

Abstract
The Pd-catalyzed coupling of N-allyl sulfamides with aryl and alkenyl triflates to afford cyclic sulfamide products is described. In contrast to other known Pd-catalyzed alkene carboamination reactions, these transformations may be selectively induced to occur by way of either anti- or syn-aminopalladation mechanistic pathways by modifying catalyst structure and reaction conditions.
Keywords: Addition Reactions, Alkenes, Heterocycles, Palladium, Stereoselective
Introduction
Cyclic sulfamides are an important class of heterocycles that have attracted attention in medicinal chemistry applications as these functional groups can serve as isosteres for cyclic ureas and are also known to form attractive electrostatic interactions with proteins and enzymes.[1] Biologically active compounds bearing these units have been examined as protease inhibitors,[2] HLE inhibitors,[3] renin inhibitors,[4] and norovirus inhibitors.[5] In addition, the SO2 unit can be cleaved from these compounds to afford synthetically useful 1,2-diamines.[6, 7] Cyclic sulfamides have also been employed as chiral auxiliaries for asymmetric aldol and alkylation reactions.[8]
Classical approaches to the synthesis of cyclic sulfamides frequently involve treatment of 1,2-diamines with sulfamide or related electrophiles and generally require relatively complex starting materials.[9] In recent years a number of metal-catalyzed alkene diamination or oxidative cyclization reactions have been described that effect the conversion of readily available substrates to cyclic sulfamide derivatives.[7]
We have previously reported a method for the construction of cyclic ureas via Pd-catalyzed alkene carboamination reactions between acyclic N-allyl ureas and aryl or alkenyl halides.[10] We felt that related transformations of N-allyl sulfamides could provide an attractive and simple approach to the generation of substituted cyclic sulfamides.[11] In addition, this strategy would complement existing methods as this would allow for the conversion of an acyclic N-allyl sulfamide to a cyclic sulfamide with generation of both a C–N and a C–C bond. Herein we describe our preliminary studies in this area, along with our findings that the stereochemistry of addition to the alkene can be controlled by the appropriate choice of catalyst and conditions, which influence syn- vs. anti-aminopalladation mechanistic pathways in the catalytic cycle.
Results and Discussion
In preliminary studies we examined the Pd-catalyzed carboamination between 1a and 4-bromobenzonitrile. After some exploration we found that use of a catalyst composed of Pd2(dba)3 and the Buchwald X-Phos[12] ligand afforded the desired product 2a in 79% isolated yield [Eq. (1)]. However, the scope of this reaction was limited to electron deficient aryl bromides, as competing Heck arylation occurred in reactions of more electron-rich electrophiles.
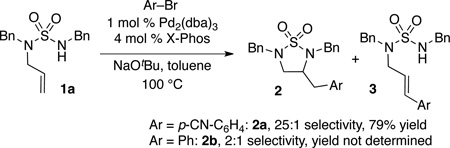 |
(1) |
We initially postulated that the mechanism of the carboamination reactions of N-allyl sulfamides was similar to that of other nucleophiles such as ureas or amines.[13] Namely, oxidative addition of the aryl bromide to Pd(0) to generate 4, which then reacts with the substrate 1 and base to afford 5.[14] Syn-aminopalladation of 5 would give 6, which could undergo reductive elimination to afford the observed product 2.
We felt that two factors could potentially be the cause of the Heck arylation side reactions observed with relatively electron-rich aryl bromides: (1) formation of the Pd–N bond (4 to 5) may be relatively slow for palladium complexes that are less electrophilic as a result of the electron-rich aryl groups bound to the metal; or (2) the aminopalladation step (5 to 6) may be reversible,[15] and competing migratory insertion of the alkene into the Pd–C bond of 4 (which leads to side product 3) may be faster with relatively electron-rich aryl groups.[16] These factors suggested that use of aryl triflate substrates could potentially lead to improved results in Pd-catalyzed carboamination reactions of N-allylsulfamides. The oxidative addition of aryl triflates to Pd(0) leads to the formation of cationic palladium complexes,[17] which should undergo more facile Pd–N bond formation due to the increased electrophilicity of these intermediates. In addition, the non-nucleophilic triflate anion is less likely to promote the formation of anionic complexes that are known to accelerate Heck reactions.[18]
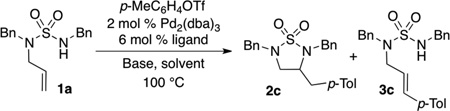
In order to test this idea we studied the coupling of 1 with p-tolyl triflate (Table 1), and our first results using X-Phos as ligand were disappointing; a 1:1 mixture of desired product:Heck side product (2c:3c) was obtained. However, after further exploration we discovered that the RuPhos ligand provided significantly better results, and a screen of bases revealed that use of LiOtBu provided further improvement. Finally, switching to the more polar solvent benzotrifluoride resulted in formation of the desired product with only a trace amount of the Heck arylation side product.
Table 1.
Optimization of Aryl Triflate Carboamination[a]
 | |||||
|---|---|---|---|---|---|
| Entry | Ligand | Base | Solvent | 2c:3c | Conversion (%)[b] |
| 1 | X-Phos | NaOtBu | toluene | 1:1 | 81 |
| 2 | RuPhos | NaOtBu | toluene | 9:1 | 90 |
| 3 | RuPhos | LiOtBu | toluene | 19:1 | 100 |
| 4 | RuPhos | LiOtBu | PhCF3 | >25:1 | 100[c] |
Reaction Conditions: 1.0 equiv 1a, 1.2 equiv p-MeC6H4OTf, 1.4 equiv base, 2 mol % Pd2(dba)3, 6 mol % ligand, solvent (0.25 M), 100 °C.
Conversion = percentage of starting material consumed.
The reaction was conducted using 2 mol % Pd(OAc)2 and 5 mol % ligand.
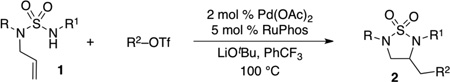
We then proceeded to examine the scope of Pd-catalyzed carboamination reactions of N-allyl sulfamides with a variety of different aryl triflates (Table 2). Both electron-withdrawing groups (entries 2, 7, 9, and 13) and electron-donating groups (entries 3, 10, and 12) were tolerated on the aryl triflate substrate. In addition, the reaction of an ortho-substituted aryl triflate also proceeded in good yield (entry 4). Alkenyl triflates were also viable substrates (entries 5–6) and the reactions proceeded with retention of alkene geometry. The RuPhos ligand provided satisfactory results with most electrophiles that were examined. However, in a few cases superior results were obtained with Brettphos (entry 5), tBu-Davephos (entry 6), or tBu-X-Phos (entry 11). In most cases the Pd-catalyzed carboamination reactions did not generate significant amounts of undesired side products. However, in a few instances side products that result from competing 6-endocyclization were observed. In addition, in some cases reactions of substrates that contain two different groups on the N-atoms (R ≠ R1) generated side products resulting from allylic transposition and cyclization. In a few instances small amounts of Heck side product 3 were also generated. In cases where this side product could not easily be separated by column chromatography the Heck side product was de-allylated (via Pd-catalyzed π-allyl formation/trapping) by addition of 1,3-bis(diphenylphosphino)propane and morpholine to the reaction mixture.
 |
(2) |
 |
(3) |
 |
(4) |
 |
(5) |
 |
(6) |
Table 2.
Pd-Catalyzed Carboamination of N-Allyl Sulfamides [a]
 | |||||
|---|---|---|---|---|---|
| Entry | R | R1 | R2 | Product | Yield (%)[b] |
| 1 | Bn | Bn | p-Me-C6H4 | 2c | 85 |
| 2 | Bn | Bn | p-NC-C6H4 | 2a | 90 |
| 3 | Bn | Bn | p-MeO-C6H4 | 2d | 90 |
| 4 | Bn | Bn | o-Me-C6H4 | 2e | 85 |
| 5 | Bn | Bn | 1-cyclohexenyl | 2f | 87[c] |
| 6 | Bn | Bn | E-1-decenyl[e] | 2g | 80[d,e,f] |
| 7 | Me | Bn | p-Cl-C6H4 | 2h | 79 |
| 8 | Bn | PMB | p-Me-C6H4 | 2i | 90 |
| 9 | Bn | Me | m-F3C-C6H4 | 2j | 86 |
| 10 | Bn | tBu | p-MeO-C6H4 | 2k | 92 |
| 11 | Bn | PMP | Ph | 2l | 90[g] |
| 12 | Me | Bn | 2m | 84 | |
| 13 | tBu | Bn | m-F3C-C6H4 | 2n | 88[h] |
| 14 | H | allyl | Ph | 2o | 51[i] |
Reaction Conditions: 1.0 equiv 1, 1.2 equiv R2OTf, 1.4 equiv LiOtBu, 2 mol % Pd(OAc)2, 5 mol % RuPhos, PhCF3 (0.25 M), 100 °C.
Isolated yield (average of two experiments).
The reaction was conducted using Brettphos as ligand.
The reaction was conducted using tBu-Davephos as ligand.
The alkenyl triflate was used as a 5:1 mixture of E:Z isomers, and the product was obtained as a 5:1 E:Z mixture.
The reaction was conducted using 1.4 equiv R2OTf and 1.6 equiv LiOtBu.
The reaction was conducted using tBu-X-Phos as ligand.
The reaction was conducted using 7.5 mol % ligand.
The reaction was conducted using 2.4 equiv R2OTf and 2.4 equiv LiOtBu.
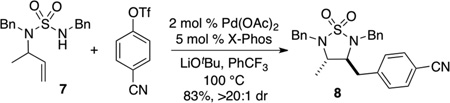
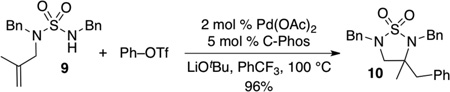
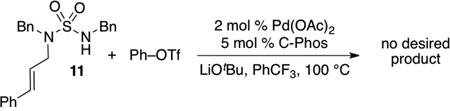
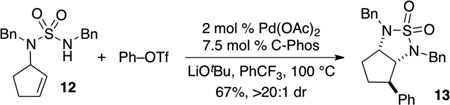
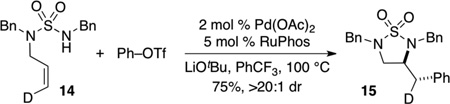
To further explore the scope of the sulfamide carboamination reactions we examined transformations of more highly substituted substrates. After some exploration we discovered that the C-Phos ligand provided higher yields and cleaner reactions than Ruphos in these transformations. Substrate 7 bearing an allylic methyl group was converted to 8 in good yield with >20:1 dr [Eq. (2)], and the presence of a methyl group on the internal alkene carbon atom was also tolerated in the conversion of 9 to 10 [Eq. (3)]. Efforts to cyclize substrate 11 bearing an E-disubstituted alkene were unsuccessful; no reaction was observed [Eq. (4)]. However, cyclopentene-derived substrate 12 was successfully coupled with phenyl triflate to afford bicyclic product 13 [Eq. (5)]. In contrast to related carboamination reactions of other nucleophiles,[13] the carboamination of 12 proceeded with anti-addition to the alkene. Similarly, deuterated substrate 14 was converted to 15, the product of anti-addition to the alkene, with high stereoselectivity [Eq. (6)].
The formation of products resulting from anti-addition to the alkene is in sharp contrast to previously reported alkene carboamination reactions, which afford syn-addition products.[11] As such, it appears that the mechanism of the Pd-catalyzed reactions of N-allyl sulfamides with aryl triflates differs from other Pd-catalyzed carboamination reactions where the Pd–N bond is generated via syn-aminopalladation of the alkene (i.e., migratory insertion of the alkene into the Pd–N bond of an intermediate palladium amido complex).[19]
The mechanism of the reactions between N-allyl sulfamides and aryl triflates most likely proceeds as illustrated in Scheme 2. Oxidative addition of the aryl triflate to Pd(0) generates cationic Pd(II) complex 16, which then binds to the alkene of substrate 14 to afford 17. A sequence of deprotonation and anti-aminopalladation then affords 18, which can undergo C–C bond-forming reductive elimination to provide the observed product and regenerate the Pd(0) catalyst.[20] Given the relatively low nucleophilicity of the sulfamide group it is likely that the aminopalladation step (17 to 18) is reversible.[15] The favorability of the anti-aminopalladation pathway may also be due in part to the low nucleophilicity of the sulfamide, which may lead to a relatively slow rate of Pd–N bond formation (to generate the palladium amido complex required for syn-aminopalladation as shown in Scheme 1).
Scheme 2.

Anti-aminopalladation mechanism
Scheme 1.

Syn Aminopalladation Mechanism
The formation of 6-endocyclization side products most likely derives from competing 6-endocyclization of 17; the formation of related side products that derive from 6-endocyclization pathways has previously been observed in other Pd-catalyzed alkene difunctionalization reactions that proceed via anti-heteropalladation.[21] The allylic transposition side product appears to be generated through oxidative addition of the N-allylsulfamide to Pd(0) to yield an intermediate allylpalladium complex and a deallylated sulfamide anion, which then recombine to the rearranged compound. Control experiments conducted in the absence of palladium did not lead to rearrangement.
In order to determine if other experimental variables also influence the anti- vs. syn-aminopalladation pathway we examined the coupling of deuterated substrate 14 with phenyl triflate under a number of different conditions (Table 3), moving from the “optimal” conditions for triflate coupling (entry 1) to those originally examined with aryl bromides (entry 6). As shown below, most conditions examined for reactions of aryl triflates favored formation of the anti-addition product 15 (entries 1, 2, and 5). However, both the ligand and the solvent polarity clearly have a significant impact on the reaction pathway, as use of X-Phos as ligand in a nonpolar solvent such as toluene or dioxane favored generation of the syn-addition product 19 (entries 3–4), whereas the anti-addition product predominated in PhCF3 solvent (entry 5). Reactions in which 19 was the major stereoisomer afforded comparatively large amounts of Heck-arylation side product 3d.
Table 3.
Influence of Reaction Conditions[a]
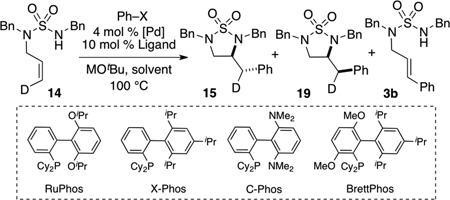 | ||||||
|---|---|---|---|---|---|---|
| Entry | X | Ligand | M | Solvent | 15:19[b] | (15+19):3b[c] |
| 1 | OTf | RuPhos | Li | PhCF3 | >20:1 | 99:1 |
| 2 | OTf | RuPhos | Na | Toluene | 7:1 | 94:6[d] |
| 3 | OTf | X-Phos | Na | Toluene | 1:7 | 72:28[d] |
| 4 | OTf | X-Phos | Li | Dioxane | 1:10 | 60:40 |
| 5 | OTf | X-Phos | Li | PhCF3 | 10:1 | 93:7 |
| 6 | Br | X-Phos | Na | Toluene | 1:4 | 70:30[d] |
| 7 | Br | RuPhos | Na | Toluene | 1:1 | 60:40[d] |
| 8 | Br | RuPhos | Na | Toluene | 1:1 | 60:40 |
| 9 | Br | RuPhos | Na | PhCF3 | 10:1 | 93:7 |
| 10 | Br | X-Phos | Na | PhCF3 | 1:1 | 60:40 |
| 11 | Br | BrettPhos | Na | PhCF3 | 10:1 | 98:2 |
| 12 | Br | C-Phos | Na | PhCF3 | >20:1 | 99:1 |
Reaction Conditions: 1.0 equiv 14, 1.2 equiv Ph–X, 1.4 equiv MOtBu, 4 mol % Pd(OAc)2, 10 mol % Ligand, Solvent (0.0625 M), 100 °C.
NMR ratio of 15:19.
NMR Ratio of (15+19):3b. In general, no significant amounts of other side products were generated in these reactions, and NMR yields are estimated to be >90% for the combined total of 15+19+3b.
Pd2(dba)3 (2 mol % complex, 4 mol % Pd) was used in place of Pd(OAc)2.
In transformations involving bromobenzene as the electrophile, the X-Phos ligand also favored formation of the syn-addition product in toluene (entry 6). Use of the RuPhos ligand in toluene afforded a 1:1 mixture of anti:syn addition products (entries 7–8). However, a survey of different but related biaryl phosphines in the polar solvent PhCF3 revealed that the anti-addition product was favored for most ligands with the exception of X-Phos (1:1 mixture, entry 10) which lacks electron donating alkoxy or amino groups on the biphenyl moiety.
Although the Pd-catalyzed carboamination of 14 is mechanistically complex, in general it appears that conditions that lead to a more electrophilic metal center and/or cationic intermediates (e.g. aryl triflate substrates, the relatively polar solvent PhCF3[22] which may facilitate generation of cationic intermediate palladium complexes) favor the anti-aminopalladation pathway. The observed influence of phosphine ligand structure also fits this general pattern, as ligands that favor anti-addition pathways contain electron donating groups on the biphenyl unit, which may be able to stabilize cationic intermediates either due to increased electron-donating ability of the phosphine (e.g., Brettphos) or through an electron-donating interaction between the biphenyl backbone and the metal (e.g., RuPhos and C-Phos).
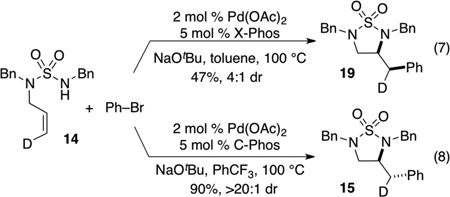
In contrast, conditions that lead to a less electrophilic metal center and/or neutral intermediates (aryl bromide substrates, nonpolar solvents) appear to favor the syn-addition pathway. Importantly, these experiments also indicate that intermolecular[23] Pd-catalyzed carboamination reactions between aryl halides and alkenes bearing pendant nucleophiles can proceed via either syn-or anti-aminopalladation pathways under appropriate reaction conditions.[24,25] As shown below [Eq. (7–8)], use of conditions that appear to be optimal for the syn-addition pathway in the coupling of 14 with bromobenzene afforded 19 in 47% yield and 4:1 dr, whereas conditions that are optimal for anti-addition afforded 15 in 90% yield and >20:1 dr.
 |
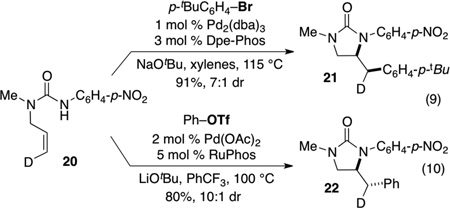
In order to determine whether this interesting influence of reaction conditions on syn- vs. anti-aminopalladation is limited solely to sulfamides, or if this can be more broadly applicable to other nucleophiles, we examined syn- vs. anti-addition reactions of related N-allylurea 20 [Eq. (9–10)]. Our prior studies have illustrated that the coupling of 20 with 4-bromo-tert-butylbenzene proceeds with syn-addition in the presence of a Pd/Dpe-Phos catalyst to generate 21.[13d] In contrast, the Pd/RuPhos-catalyzed coupling of 20 with phenyl triflate proceeds with net anti-addition to afford 22. On the basis of this result it appears that it will likely be possible to control syn- vs. anti-aminopalladation pathways in carboamination reactions of other nucleophilic species. However, further catalyst development will be necessary to broaden the scope to include internal alkene substrates, as efforts to apply optimized syn-addition conditions to cyclopentene-derived substrate afforded a complex mixture of products (although anti-addition product 13 was not formed).
 |
Conclusion
In conclusion, we have developed a new approach to the construction of cyclic sulfamides via Pd-catalyzed alkene carboamination of N-allyl sulfamide derivatives. The mechanism of these reactions is dependent on conditions, and can selectively proceed via either syn- or anti-aminopalladation pathways. Preliminary experiments suggest the use of different catalysts or reaction conditions to control mechanistic pathways will extend beyond sulfamide substrates, which could have broadly significant implications to a number of different Pd-catalyzed alkene difunctionalization reactions. Further studies on the development of other alkene carboheterofunctionalization reactions that proceed through anti-heteropalladation processes are currently underway.
Experimental Section
General Considerations
All reactions were carried out under a nitrogen atmosphere in flame-dried glassware unless otherwise noted. Palladium precatalysts and phosphine ligands were purchased from commercial sources and used without purification. All other reagents were obtained from commercial sources and were used as obtained unless otherwise noted. Bulk quantities of lithium tert-butoxide and sodium tert-butoxide were stored in a glove box and removed in small amounts (ca. 1–2 g) that were consumed within a few days. Toluene, THF, diethyl ether and dichloromethane were purified using a GlassContour solvent purification system. Anhydrous benzotrifluoride was obtained from commercial sources and was used without further purification. Yields refer to isolated yields of compounds estimated to be ≥95% pure as determined by 1H NMR analysis unless otherwise noted. The yields reported in the experimental section describe the result of a single experiment, whereas isolated yields reported in Tables 1–3 and equations 1–10 are averages of yields for two or more experiments. Thus, the yields reported in the experimental section may differ from those shown in Tables 1–3 and equations 1–10.
General Procedure for Pd-catalyzed carboamination reactions of N-allylsulfamide and N-allylurea derivatives with aryl trifluoromethanesulfonates
A test tube was charged with Pd(OAc)2 (2 mol %), a phosphine ligand (5 mol %), the sulfamide substrate (1.0 equiv), and LiOtBu (1.4 equiv unless otherwise noted). The test tube was purged with N2 then benzotrifluoride was added (reactions were conducted at 0.25 M substrate concentration unless specified otherwise), followed by the aryl trifluoromethanesulfonate (1.2 equiv). The resulting mixture was heated to 100 °C and stirred overnight. The reaction mixture was then cooled to rt, quenched with saturated aqueous ammonium chloride, and extracted with dichloromethane. The combined organics were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was then purified by flash chromatography on silica gel.
4-[(2,5-Dibenzyl-1,1-dioxido-1,2,5-thiadiazolidin-3-yl)methyl]benzonitrile (2a)
The General Procedure was employed for the coupling of 1-allyl-1,3-bis-benzylsulfamide 1a (79 mg, 0.25 mmol) and 4-cyanophenyl trifluoromethanesulfonate (75 mg, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 94 mg (90%) of the title compound as a white solid: mp = 107–108 °C. 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 7.9 Hz, 2 H), 7.43–7.28 (m, 10 H), 6.99 (d, J = 7.9 Hz, 2 H), 4.43 (d, J = 14.7 Hz, 1 H), 4.28 (d, J = 13.6 Hz, 1 H), 4.23 (d, J = 14.7 Hz, 1 H), 4.07 (d, J = 13.7 Hz, 1 H), 3.50 (td, J = 3.9, 7.7 Hz, 1 H), 3.13 (dd, J = 7.2, 9.4 Hz, 1 H), 2.91 (dd, J = 5.7, 13.6 Hz, 1 H), 2.75 (dd, J = 5.7, 9.5 Hz, 1 H), 2.70 (dd, J = 8.4, 13.6 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 141.7, 135.1, 134.7, 132.4, 130.0, 128.9, 128.8, 128.6, 128.3, 118.6, 110.9, 57.0, 51.7, 50.4, 49.3, 39.7; IR (film) 2228, 1321, 1158 cm−1. MS (ESI) 418.1581 (418.1584 calcd for C24H23N3O2S, M + H+).
2,5-Dibenzyl-3-(4-methylbenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (2c)
The General Procedure was employed for the coupling of 1-allyl-1,3-bis-benzylsulfamide 1a (79 mg, 0.25 mmol) and p-tolyl trifluoromethanesulfonate (54 µL, 0.30 mmol) using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 84 mg (83%) of the title compound as a yellow solid: mp = 96–99 °C. 1H NMR (500 MHz, CDCl3) δ 7.49–7.44 (m, 2 H), 7.44–7.30 (m, 8 H), 7.03 (d, J = 7.8 Hz, 2 H), 6.81 (d, J = 7.9 Hz, 2 H), 4.46 (d, J = 14.9 Hz, 1 H), 4.33 (dd, J = 5.8, 14.3 Hz, 2 H), 4.06 (d, J = 13.9 Hz, 1 H), 3.49 (dtd, J = 4.9, 6.6, 9.5 Hz, 1 H), 3.08 (dd, J = 6.9, 9.4 Hz, 1 H), 2.89 (dd, J = 4.9, 13.5 Hz, 1 H), 2.84 (dd, J = 6.3, 9.5 Hz, 1 H), 2.61 (dd, J = 9.6, 13.5 Hz, 1 H), 2.29 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 136.6, 135.5, 135.0, 132.9, 129.4, 129.0, 128.7, 128.7, 128.6, 128.1, 57.6, 50.9, 50.7, 49.6, 39.0, 21.0; IR (film) 1286, 1160 cm−1. MS (ESI) 407.1791 (407.1788 calcd for C24H26N2O2S, M + H+).
2,5-Dibenzyl-3-(4-methoxybenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (2d)
The General Procedure was employed for the coupling of 1-allyl-1,3-bis-benzylsulfamide 1a (79 mg, 0.25 mmol) and 4-methoxyphenyl trifluoromethanesulfonate (54 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 93 mg (88%) of the title compound as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.45 (dd, J = 1.7, 7.8 Hz, 2 H), 7.44–7.30 (m, 8 H), 6.83 (d, J = 8.6 Hz, 2 H), 6.75 (d, J = 8.5 Hz, 2 H), 4.44 (d, J = 14.8 Hz, 1 H), 4.32 (d, J = 14.5 Hz, 2 H), 4.05 (d, J = 13.8 Hz, 1 H), 3.76 (s, 3 H), 3.46 (ddd, J = 5.0, 6.9, 9.7 Hz, 1 H), 3.07 (ddd, J = 1.4, 7.0, 8.4 Hz, 1 H), 2.86 (dd, J = 5.0, 13.6 Hz, 1 H), 2.82 (dd, J = 6.4, 9.4 Hz, 1 H), 2.58 (dd, J = 9.4, 13.6 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 188.0, 158.6, 135.5, 135.0, 130.1, 129.0, 128.7, 128.7, 128.6, 128.1, 128.0, 114.1, 57.6, 55.2, 50.9, 50.7, 49.6, 38.6; IR (film) 1246, 1160 cm−1. MS (ESI) 423.1739 (423.1737 calcd for C24H26N2O3S, M + H+).
2,5-Dibenzyl-3-(2-methylbenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (2e)
The General Procedure was employed for the coupling of 1-allyl-1,3-bis-benzylsulfamide 1a (79 mg, 0.25 mmol) and o-tolyl trifluoromethanesulfonate (54 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 86 mg (85%) of the title compound as a white solid: mp = 120–122 °C. 1H NMR (500 MHz, CDCl3) δ 7.46–7.30 (m, 10 H), 7.07 (ddd, J = 7.2, 14.7, 25.7 Hz, 3 H), 6.86 (dd, J = 1.4, 7.5 Hz, 1 H), 4.37 (s, 2 H), 4.30 (d, J = 13.7 Hz, 1 H), 4.13 (d, J = 13.7 Hz, 1 H), 3.51 (ddt, J = 5.5, 6.8, 9.4 Hz, 1 H), 3.09 (dd, J = 6.9, 9.4 Hz, 1 H), 2.98 (dd, J = 5.2, 13.6 Hz, 1 H), 2.87 (dd, J = 5.8, 9.4 Hz, 1 H), 2.69 (dd, J = 9.6, 13.6 Hz, 1 H), 1.99 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 136.3, 135.4, 135.0, 134.4, 130.6, 130.1, 128.9, 128.7, 128.7, 128.7, 128.2, 128.1, 127.1, 126.1, 55.7, 50.7, 50.6, 49.6, 36.8, 19.1; IR (film) 1283, 1164 cm−1. MS (ESI) 407.1790 (407.1788 calcd for C24H26N2O2S, M + H+).
2,5-Dibenzyl-3-(cyclohex-1-en-1-ylmethyl)-1,2,5-thiadiazolidine-1,1-dioxide (2f)
The General Procedure was employed for the coupling of 1-allyl-1,3-bis-benzylsulfamide 1a (79 mg, 0.25 mmol) and 1-cyclohexenyl trifluoromethanesulfonate (52 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and BrettPhos (6.7 mg, 0.0125 mmol). This procedure afforded 89 mg (90%) of the title compound as a pale yellow solid: mp = 66–68 °C. 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 7.0 Hz, 2 H), 7.42–7.29 (m, 8 H), 5.33 (s, 1 H), 4.49 (d, J = 15.0 Hz, 1 H), 4.36 (d, J = 13.8 Hz, 1 H), 4.26 (d, J = 15.0 Hz, 1 H), 4.05 (d, J = 13.8 Hz, 1 H), 3.47–3.36 (m, 1 H), 3.21 (dd, J = 7.0, 9.4 Hz, 1 H), 2.81 (dd, J = 6.6, 9.4 Hz, 1 H), 2.23 (dd, J = 2.2, 14.0 Hz, 1 H), 2.04 (dd, J = 9.7, 13.6 Hz, 1 H), 1.94–1.80 (m, 2 H), 1.73–1.59 (m, 1 H), 1.59–1.33 (m, 5 H); 13C NMR (126 MHz, CDCl3) δ 135.8, 135.7, 135.1, 132.2, 128.7, 128.7, 128.6, 128.0, 127.9, 125.5, 54.8, 50.8, 50.6, 49.9, 41.9, 28.3, 25.1, 22.6, 22.0; IR (film) 1286, 1154 cm−1. MS (ESI) 397.1949 (397.1944 calcd for C23H28N2O2S, M + H+).
(E)-2,5-Dibenzyl-3-(undec-2-en-1-yl)-1,2,5-thiadiazolidine-1,1-dioxide (2g)
The General Procedure was employed for the coupling of 1-allyl-1,3-bis-benzylsulfamide 1a (79 mg, 0.25 mmol) and (E)-dec-1-en-1-yl trifluoromethanesulfonate (101 µL, 0.35 mmol, 5:1 mixture of E:Z isomers), using LiOtBu (32 mg, 0.40 mmol) and a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and tBuDavePhos (4.3 mg, 0.0125 mmol). This procedure afforded 90 mg (79%) of the title compound as a yellow oil. The compound was judged to be a 5:1 mixture of E:Z isomers by 1H NMR analysis. Data are for the major (E) isomer. 1H NMR (500 MHz, CDCl3) δ 7.51–7.42 (m, 2 H), 7.42–7.29 (m, 8 H), 5.44 (dt, J = 7.4, 11.0 Hz, 1 H), 5.16–5.06 (m, 1 H), 4.51 (d, J = 15.1 Hz, 1 H), 4.36 (d, J = 13.7 Hz, 1 H), 4.26 (d, J = 14.8 Hz, 1 H), 4.02 (d, J = 13.7 Hz, 1 H), 3.33 (ddd, J = 5.6, 9.2, 11.1 Hz, 1 H), 3.22 (dd, J = 7.0, 9.3 Hz, 1 H), 2.79 (dd, J = 7.1, 9.3 Hz, 1 H), 2.27 (dddd, J = 5.5, 7.5, 13.5, 15.9 Hz, 1 H), 2.16 (dt, J = 8.4, 15.2 Hz, 1 H), 1.81 (tt, J = 6.9, 12.9 Hz, 2 H), 1.35–1.18 (m, 12 H), 0.91 (q, J = 6.5 Hz, 3 H); 13C NMR (126 MHz, CDCl3) δ 135.7, 134.9, 134.4, 128.7, 128.7, 128.7, 128.6, 128.1, 128.0, 122.1, 56.3, 50.9, 50.6, 49.6, 31.9, 30.5, 29.5, 29.4, 29.3, 29.3, 27.4, 22.7, 14.1; IR (film) 1304, 1164 cm−1. MS (ESI) 455.2735 (455.2727 calcd for C27H38N2O2S, M + H+).
2-Benzyl-3-(4-chlorobenzyl)-5-methyl-1,2,5-thiadiazolidine-1,1-dioxide (2h)
The General Procedure was employed for the coupling of 1b (60 mg, 0.25 mmol) and 4-chlorophenyl trifluoromethanesulfonate (52 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 68 mg (78%) of the title compound as a white solid: mp = 133–135 °C. 1H NMR (500 MHz, CDCl3) δ 7.41–7.36 (m, 4 H), 7.36–7.31 (m, 1 H), 7.23 (d, J = 8.3 Hz, 2 H), 6.92 (d, J = 8.3 Hz, 2 H), 4.43 (d, J = 14.9 Hz, 1 H), 4.25 (d, J = 14.9 Hz, 1 H), 3.51 (dtd, J = 5.2, 6.9, 9.3 Hz, 1 H), 3.17 (dd, J = 7.0, 9.3 Hz, 1 H), 2.89 (dd, J = 5.2, 13.6 Hz, 1 H), 2.86 (dd, J = 6.8, 9.3 Hz, 1 H), 2.73 (s, 3 H), 2.61 (dd, J = 9.3, 13.6 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 135.3, 134.5, 132.9, 130.4, 128.9, 128.8, 128.7, 128.1, 57.1, 52.5, 51.2, 39.0, 33.2; IR (film) 1297, 1149 cm−1. MS (ESI) 351.0926 (351.0929 calcd for C17H19ClN2O2S, M + H+).
5-Benzyl-2-(4-methoxybenzyl)-3-(4-methylbenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (2i)
The General Procedure was employed for the coupling of 1c (87 mg, 0.25 mmol) and p-tolyl trifluoromethanesulfonate (54 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 95 mg (87%) of the title compound as a pale yellow solid: mp = 109–113 °C. 1H NMR (400 MHz, CDCl3) δ 7.42–7.25 (m, 7 H), 6.99 (d, J = 7.8 Hz, 2 H), 6.92–6.86 (m, 2 H), 6.79 (d, J = 7.9 Hz, 2 H), 4.36–4.21 (m, 3 H), 4.01 (d, J = 13.8 Hz, 1 H), 3.81 (s, 3 H), 3.49–3.37 (m, 1 H), 3.02 (dd, J = 7.0, 9.4 Hz, 1 H), 2.86 (dd, J = 5.0, 13.5 Hz, 1 H), 2.78 (dd, J = 6.2, 9.4 Hz, 1 H), 2.56 (dd, J = 9.5, 13.5 Hz, 1 H), 2.26 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 159.5, 136.5, 135.0, 133.0, 130.4, 129.4, 129.0, 128.7, 128.6, 128.1, 127.3, 114.0, 57.1, 55.3, 50.6, 50.3, 49.5, 39.0, 21.0; IR (film) 1281, 1158 cm−1. MS (ESI) 437.1885 (437.1893 calcd for C25H28N2O3S, M + H+).
5-Benzyl-2-methyl-3-(3-(trifluoromethyl)benzyl)-1,2,5-thiadiazolidine-1,1-dioxide (2j)
The General Procedure was employed for the coupling of 1d (60 mg, 0.25 mmol) and 3-trifluoromethylphenyl trifluoromethanesulfonate (60 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 78 mg (81%) of the title compound as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 7.8 Hz, 1 H), 7.43 (t, J = 7.7 Hz, 1 H), 7.40 (d, J = 1.6 Hz, 1 H), 7.38–7.29 (m, 6 H), 4.31 (d, J = 13.9 Hz, 1 H), 3.99 (d, J = 13.9 Hz, 1 H), 3.44 (m, 1 H), 3.16 (dd, J = 6.9, 9.4 Hz, 1 H), 3.12 (dd, J = 5.7, 13.6 Hz, 1 H), 2.85–2.78 (m, 2 H), 2.75 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 136.9, 134.8, 132.7, 131.2 (q, J = 33.8 Hz), 129.3, 128.7, 128.6, 128.2, 125.8, 124.1, 123.9 (q, J = 272.4 Hz), 59.6, 50.8, 49.5, 38.8, 33.6; IR (film) 1242, 1127 cm−1. MS (ESI) 385.1193 (385.1192 calcd for C18H19F3N2O2S, M + H+).
5-Benzyl-2-(tert-butyl)-3-(4-methoxybenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (2k)
The General Procedure was employed for the coupling of 1e (71 mg, 0.25 mmol) and 4-methoxyphenyl trifluoromethanesulfonate (54 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 90 mg (93%) of the title compound as a white solid: mp = 104–106 °C. 1H NMR (500 MHz, CDCl3) δ 7.47–7.32 (m, 5 H), 6.79 (d, J = 8.6 Hz, 2 H), 6.71 (d, J = 8.6 Hz, 2 H), 4.46 (d, J = 13.5 Hz, 1 H), 3.77 (d, J = 13.6 Hz, 1 H), 3.75 (s, 3 H), 3.54 (dddd, J = 1.3, 4.1, 5.8, 10.4 Hz, 1 H), 2.98–2.78 (m, 4 H), 1.53 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 158.4, 135.4, 130.2, 129.3, 128.8, 128.7, 128.1, 114.0, 57.8, 55.4, 55.2, 49.0, 47.1, 40.8, 28.2; IR (film) 1279, 1142 cm−1. MS (ESI) 389.1893 (389.1893 calcd for C21H28N2O3S, M + H+).
3,5-Dibenzyl-2-(4-methoxyphenyl)-1,2,5-thiadiazolidine-1,1-dioxide (2l)
The General Procedure was employed for the coupling of 1-allyl-1-benzyl-3-(4-methoxyphenyl)sulfamide 1f (83 mg, 0.25 mmol) and phenyl trifluoromethanesulfonate (49 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and tBuXPhos (5.3 mg, 0.0125 mmol). This procedure afforded 91 mg (89%) of the title compound as a yellow solid: mp = 95–97 °C. 1H NMR (500 MHz, CDCl3) δ 7.45–7.30 (m, 7 H), 7.29–7.19 (m, 3 H), 7.07–6.97 (m, 4 H), 4.41 (d, J = 14.0 Hz, 1 H), 4.21–4.12 (m, 1 H), 4.06 (d, J = 14.0 Hz, 1 H), 3.85 (s, 3 H), 3.24 (dd, J = 6.5, 9.2 Hz, 1 H), 3.03 (dd, J = 7.6, 9.4 Hz, 1 H), 3.00 (dd, J = 4.2, 13.8 Hz, 1 H), 2.71 (dd, J = 9.5, 13.7 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 159.2, 135.5, 134.9, 129.2, 128.8, 128.7, 128.7, 128.6, 128.2, 128.1, 127.0, 115.0, 58.6, 55.5, 51.2, 49.7, 38.7; IR (film) 1289, 1157 cm−1. MS (ESI) 409.1577 (409.1580 calcd for C23H24N2O3S, M + H+).
3-(Benzo[d][1,3]dioxol-5-ylmethyl)-2-benzyl-5-methyl-1,2,5-thiadiazolidine-1,1-dioxide (2m)
The General Procedure was employed for the coupling of 1b (60 mg, 0.25 mmol) and 3,4-methylenedioxyphenyl trifluoromethanesulfonate (52 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 76 mg (84%) of the title compound as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.45–7.29 (m, 5 H), 6.69 (d, J = 7.8 Hz, 1 H), 6.49–6.42 (m, 2 H), 5.91 (m, 2 H), 4.43 (d, J = 15.0 Hz, 1 H), 4.25 (d, J = 14.9 Hz, 1 H), 3.47 (dtd, J = 4.9, 6.9, 9.7 Hz, 1 H), 3.17 (dd, J = 6.9, 9.4 Hz, 1 H), 2.88 (dd, J = 6.9, 9.4 Hz, 1 H), 2.83 (dd, J = 5.0, 13.5 Hz, 1 H), 2.72 (s, 3 H), 2.53 (dd, J = 9.7, 13.5 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 147.8, 146.6, 135.4, 129.6, 128.8, 128.7, 128.1, 122.1, 109.2, 108.4, 101.0, 57.4, 52.5, 51.0, 39.2, 33.2; IR (film) 1246, 1150 cm−1. MS (ESI) 361.1219 (361.1217 calcd for C18H20N2O4S, M + H+).
2-Benzyl-5-(tert-butyl)-3-[3-(trifluoromethyl)benzyl]-1,2,5-thiadiazolidine-1,1-dioxide (2n)
The General Procedure was employed for the coupling of 1g (71 mg, 0.25 mmol) and 3-trifluoromethylphenyl trifluoromethanesulfonate (60 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 91 mg (85%) of the title compound as a yellow solid: mp = 104–106 °C. 1H NMR (500 MHz, CDCl3) δ 7.49 (d, J = 7.8 Hz, 1 H), 7.42–7.29 (m, 6 H), 7.26–7.19 (m, 2 H), 4.38 (d, J = 14.8 Hz, 1 H), 4.19 (d, J = 14.8 Hz, 1 H), 3.53–3.43 (m, 1 H), 3.25 (dd, J = 6.6, 8.9 Hz, 1 H), 3.10–2.98 (m, 2 H), 2.74 (dd, J = 9.1, 13.6 Hz, 1 H), 1.42 (s, 9 H); 13C NMR (126 MHz, CDCl3) δ 137.6, 135.5, 132.6, 131.0 (q, J = 32.1 Hz), 129.2, 128.8, 128.6, 128.0, 125.7 (q, J = 3.7 Hz), 123.9 (q, J = 272.3 Hz), 123.8 (q, J = 3.8 Hz), 56.2, 56.1, 50.6, 45.6, 38.5, 27.4; IR (film) 1302, 1120 cm−1. MS (ESI) 427.1663 (427.1662 calcd for C21H25F3N2O2S, M + H+).
2-Allyl-3-benzyl-1,2,5-thiadiazolidine-1,1-dioxide (2o)
The General Procedure was employed for the coupling of 1h (44 mg, 0.25 mmol) and phenyl trifluoromethanesulfonate (98 µL, 0.60 mmol), using LiOtBu (48 mg, 0.60 mmol) and a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 30 mg (48%) of the title compound as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.37–7.24 (m, 3 H), 7.24–7.15 (m, 2 H), 5.94 (dddd, J = 5.6, 7.7, 10.1, 17.5 Hz, 1 H), 5.36–5.25 (m, 2 H), 4.44 (t, J = 7.5 Hz, 1 H), 3.78 (ddt, J = 1.5, 5.6, 15.1 Hz, 1 H), 3.72 (ddt, J = 5.1, 6.8, 8.4 Hz, 1 H), 3.67 (ddt, J = 1.2, 7.7, 15.1 Hz, 1 H), 3.39 (dt, J = 6.9, 11.7 Hz, 1 H), 3.22 (ddd, J = 4.7, 6.7, 11.6 Hz, 1 H), 3.07 (dd, J = 5.4, 13.5 Hz, 1 H), 2.77 (dd, J = 8.3, 13.5 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 136.0, 132.5, 129.3, 128.8, 127.2, 120.0, 61.1, 48.7, 44.9, 39.2; IR (film) 3242, 1296, 1159 cm−1. MS (ESI) 253.1005 (253.1005 calcd for C12H16N2O2S, M + H+).
2-Benzyl-6-(4-methoxyphenyl)-4-phenyl-1,2,6-thiadiazinane-1,1-dioxide (S2)
The General Procedure was employed for the coupling of 1-allyl-1-benzyl-3-(4-methoxyphenyl)sulfamide 1f (83 mg, 0.25 mmol) and phenyl trifluoromethanesulfonate (49 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). The major product generated in this reaction was 2l (described above), and a small amount of side product S2 was also formed. Upon careful chromatographic purification, S2 was isolated (7 mg, 7%) as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.47–7.42 (m, 4 H), 7.42–7.36 (m, 2 H), 7.33 (tq, J = 1.5, 8.3 Hz, 3 H), 7.30–7.25 (m, 1 H), 7.25–7.21 (m, 2 H), 6.96–6.92 (m, 2 H), 4.69 (d, J = 14.0 Hz, 1 H), 4.48 (d, J = 14.0 Hz, 1 H), 4.24 (t, J = 11.8 Hz, 1 H), 4.04–3.94 (m, 1 H), 3.83 (s, 3 H), 3.61–3.47 (m, 2 H), 3.29 (ddd, J = 2.3, 4.1, 14.1 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 158.9, 137.8, 135.5, 134.4, 128.9, 128.8, 128.1, 128.0, 127.7, 127.5, 114.5, 59.1, 55.5, 53.2, 52.7, 36.9; IR (film) 1344, 1157 cm−1. MS (ESI) 409.1581 (409.1580 calcd for C23H24N2O3S, M + H+).
(±)-(3R,4R)-4-[(2,5-Dibenzyl-4-methyl-1,1-dioxido-1,2,5-thiadiazolidin-3-yl)methyl]benzonitrile (8)
The General Procedure was employed for the coupling of 7 (83 mg, 0.25 mmol) and 4-cyanophenyl trifluoromethanesulfonate (75 mg, 0.30 mmol) using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol), and XPhos (6.0 mg, 0.0125 mmol). This procedure afforded 86 mg (80%) of the title compound as a white solid: mp = 102–108 °C. This compound was obtained as a >20:1 mixture of diastereomers as judged by 1H NMR analysis. 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 8.2 Hz, 2 H), 7.43–7.31 (m, 8 H), 7.30–7.24 (m, 2 H), 6.96 (d, J = 8.2 Hz, 2 H), 4.36–4.16 (m, 4 H), 3.12 (td, J = 3.6, 7.1 Hz, 1 H), 3.03 (qd, J = 3.6, 6.3 Hz, 1 H), 2.91 (dd, J = 6.5, 13.6 Hz, 1 H), 2.73 (dd, J = 7.4, 13.6 Hz, 1 H), 0.95 (d, J = 6.3 Hz, 3 H); 13C NMR (126 MHz, CDCl3) δ 142.2, 135.5, 134.9, 132.3, 130.1, 129.1, 128.8, 128.7, 128.7, 128.2, 128.1, 118.6, 110.8, 64.0, 56.7, 51.9, 48.6, 39.1, 18.6; IR (film) 1294, 1132 cm–1. MS (ESI) 432.1740 (432.1740 calcd for C25H25N3O2S, M + H+).
2,3,5-Tribenzyl-3-methyl-1,2,5-thiadiazolidine-1,1-dioxide (10)
The General Procedure was employed for the coupling of 9 (83 mg, 0.25 mmol) and phenyl trifluoromethanesulfonate (49 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and CPhos (5.5 mg, 0.0125 mmol). This procedure afforded 97 mg (95%) of the title compound as an off-white solid: mp = 129–131 °C. 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 7.2 Hz, 2 H), 7.45–7.35 (m, 7 H), 7.32 (t, J = 7.4 Hz, 1 H), 7.22–7.10 (m, 2 H), 6.88 (d, J = 6.6 Hz, 1 H), 4.42 (s, 2 H), 4.38 (d, J = 13.6 Hz, 1 H), 4.02 (d, J = 13.6 Hz, 1 H), 3.17 (d, J = 9.3 Hz, 1 H), 3.01 (d, J = 13.1 Hz, 1 H), 2.78 (d, J = 13.1 Hz, 1 H), 2.70 (d, J = 9.2 Hz, 1 H), 1.21 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 137.0, 135.7, 135.0, 130.3, 129.0, 128.7, 128.6, 128.4, 128.3, 128.2, 127.8, 126.9, 61.8, 54.4, 50.2, 44.9, 43.0, 22.3; IR (film) 1299, 1167 cm−1. MS (ESI) 407.1789 (407.1788 calcd for C24H26N2O2S, M + H+).
(±)-(3aR,4R,6aS)-1,3-Dibenzyl-4-phenylhexahydro-1H-cyclopenta[c][1,2,5]thiadiazole-2,2-dioxide (13)
The General Procedure was employed for the coupling of 1,3-bis-benzyl-1-cyclopent-2-enylsulfamide 12 (86 mg, 0.25 mmol) and phenyl trifluoromethanesulfonate (81 µL, 0.50 mmol) using LiOtBu (44 mg, 0.55 mmol) and a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol), and CPhos (8.2 mg, 0.01875 mmol). This procedure afforded 70 mg (67%) of the title compound as an off-white solid: mp = 118–120 °C. This compound was obtained as a >20:1 mixture of diastereomers as judged by 1H NMR analysis. 1H NMR (500 MHz, C6D6) δ 7.28–7.21 (m, 2 H), 7.16–7.02 (m, 4 H), 6.99–6.89 (m, 7 H), 6.65–6.57 (m, 2 H), 4.24 (d, J = 14.2, 1 H), 4.15 (d, J = 14.8, 1 H), 4.08 (d, J = 14.2, 1 H), 4.04 (d, J = 14.8, 1 H), 3.38 (dd, J = 6.9, 9.2, 1 H), 3.27 (dt, J = 6.9, 9.2, 1 H), 3.11 (dt, J = 6.7, 10.8, 1 H), 1.74–1.51 (m, 2 H), 1.32 (dtd, J = 2.8, 6.7, 13.2, 1 H), 0.93 (dtd, J = 6.4, 10.7, 12.3, 1 H); 13C NMR (126 MHz, CDCl3) δ 142.0, 135.2, 134.9, 129.0, 128.8, 128.7, 128.7, 128.3, 128.1, 127.7, 127.2, 126.7, 66.1, 60.2, 51.0, 49.8, 49.6, 33.0, 30.9; IR (film) 1310, 1156 cm−1. MS (ESI) 419.1784 (419.1788 calcd for C25H26N2O2S, M + H+).
(±)-(1’S,3S)-2,5-Dibenzyl-1’-deuterio-3-(4-methylbenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (15)
The General Procedure was employed for the coupling of 14 (79 mg, 0.25 mmol) and phenyl trifluoromethanesulfonate (49 µL, 0.30 mmol), using a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and RuPhos (5.8 mg, 0.0125 mmol). This procedure afforded 75 mg (76%) of the title compound as a yellow solid: mp = 74–76 °C. This compound was obtained as a >20:1 mixture of diastereomers as judged by 1H NMR analysis. Data are for the major isomer. 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (m, 2 H), 7.43–7.29 (m, 8 H), 7.26–7.15 (m, 3 H), 6.92 (dd, J = 1.9, 7.6 Hz, 2 H), 4.44 (d, J = 14.9 Hz, 1 H), 4.34 (d, J = 14.8 Hz, 1 H), 4.32 (d, J = 13.8 Hz, 1 H), 4.07 (d, J = 13.8 Hz, 1 H), 3.51 (td, J = 5.0, 6.6 Hz, 1 H), 3.08 (dd, J = 7.0, 9.4 Hz, 1 H), 2.90 (d, J = 5.0 Hz, 1 H), 2.84 (dd, J = 6.2, 9.5 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 136.0, 135.4, 134.9, 129.0, 128.9, 128.7, 128.6, 128.1, 128.1, 127.0, 57.3, 50.9, 50.6, 49.5, 39.1 (t, J = 19.4 Hz); IR (film) 1324, 1154 cm−1. MS (ESI) 394.1698 (394.1694 calcd for C23H23DN2O2S, M + H+).
(±)-(1’S,3S)-2,5-Dibenzyl-1’-deuterio-3-(4-methylbenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (15)
The General Procedure was employed for the coupling of 14 (79 mg, 0.25 mmol) and bromobenzene (32 µL, 0.30 mmol), except using NaOtBu (34 mg, 0.35 mmol) in place of LiOtBu and a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and CPhos (5.5 mg, 0.0125 mmol). This procedure afforded 91 mg (92%) of the title compound as a yellow solid. This compound was obtained as a >20:1 mixture of diastereomers as judged by 1H NMR analysis. Physical properties and spectroscopic data were identical to those provided above.
2,5-Dibenzyl-(1’R,3S)-1’-deuterio-3-(4-methylbenzyl)-1,2,5-thiadiazolidine-1,1-dioxide (19)
The General Procedure was employed for the coupling of 14 (79 mg, 0.25 mmol) and bromobenzene (32 µL, 0.30 mmol) except using NaOtBu (34 mg, 0.35 mmol) in place of LiOtBu, toluene in place of PhCF3, and a catalyst composed of Pd(OAc)2 (1.1 mg, 0.005 mmol) and XPhos (6.0 mg, 0.0125 mmol). After the starting material had been completely consumed, DPPP (2.1 mg, 0.0125 mmol) and morpholine (65 µL, 0.75 mmol) in 1 mL xylene was added, and heated to 120 °C for two hours (this step was employed to facilitate purification by de-allylating small amounts of a side product resulting from competing Heck-arylation), then worked up according to the general procedure. This procedure afforded 46 mg (47%) of the title compound as a yellow solid: mp = 74–76 °C. This compound was obtained as a 4:1 mixture of diastereomers as judged by 1H NMR analysis. Data are for the major isomer. 1H NMR (500 MHz, CDCl3) δ 7.49–7.43 (m, 2 H), 7.43–7.30 (m, 8 H), 7.25–7.17 (m, 3 H), 6.96–6.89 (m, 2 H), 4.44 (d, J = 14.8 Hz, 1 H), 4.34 (d, J = 14.8 Hz, 1 H), 4.32 (d, J = 13.8 Hz, 1 H), 4.07 (d, J = 13.8 Hz, 1 H), 3.51 (dt, J = 6.5, 9.3 Hz, 1 H), 3.09 (dd, J = 7.0, 9.4 Hz, 1 H), 2.85 (dd, J = 6.2, 9.4 Hz, 1 H), 2.63 (d, J = 9.5 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 136.0, 135.4, 134.9, 129.1, 128.9, 128.7, 128.6, 128.1, 128.1, 127.0, 57.3, 50.9, 50.6, 49.5, 39.2 (t, J = 19.6 Hz); IR (film) 1323, 1155 cm−1. MS (ESI) 394.1699 (394.1694 calcd for C23H23DN2O2S, M + H+).
(1’S,4S)-1’-Deuterio-4-benzyl-1-methyl-3-(4-nitrophenyl)imidazolidin-2-one (22)
The General Procedure was employed for the coupling of (Z)-1-(3-d-allyl)-1-methyl-3-(4-nitrophenyl)urea (30 mg, 0.125 mmol) and phenyl trifluoromethanesulfonate (25 µL, 0.15 mmol) using a catalyst composed of Pd(OAc)2 (0.6 mg, 0.0025 mmol), and RuPhos (2.9 mg, 0.00625 mmol). This procedure afforded 33 mg (85%) of the title compound as a bright yellow solid: mp = 167–168 °C. This compound was obtained as a 10:1 mixture of diastereomers as judged by 1H NMR analysis. Data are for the major isomer. 1H NMR (500 MHz, C6D6) δ 7.99 (d, J = 9.3, 2 H), 7.55 (d, J = 9.3, 2 H), 7.10–7.00 (m, 3 H), 6.72 (d, J = 7.4, 2 H), 3.58 (dt, J = 3.3, 8.6, 1 H), 2.49 (dt, J = 1.7, 3.2, 1 H), 2.40 (dd, J = 3.1, 8.9, 1 H), 2.30 (s, 4 H); 13C NMR (126 MHz, CDCl3) δ 156.7, 145.1, 141.9, 135.5, 129.1, 128.9, 127.3, 125.0, 117.7, 53.6, 48.4, 37.4 (t, J = 19.2 Hz), 30.8; IR (film) 1702 cm−1. MS (ESI) 313.1407 (313.1405 calcd for C17H16DN3O3, M + H+).
Supplementary Material
Acknowledgements
The authors acknowledge the NIH-NIGMS (GM 098314) for financial support of this work.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.Reitz AB, Smith GR, Parker MH. Expert Opin. Ther. Patents. 2009;19:1449–1453. doi: 10.1517/13543770903185920. [DOI] [PubMed] [Google Scholar]
- 2.Spaltenstein A, Almond MR, Bock WJ, Cleary DG, Furfine ES, Hazen RJ, Kazmierski WM, Salituro FG, Tung RD, Wright LL. Bioorg. Med. Chem. Lett. 2000;10:1159–1162. [PubMed] [Google Scholar]
- 3.Zhong J, Gan X, Alliston KR, Groutas WC. Bioorg. Med. Chem. 2004;12:589–593. doi: 10.1016/j.bmc.2003.10.059. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SH, Dellaria JF, Kempf DJ, Hutchins CW, Woods KW, Maki RG, de Lara E, Spina KP, Stein HH, Cohen J, Baker WR, Plattner JJ, Kleinert HD, Perun TJ. J. Med. Chem. 1990;33:1582–1590. doi: 10.1021/jm00168a009. [DOI] [PubMed] [Google Scholar]
- 5.Dou D, Mandadapu SR, Alliston KR, Kim Y, Chang K-O, Groutas WC. Eur. J. Med. Chem. 2012;47:59–64. doi: 10.1016/j.ejmech.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pansare SV, Rai AN, Kate SN. Synlett. 1998:623–624. [Google Scholar]
- 7. McDonald RI, Stahl SS. Angew. Chem. Angew. Chem., Int. Ed. 2010;2010;12249:5661–5664. 5529–5532. doi: 10.1002/ange.200906342. Muñiz K, Streuff J, Hövelmann CH, Núñez A. Angew. Chem. 2007;119:7255–7258. doi: 10.1002/anie.200702160. Angew. Chem., Int. Ed. 2007;46:7125–7127. doi: 10.1002/anie.200702160. Muñiz K, Hövelmann CH, Campos-Gómez E, Barluenga J, González JM, Streuff J, Nieger M. Chem. Asian. J. 2008;3:776–788. doi: 10.1002/asia.200700373. Hamaguchi H, Kosaka S, Ohno H, Fujii N, Tanaka T. Chem. Eur. J. 2007;13:1692–1708. doi: 10.1002/chem.200601373. Zhao B, Yuan W, Du H, Shi Y. Org. Lett. 2007;9:4943–4945. doi: 10.1021/ol702061s. Wang B, Du H, Shi Y. Angew. Chem. 2008;120:8348–8351. Angew. Chem., Int. Ed. 2008;47:8224–8227. doi: 10.1002/anie.200803184. Cornwall RG, Zhao B, Shi Y. Org. Lett. 2013;15:796–799. doi: 10.1021/ol303469a. Zabawa TP, Kasi D, Chemler SR. J. Am. Chem. Soc. 2005;127:11250–11251. doi: 10.1021/ja053335v.
- 8.Fecourt F, Lopez G, Van Der Lee A, Martinez J, Dewynter G. Tetrahedron: Asymmetry. 2010;21:2361–2366. [Google Scholar]
- 9.For representative examples, see: Dou D, Tiew K-C, He G, Mandadapu SR, Aravapalli S, Alliston KR, Kim Y, Chang K-O, Groutas WC. Bioorg. Med. Chem. 2011;19:5975–5983. doi: 10.1016/j.bmc.2011.08.054. Kim SJ, Jung M-H, Yoo KH, Cho J-H, Oh C-H. Bioorg. Med. Chem. Lett. 2008;18:5815–5818. doi: 10.1016/j.bmcl.2008.09.034.
- 10.(a) Fritz JA, Nakhla JS, Wolfe JP. Org. Lett. 2006;8:2531–2534. doi: 10.1021/ol060707b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fritz JA, Wolfe JP. Tetrahedron. 2008;64:6838–6852. doi: 10.1016/j.tet.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For recent reviews, see: Wolfe JP. Top. Heterocycl. Chem. 2013;32:1–38. Schultz DM, Wolfe JP. Synthesis. 2012;44:351–361. doi: 10.1055/s-0031-1289668.
- 12. Surry DS, Buchwald SL. Chem. Sci. 2011;2:27–50. doi: 10.1039/C0SC00331J. Surry DS, Buchwald SL. Angew. Chem. 2008;120:6438–6461. doi: 10.1002/anie.200800497. Angew. Chem., Int. Ed. 2008;47:6338–6361. doi: 10.1002/anie.200800497.
- 13.Our prior studies have illustrated that Pd-catalyzed carboamination reactions between aryl bromides and alkenes bearing pendant nucleophiles such as anilines, carbamates, ureas, and hydroxylamines proceed via syn-addition pathways. See: Ney JE, Wolfe JP. Angew. Chem. 2004;116:3689–3692. Angew. Chem., Int. Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060. Bertrand MB, Neukom JD, Wolfe JP. J. Org. Chem. 2008;73:8851–8860. doi: 10.1021/jo801631v. Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J. Org. Chem. 2009;74:2533–2540. doi: 10.1021/jo8027399. Hopkins BA, Wolfe JP. Angew. Chem. 2012;124:10024–10028. Angew. Chem. Int. Ed. 2012;51:9886–9890. doi: 10.1002/anie.201205233.
- 14.Palladium-catalyzed N-arylation reactions of sulfamides are believed to proceed through intermediates similar to 5. See: Alcaraz L, Bennion C, Morris J, Meghani P, Thom SM. Org. Lett. 2004;6:2705–2708. doi: 10.1021/ol049091l.
- 15.Stahl has illustrated that aminopalladation reactions of relatively electron-poor nucleophiles are reversible. See: White PB, Stahl SS. J. Am. Chem. Soc. 2011;133:18594–18597. doi: 10.1021/ja208560h.
- 16.(a) Rauf W, Brown JM. Chem. Comm. 2013;49:8430–8440. doi: 10.1039/c3cc44842h. [DOI] [PubMed] [Google Scholar]; (b) Dodds DL, Boele MDK, van Strijdonck GPF, de Vries JG, van Leeuwen PWNM, Kamer PCJ. Eur. J. Inorg. Chem. 2012:1660–1671. [Google Scholar]
- 17.In nonpolar solvents LnPd(Ar)(OTf) complexes exist as tight ion pairs, and these complexes are fully dissociated into solvent-separated ions in polar solvents. See: Jutand A, Mosleh A. Organometallics. 1995;14:1810–1817.
- 18.Carrow BP, Hartwig JF. J. Am. Chem. Soc. 2010;132:79–81. doi: 10.1021/ja909306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For studies on the mechanism of syn-migratory insertion of alkenes into Pd–N bonds, see: Neukom JD, Perch NS, Wolfe JP. J. Am. Chem. Soc. 2010;132:6276–6277. doi: 10.1021/ja9102259. Hanley PS, Marković D, Hartwig JF. J. Am. Chem. Soc. 2010;132:6302–6303. doi: 10.1021/ja102172m. Neukom JD, Perch NS, Wolfe JP. Organometallics. 2011;30:1269–1277. Hanley PS, Hartwig JF. J. Am. Chem. Soc. 2011;133:15661–15673. doi: 10.1021/ja205722f. White PB, Stahl SS. J. Am. Chem. Soc. 2011;133:18594–18597. doi: 10.1021/ja208560h.
- 20.Michael has previously illustrated that Pd-catalyzed arene C–H functionalization/alkene carboamination reactions of N-pentenyl amides proceed via anti-aminopalladation pathways. These transformations involve a Pd(II)/Pd(IV) catalytic cycle as opposed to the Pd(0)/Pd(II) cycle for the sulfamides described herein. See: Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J. Am. Chem. Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w.
- 21.Semmelhack MF, Bodurow C. J. Am. Chem. Soc. 1984;106:1496–1498. [Google Scholar]
- 22.PhCF3 is more polar than toluene and dioxane as measured by dielectric constant (toluene: 2.38; dioxane: 2.21; PhCF3: 9.4) or EN values (toluene: 0.099, dioxane: 0.164, PhCF3: 0.241). See: Reichardt C. Chem. Rev. 1994;94:2319–2358. Ogawa A, Tsuchii K. “α,α,α-Trifluorotoluene”. The Electronic Encyclopedia of Reagents for Organic Synthesis. http://mrw.interscience.wiley.com/eros/
- 23.Although there is an intramolecular component to the reactions described herein, the aryl/alkenyl halide and urea substrate are separate components coupled in an intermolecular process.
- 24.We have previously demonstrated that ligand effects can influence alkene syn- vs. anti-heteropalladation pathways in intramolecular Pd-catalyzed carboalkoxylation and carboamination reactions See: See: Nakhla JS, Kampf JW, Wolfe JP. J. Am. Chem. Soc. 2006;128:2893–2901. doi: 10.1021/ja057489m.
- 25.Stahl has shown that ligands, bases, and oxidants influence syn- vs. anti-aminopalladation pathways in Wacker-type oxidative cyclizations of aminoalkenes. See: Liu G, Stahl SS. J. Am. Chem. Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u. Weinstein AB, Stahl SS. Angew. Chem. 2012;124:11673–11677. Angew. Chem. Int. Ed. 2012;51:11505–11509. doi: 10.1002/anie.201206702. Ye X, White PB, Stahl SS. J. Org. Chem. 2013;78:2083–2090. doi: 10.1021/jo302266t. Martinez C, Wu Y, Weinstein AB, Stahl SS, Liu G, Muniz K. J. Org. Chem. 2013;78:6309–6315. doi: 10.1021/jo400671q.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.