]
- 4.Gorlick R. Current concepts on the molecular biology of osteosarcoma. Cancer Treat Res (2009) 152:467–78 10.1007/978-1-4419-0284-9_27 [DOI] [PubMed] [Google Scholar]
- 5.Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment – where do we stand? A state of the art review. Cancer Treat Rev (2014) 40(4):523–32 10.1016/j.ctrv.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 6.Janeway KA, Grier HE. Sequelae of osteosarcoma medical therapy: a review of rare acute toxicities and late effects. Lancet Oncol (2010) 11(7):670–8 10.1016/S1470-2045(10)70062-0 [DOI] [PubMed] [Google Scholar]
- 7.Picci P, Ferrari S, Bacci G, Gherlinzoni F. Treatment recommendations for osteosarcoma and adult soft tissue sarcomas. Drugs (1994) 47(1):82–92 10.2165/00003495-199447010-00006 [DOI] [PubMed] [Google Scholar]
- 8.Sloet van Oldruitenborgh-Oosterbaan MM, Klein WR, Misdorp W. Differential diagnosis of non-healing ‘fungal’ patches in horses. Tijdschr Diergeneeskd (1994) 119(24):756–9 [PubMed] [Google Scholar]
- 9.Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet (2013) 45(7):799–803 10.1038/ng.2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: osteosarcoma and related tumors. Cancer Genet Cytogenet (2003) 145(1):1–30 10.1016/S0165-4608(02)00848-8 [DOI] [PubMed] [Google Scholar]
- 11.Pasic I, Shlien A, Durbin AD, Stavropoulos DJ, Baskin B, Ray PN, et al. Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Res (2010) 70(1):160–71 10.1158/0008-5472.CAN-09-1902 [DOI] [PubMed] [Google Scholar]
- 12.Fletcher JA, Gebhardt MC, Kozakewich HP. Cytogenetic aberrations in osteosarcomas. Nonrandom deletions, rings, and double-minute chromosomes. Cancer Genet Cytogenet (1994) 77(1):81–8 10.1016/0165-4608(94)90154-6 [DOI] [PubMed] [Google Scholar]
- 13.Overholtzer M, Rao PH, Favis R, Lu XY, Elowitz MB, Barany F, et al. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci U S A (2003) 100(20):11547–52 10.1073/pnas.1934852100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller CW, Aslo A, Tsay C, Slamon D, Ishizaki K, Toguchida J, et al. Frequency and structure of p53 rearrangements in human osteosarcoma. Cancer Res (1990) 50(24):7950–4 [PubMed] [Google Scholar]
- 15.Miller CW, Aslo A, Won A, Tan M, Lampkin B, Koeffler HP. Alterations of the p53, Rb and MDM2 genes in osteosarcoma. J Cancer Res Clin Oncol (1996) 122(9):559–65 10.1007/BF01213553 [DOI] [PubMed] [Google Scholar]
- 16.Toguchida J, Yamaguchi T, Ritchie B, Beauchamp RL, Dayton SH, Herrera GE, et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res (1992) 52(22):6194–9 [PubMed] [Google Scholar]
- 17.Hansen MF, Koufos A, Gallie BL, Phillips RA, Fodstad O, Brogger A, et al. Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proc Natl Acad Sci U S A (1985) 82(18):6216–20 10.1073/pnas.82.18.6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wadayama B, Toguchida J, Shimizu T, Ishizaki K, Sasaki MS, Kotoura Y, et al. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res (1994) 54(11):3042–8 [PubMed] [Google Scholar]
- 19.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature (1986) 323(6089):643–6 10.1038/323643a0 [DOI] [PubMed] [Google Scholar]
- 20.Maitra A, Roberts H, Weinberg AG, Geradts J. Loss of p16(INK4a) expression correlates with decreased survival in pediatric osteosarcomas. Int J Cancer (2001) 95(1):34–8 [DOI] [PubMed] [Google Scholar]
- 21.Miller CW, Aslo A, Campbell MJ, Kawamata N, Lampkin BC, Koeffler HP. Alterations of the p15, p16, and p18 genes in osteosarcoma. Cancer Genet Cytogenet (1996) 86(2):136–42 10.1016/0165-4608(95)00216-2 [DOI] [PubMed] [Google Scholar]
- 22.Wei G, Lonardo F, Ueda T, Kim T, Huvos AG, Healey JH, et al. CDK4 gene amplification in osteosarcoma: reciprocal relationship with INK4A gene alterations and mapping of 12q13 amplicons. Int J Cancer (1999) 80(2):199–204 [DOI] [PubMed] [Google Scholar]
- 23.Misdorp W. Skeletal osteosarcoma. Animal model: canine osteosarcoma. Am J Pathol (1980) 98(1):285–8 [PMC free article] [PubMed] [Google Scholar]
- 24.Selvarajah GT, Kirpensteijn J, van Wolferen ME, Rao NA, Fieten H, Mol JA. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Mol Cancer (2009) 8:72. 10.1186/1476-4598-8-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paoloni M, Davis S, Lana S, Withrow S, Sangiorgi L, Picci P, et al. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics (2009) 10:625. 10.1186/1471-2164-10-625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller F, Fuchs B, Kaser-Hotz B. Comparative biology of human and canine osteosarcoma. Anticancer Res (2007) 27(1A):155–64 [PubMed] [Google Scholar]
- 27.Rankin KS, Starkey M, Lunec J, Gerrand CH, Murphy S, Biswas S. Of dogs and men: comparative biology as a tool for the discovery of novel biomarkers and drug development targets in osteosarcoma. Pediatr Blood Cancer (2012) 58(3):327–33 10.1002/pbc.23341 [DOI] [PubMed] [Google Scholar]
- 28.Kirpensteijn J, Teske E, Kik M, Klenner T, Rutteman GR. Lobaplatin as an adjuvant chemotherapy to surgery in canine appendicular osteosarcoma: a phase II evaluation. Anticancer Res (2002) 22(5):2765–70 [PubMed] [Google Scholar]
- 29.Kirpensteijn J, Timmermans-Sprang EP, van Garderen E, Rutteman GR, Lantinga-van Leeuwen IS, Mol JA. Growth hormone gene expression in canine normal growth plates and spontaneous osteosarcoma. Mol Cell Endocrinol (2002) 197(1–2):179–85 10.1016/S0303-7207(02)00269-1 [DOI] [PubMed] [Google Scholar]
- 30.Kirpensteijn J, Kik M, Rutteman GR, Teske E. Prognostic significance of a new histologic grading system for canine osteosarcoma. Vet Pathol (2002) 39(2):240–6 10.1354/vp.39-2-240 [DOI] [PubMed] [Google Scholar]
- 31.Lascelles BD, Dernell WS, Correa MT, Lafferty M, Devitt CM, Kuntz CA, et al. Improved survival associated with postoperative wound infection in dogs treated with limb-salvage surgery for osteosarcoma. Ann Surg Oncol (2005) 12(12):1073–83 10.1245/ASO.2005.01.011 [DOI] [PubMed] [Google Scholar]
- 32.Kirpensteijn J, Kik M, Teske E, Rutteman GR. TP53 gene mutations in canine osteosarcoma. Vet Surg (2008) 37(5):454–60 10.1111/j.1532-950X.2008.00407.x [DOI] [PubMed] [Google Scholar]
- 33.van Leeuwen IS, Cornelisse CJ, Misdorp W, Goedegebuure SA, Kirpensteijn J, Rutteman GR. P53 gene mutations in osteosarcomas in the dog. Cancer Lett (1997) 111(1–2):173–8 10.1016/S0304-3835(96)04529-6 [DOI] [PubMed] [Google Scholar]
- 34.Johnson AS, Couto CG, Weghorst CM. Mutation of the p53 tumor suppressor gene in spontaneously occurring osteosarcomas of the dog. Carcinogenesis (1998) 19(1):213–7 10.1093/carcin/19.1.213 [DOI] [PubMed] [Google Scholar]
- 35.Levine RA, Fleischli MA. Inactivation of p53 and retinoblastoma family pathways in canine osteosarcoma cell lines. Vet Pathol (2000) 37(1):54–61 10.1354/vp.37-1-54 [DOI] [PubMed] [Google Scholar]
- 36.Levine RA, Forest T, Smith C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol (2002) 39(3):372–8 10.1354/vp.39-3-372 [DOI] [PubMed] [Google Scholar]
- 37.Liao AT, McMahon M, London CA. Identification of a novel germline MET mutation in dogs. Anim Genet (2006) 37(3):248–52 10.1111/j.1365-2052.2006.01415.x [DOI] [PubMed] [Google Scholar]
- 38.Martin TJ, Ingleton PM, Underwood JC, Michelangeli VP, Hunt NH, Melick RA. Parathyroid hormone-responsive adenylate cyclase in induced transplantable osteogenic rat sarcoma. Nature (1976) 260(5550):436–8 10.1038/260436a0 [DOI] [PubMed] [Google Scholar]
- 39.Ingleton PM, Coulton LA, Preston CJ, Martin TJ. Alkaline phosphatase in serum and tumour of rats bearing a hormone-responsive transplantable osteogenic sarcoma. Eur J Cancer (1979) 15(5):685–91 10.1016/0014-2964(79)90142-7 [DOI] [PubMed] [Google Scholar]
- 40.Underwood JC, Melick RA, Loomes RS, Dangerfield VM, Crawford A, Coulton L, et al. Structural and functional correlations in parathyroid hormone responsive transplantable osteogenic sarcomas. Eur J Cancer (1979) 15(9):1151–8 10.1016/0014-2964(79)90131-2 [DOI] [PubMed] [Google Scholar]
- 41.Bensted JP, Blackett NM, Lamerton LF. Studies on the development of radiation-induced bone tumours. Acta Unio Int Contra Cancrum (1959) 15:559–60 [PubMed] [Google Scholar]
- 42.Ek ET, Dass CR, Choong PF. Commonly used mouse models of osteosarcoma. Crit Rev Oncol Hematol (2006) 60(1):1–8 10.1016/j.critrevonc.2006.03.006 [DOI] [PubMed] [Google Scholar]
- 43.Dass CR, Ek ET, Choong PF. Human xenograft osteosarcoma models with spontaneous metastasis in mice: clinical relevance and applicability for drug testing. J Cancer Res Clin Oncol (2007) 133(3):193–8 10.1007/s00432-006-0157-x [DOI] [PubMed] [Google Scholar]
- 44.Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res (2006) 66(7):3355–8 discussion 8-9, 10.1158/0008-5472.CAN-05-3827 [DOI] [PubMed] [Google Scholar]
- 45.Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov (2006) 5(9):741–54 10.1038/nrd2110 [DOI] [PubMed] [Google Scholar]
- 46.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer (2007) 7(9):645–58 10.1038/nrc2192 [DOI] [PubMed] [Google Scholar]
- 47.Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clin Exp Metastasis (2000) 18(3):261–71 10.1023/A:1006767007547 [DOI] [PubMed] [Google Scholar]
- 48.Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, et al. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res (2001) 61(9):3750–9 [PubMed] [Google Scholar]
- 49.Sampson VB, Gorlick R, Kamara D, Anders Kolb E. A review of targeted therapies evaluated by the pediatric preclinical testing program for osteosarcoma. Front Oncol (2013) 3:132. 10.3389/fonc.2013.00132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kresse SH, Meza-Zepeda LA, Machado I, Llombart-Bosch A, Myklebost O. Preclinical xenograft models of human sarcoma show nonrandom loss of aberrations. Cancer (2012) 118(2):558–70 10.1002/cncr.26276 [DOI] [PubMed] [Google Scholar]
- 51.Berlin O, Samid D, Donthineni-Rao R, Akeson W, Amiel D, Woods VL., Jr Development of a novel spontaneous metastasis model of human osteosarcoma transplanted orthotopically into bone of athymic mice. Cancer Res (1993) 53(20):4890–5 [PubMed] [Google Scholar]
- 52.Crnalic S, Hakansson I, Boquist L, Lofvenberg R, Brostrom LA. A novel spontaneous metastasis model of human osteosarcoma developed using orthotopic transplantation of intact tumor tissue into tibia of nude mice. Clin Exp Metastasis (1997) 15(2):164–72 10.1023/A:1018456911823 [DOI] [PubMed] [Google Scholar]
- 53.Yuan J, Ossendorf C, Szatkowski JP, Bronk JT, Maran A, Yaszemski M, et al. Osteoblastic and osteolytic human osteosarcomas can be studied with a new xenograft mouse model producing spontaneous metastases. Cancer Invest (2009) 27(4):435–42 10.1080/07357900802491477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janeway KA, Walkley CR. Modeling human osteosarcoma in the mouse: from bedside to bench. Bone (2010) 47(5):859–65 10.1016/j.bone.2010.07.028 [DOI] [PubMed] [Google Scholar]
- 55.Ng AJ, Mutsaers AJ, Baker EK, Walkley CR. Genetically engineered mouse models and human osteosarcoma. Clin Sarcoma Res (2012) 2(1):19. 10.1186/2045-3329-2-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature (1992) 356(6366):215–21 10.1038/356215a0 [DOI] [PubMed] [Google Scholar]
- 57.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol (1994) 4(1):1–7 10.1016/S0960-9822(00)00002-6 [DOI] [PubMed] [Google Scholar]
- 58.Lavigueur A, Maltby V, Mock D, Rossant J, Pawson T, Bernstein A. High incidence of lung, bone, and lymphoid tumors in transgenic mice overexpressing mutant alleles of the p53 oncogene. Mol Cell Biol (1989) 9(9):3982–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell (2004) 119(6):861–72 10.1016/j.cell.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 60.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell (2004) 119(6):847–60 10.1016/j.cell.2004.11.004 [DOI] [PubMed] [Google Scholar]
- 61.Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet (1994) 7(4):480–4 10.1038/ng0894-480 [DOI] [PubMed] [Google Scholar]
- 62.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature (1992) 359(6393):288–94 10.1038/359288a0 [DOI] [PubMed] [Google Scholar]
- 63.VanKoevering KK, Williams BO. Transgenic mouse strains for conditional gene deletion during skeletal development. IBMS boneKEy (2008) 5:151–70 10.1138/20080312 [DOI] [Google Scholar]
- 64.Mohseny AB, Hogendoorn PC, Cleton-Jansen AM. Osteosarcoma models: from cell lines to zebrafish. Sarcoma (2012) 2012:417271. 10.1155/2012/417271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin PP, Pandey MK, Jin F, Raymond AK, Akiyama H, Lozano G. Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis (2009) 30(10):1789–95 10.1093/carcin/bgp180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. Rb regulates fate choice and lineage commitment in vivo. Nature (2010) 466(7310):1110–4 10.1038/nature09264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A (2008) 105(33):11851–6 10.1073/pnas.0805462105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev (2008) 22(12):1662–76 10.1101/gad.1656808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mutsaers AJ, Ng AJ, Baker EK, Russell MR, Chalk AM, Wall M, et al. Modeling distinct osteosarcoma subtypes in vivo using Cre:lox and lineage-restricted transgenic shRNA. Bone (2013) 55(1):166–78 10.1016/j.bone.2013.02.016 [DOI] [PubMed] [Google Scholar]
- 70.Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, et al. Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J Cell Biol (2006) 172(6):909–21 10.1083/jcb.200508130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J, et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest (2010) 120(9):3310–25 10.1172/JCI42391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis (2002) 33(2):77–80 10.1002/gene.10092 [DOI] [PubMed] [Google Scholar]
- 73.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development (2006) 133(16):3231–44 10.1242/dev.02480 [DOI] [PubMed] [Google Scholar]
- 74.Frendo JL, Xiao G, Fuchs S, Franceschi RT, Karsenty G, Ducy P. Functional hierarchy between two OSE2 elements in the control of osteocalcin gene expression in vivo. J Biol Chem (1998) 273(46):30509–16 10.1074/jbc.273.46.30509 [DOI] [PubMed] [Google Scholar]
- 75.Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell (2001) 8(2):303–16 10.1016/S1097-2765(01)00327-6 [DOI] [PubMed] [Google Scholar]
- 76.Kuijjer ML, Namlos HM, Hauben EI, Machado I, Kresse SH, Serra M, et al. mRNA expression profiles of primary high-grade central osteosarcoma are preserved in cell lines and xenografts. BMC Med Genomics (2011) 4:66. 10.1186/1755-8794-4-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weisstein JS, Majeska RJ, Klein MJ, Einhorn TA. Detection of c-fos expression in benign and malignant musculoskeletal lesions. J Orthop Res (2001) 19(3):339–45 10.1016/S0736-0266(00)90020-2 [DOI] [PubMed] [Google Scholar]
- 78.Ruther U, Komitowski D, Schubert FR, Wagner EF. C-Fos expression induces bone tumors in transgenic mice. Oncogene (1989) 4(7):861–5 [PubMed] [Google Scholar]
- 79.Entz-Werle N, Choquet P, Neuville A, Kuchler-Bopp S, Clauss F, Danse JM, et al. Targeted apc;twist double-mutant mice: a new model of spontaneous osteosarcoma that mimics the human disease. Transl Oncol (2010) 3(6):344–53 10.1593/tlo.10169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, et al. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature (2007) 448(7156):943–6 10.1038/nature06084 [DOI] [PubMed] [Google Scholar]
- 81.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature (2001) 413(6851):86–91 10.1038/35092592 [DOI] [PubMed] [Google Scholar]
- 82.Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res (2001) 61(16):6234–8 [PubMed] [Google Scholar]
- 83.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer (2007) 49(7):928–40 10.1002/pbc.21078 [DOI] [PubMed] [Google Scholar]
- 84.Chawla SP, Staddon AP, Baker LH, Schuetze SM, Tolcher AW, D’Amato GZ, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol (2012) 30(1):78–84 10.1200/JCO.2011.35.6329 [DOI] [PubMed] [Google Scholar]
- 85.Grignani G, Palmerini E, Dileo P, Asaftei SD, D’Ambrosio L, Pignochino Y, et al. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol (2012) 23(2):508–16 10.1093/annonc/mdr151 [DOI] [PubMed] [Google Scholar]
- 86.Malempati S, Weigel B, Ingle AM, Ahern CH, Carroll JM, Roberts CT, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol (2012) 30(3):256–62 10.1200/JCO.2011.37.4355 [DOI] [PMC free article] [PubMed] [Google Scholar]