

Abstract
Lactic acidosis is a build-up of lactic acid in the blood and tissues, which can be due to several inborn errors of metabolism as well as nongenetic conditions. Deficiency of pyruvate dehydrogenase complex (PDHC) is the most common genetic disorder leading to lactic acidosis. Phosphorylation of specific serine residues of the E1α subunit of PDHC by pyruvate dehydrogenase kinase (PDK) inactivates the enzyme, whereas dephosphorylation restores PDHC activity. We found that phenylbutyrate enhances PDHC enzymatic activity in vitro and in vivo by increasing the proportion of unphosphorylated enzyme through inhibition of PDK. Phenylbutyrate given to C57B6/L wild-type mice results in a significant increase in PDHC enzyme activity and a reduction of phosphorylated E1α in brain, muscle, and liver compared to saline-treated mice. By means of recombinant enzymes, we showed that phenylbutyrate prevents phosphorylation of E1α through binding and inhibition of PDK, providing a molecular explanation for the effect of phenylbutyrate on PDHC activity. Phenylbutyrate increases PDHC activity in fibroblasts from PDHC-deficient patients harboring various molecular defects and corrects the morphological, locomotor, and biochemical abnormalities in the noam631 zebrafish model of PDHC deficiency. In mice, phenylbutyrate prevents systemic lactic acidosis induced by partial hepatectomy. Because phenylbutyrate is already approved for human use in other diseases, the findings of this study have the potential to be rapidly translated for treatment of patients with PDHC deficiency and other forms of primary and secondary lactic acidosis.
Introduction
Lactic acid is the product of anaerobic metabolism of glucose and is generated by reduction of pyruvate. Patients with inborn errors of metabolism, including mitochondrial oxidative phosphorylation defects and disorders of gluconeogenesis, as well as several nongenetic diseases, present with lactic acidosis. Pyruvate dehydrogenase complex (PDHC), an enzyme complex that catalyzes the irreversible conversion of pyruvate into acetyl-CoA (coenzyme A), plays a central role in linking glycolysis to the tricarboxylic acid (TCA) cycle and lipogenic pathways (1). The oxidative metabolism of pyruvate, the end product of glycolysis, proceeds through PDHC into the TCA cycle and the mitochondrial respiratory chain. A deficiency of any enzyme in these pathways results in inadequate removal of pyruvate and lactate from blood and tissues and causes lactic acidosis. Deficiency of the nuclear-encoded PDHC is one of the most common inborn errors of mitochondrial energy metabolism. Most patients show progressive neurological degeneration and either persistent or episodic elevation of lactate in blood, cerebrospinal fluid (CSF), or both. These patients present with a graded spectrum of severity ranging from the most severe Leigh syndrome with overwhelming lactic acidemia and death in the neonatal period to a milder form with carbohydrate induced ataxia (2-3). The severity of the disease is a function of the severity of lactic acidosis and degree of residual enzymatic activity (2, 4). Currently, there are no proven treatments for PDHC deficiency. Anecdotal studies have reported beneficial effects from the use of high-fat, ketogenic diets, which provide an alternative energy source to carbohydrate (5-7). Availability of PDHC substrates [pyruvate and adenosine diphosphate (ADP)] results in inhibition of pyruvate dehydrogenase kinase (PDK) (8) and PDHC activation. On the basis of this inhibitory role, pyruvate administration has been attempted in patients with PDHC deficiency (9-11). Dichloroacetate (DCA), a pyruvate analogue, also increases PDHC activity and is effective in reducing blood, CSF, and brain lactate, thus improving morbidity in patients with deficiency of PDHC, respiratory chain defects, or mitochondrial DNA (mtDNA) mutations (12). However, in a controlled trial in children with various forms of congenital lactic acidosis, chronic DCA administration did not result in improvements of neurologic problems and other clinical outcomes (13). Moreover, DCA has raised some concerns because it has been associated with hepatocellular and peripheral nerve toxicity, particularly in adults (14). Therefore, investigations of effective treatments for PDHC deficiency are highly needed.
PDHC consists of three different enzymes: (i) thiamine diphosphate (ThDP)-dependent pyruvate dehydrogenase (E1), a heterotetrameric (α2β2) protein that irreversibly oxidizes pyruvate to acetyl CoA; (ii) dihydrolipoamide acetyltransferase (E2), which forms the structural core of the complex; and (iii) FAD-containing dihydrolipoamide dehydrogenase (E3), which is integrated into the complex by an E3-binding protein (E3BP). In humans, the disease due to PDHC deficiency may be due to defects in E1, E2, or E3 components of the complex or to a deficiency of E3BP or PDHC phosphatase (2-3).
PDHC is one of the highly conserved mitochondrial α-ketoacid dehydrogenase complexes, which include the branched chain α-ketoacid dehydrogenase (BCKDC), the α-ketoglutarate dehydrogenase complex, and PDHC (1). Phosphorylation of PDHC by PDK was discovered in the late 1960s and is among the first examples of enzyme regulation dependent upon reversible phosphorylation (15). Phosphorylation of E1α occurs at three serine residues (Ser264-α, site 1; Ser271-α, site 2, and Ser203-α, site 3). Phosphorylation at any of these sites inactivates PDHC (16). Four isoforms of PDK (PDK1, PDK2, PDK3, and PDK4) are known to phosphorylate the E1α subunit of PDHC, thereby reducing the activity of the complex (17-19). Conversely, dephosphorylation of these residues by pyruvate dehydrogenase phosphatases (PDP1 and PDP2) restores PDHC activity (19).
We have recently discovered that phenylbutyrate is an effective treatment for patients with maple syrup urine disease (MSUD), which is caused by mutations in one of the components of BCKDC (20). We found that phenylbutyrate significantly reduces the plasma concentrations of branched chain amino acids and their corresponding branched chain ketoacids in patients with MSUD and inhibits the kinase of BCKDC (BDK) (20). Similarly to PDHC, BCKDC is tightly regulated by reversible phosphorylation of the E1 subunit. Moreover, mitochondrial protein kinases, including PDKs and BDK, are members of the same family of protein kinases (21). On the basis of these observations, we hypothesized that phenylbutyrate would enhance PDHC enzyme activity by increasing the proportion of unphosphorylated enzyme and improve the phenotype of PDHC deficiency.
Results
Phenylbutyrate increases unphosphorylated PDH-E1α and PDHC enzyme activity in human fibroblasts
To investigate the efficacy of phenylbutyrate in altering PDHC phosphorylation and activity, we treated wild-type human fibroblasts with different concentrations of phenylbutyrate (0.25, 0.5, and 1 mM) for up to 24 hours. By Western blot analysis with an antibody specific for phosphorylated E1α subunit of PDHC, we observed a significant (p<0.05) reduction in the amount of phosphorylated E1α compared to the untreated cells, with all three concentrations of phenylbutyrate tested for 24 hours (Fig. 1A and B). No changes were observed in the amount of phosphorylated E1α after 2 and 6 hours of incubation with phenylbutyrate (supplementary Fig. 1). The results were confirmed on a second independent fibroblast cell line (supplementary Fig. 2). We next determined PDHC enzyme activity in fibroblasts incubated with phenylbutyrate. PDHC activity was found to be increased 1.4 and 2.2 fold over baseline untreated levels in two independent cell lines (p<0.05) (Fig. 1C). Because of reports that phenylbutyrate can function as a histone deacetylase (HDAC) inhibitor (22), we studied phenylbutyrate-treated fibroblasts and measured the RNA levels of PDHC subunits, testis-specific PDHA2 (23), PDPs, and PDKs to determine whether increased transcription of PDHC or its regulatory enzymes contributes to elevated enzyme activity. We found that concentrations of mRNA transcribed from the genes PDHA1, PDHA2, pyruvate dehydrogenase E1β (PDHB), dihydrolipoamide S-acetyltransferase (DLAT), dihydrolipoamide dehydrogenase (DLD), PDP1, PDP2, component X of the PDHC (PDHX), PDK1, PDK2, PDK3, and PDK4 were unchanged in fibroblasts treated with 1 mM phenylbutyrate compared to untreated cells (supplementary Fig. 3). These results indicate that the effect of phenylbutyrate on enzyme activity and inhibition of E1α phosphorylation is not mediated by increased RNA expression of genes encoding subunits of the complex or its regulatory enzymes.
Fig. 1. PDHC phosphorylation and activity after drug treatment of human wild-type (WT) fibroblasts.
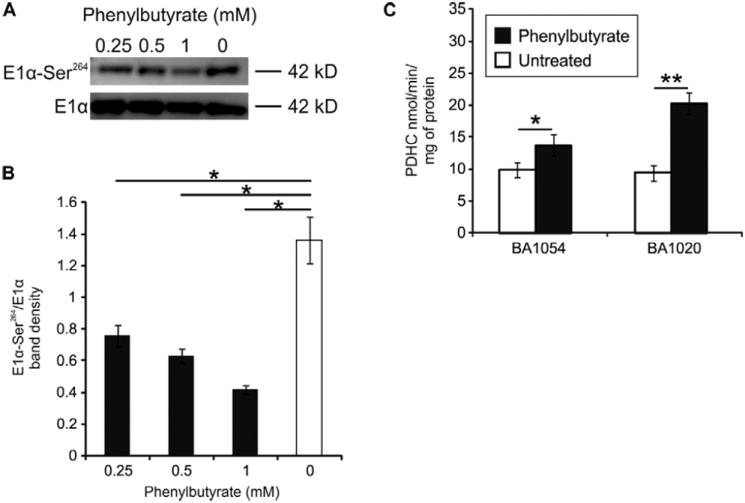
A. Western blotting of BA1054 fibroblasts treated with 0.25, 0.5, or 1 mM of phenylbutyrate for 24 hours, or untreated. The images are representative of two independent experiments. B. The average band intensities of phosphorylated E1 normalized for total E1 from two independent experiments. C. Two independent fibroblast cell lines (BA1054 and BA1020) were treated with phenylbutyrate at 1 mM for 24 hours before measurement of PDH enzyme activity. *: p<0.05; **: p<0.01.
Phenylbutyrate increases unphosphorylated PDH-E1α and PDHC enzyme activity in mouse brain, muscle, and liver
To investigate the effect of phenylbutyrate in vivo, C57B6 wild-type mice (at least n=5 per group) were given either saline or phenylbutyrate (250 or 500 mg/kg/day, divided into three administrations) orally by gavage, and after 3 days of treatment they were sacrificed for analyses. Both phenylbutyrate and phenylacetate (a metabolite of phenylbutyrate) were undetectable by tandem mass spectrometry on serum samples collected at the time of sacrifice (1.5 hours after the last drug administration) in mice treated with 250 mg/kg/day. Phenylacetate and trace amounts of phenylbutyrate were, however, detected in the sera of mice treated with 500 mg/kg/day of phenylbutyrate (supplementary table 1). Western blot analysis of brain and muscle mitochondrial extracts showed that both doses of phenylbutyrate resulted in a significant (p<0.05) reduction of phosphorylated E1α subunit of PDHC (PDH-E1α-Ser203, -Ser264, -Ser271) compared to saline-treated mice (Fig. 2 and supplementary Fig. 4). Reduction of phosphorylated E1α subunit of PDHC (PDH-E1α-Ser203 and Ser271) was also detected in the livers of treated animals compared to saline-treated mice at the dose of 250 mg/kg/day, but PDH-E1α-Ser264 amount was reduced in the liver only at the higher dose of 500 mg/kg/day (Fig. 2 and supplementary Fig. 4). Phenylbutyrate did not have an effect on total E1α subunit protein (PDH-E1α; Fig. 2 and supplementary Fig. 4). The less prominent effect on phosphorylated E1α in the liver compared to brain and muscle is likely dependent on the hepatic inactivation of phenylbutyrate. In humans, phenylbutyrate is rapidly converted to phenylacetate by hepatic β-oxidation and is undetectable in the serum by 6 hours after oral administration (24-25). Taken together, these results show that phenylbutyrate increases the proportion of active and unphosphorylated enzyme but does not affect total E1 levels in vivo. The increase in the active form of PDHC was detected in brain and muscle, the tissues most affected by PDHC deficiency disease.
Fig. 2. PDHC phosphorylation status in mouse tissues.
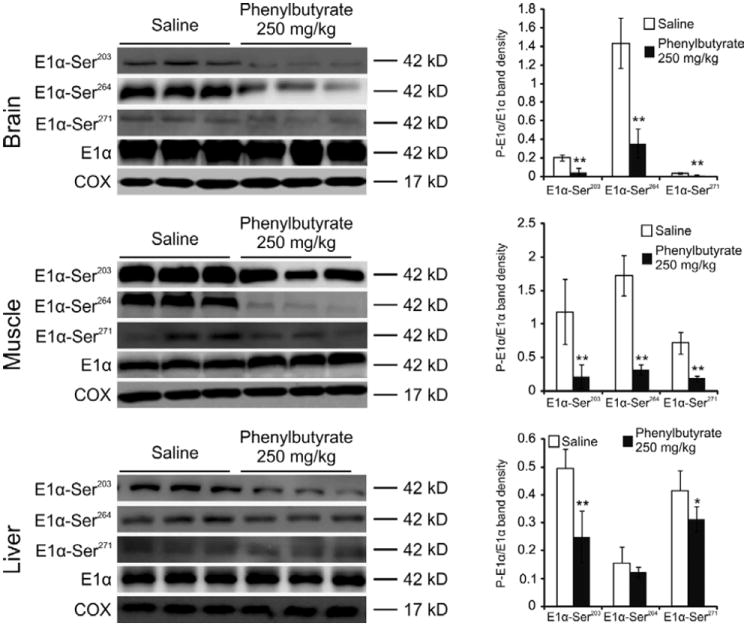
Western blot analysis of brain, muscle, and liver mitochondrial extract was performed with antibodies against the phosphorylated form of E1α (P-E1α). Each lane corresponds to brain, muscle, or liver mitochondrial extracts from an independent, representative mouse. Total E1α protein and cytochrome c oxidase (COX) are shown as controls. The graphs present densitometric quantification of n=5 mice from each treatment group. Means ± SDs are shown. *p<0.05; **p<0.01.
Compared to saline controls (n=16), PDHC enzyme activity in brain mitochondrial extracts of mice treated with phenylbutyrate [250 mg/kg/day (n=10) and 500 mg/kg/day (n=8)] was found to be increased 1.5 fold (p<0.01) and 2.8 fold (p<0.01), respectively (Fig. 3).
Fig. 3. Analysis of PDHC activity in mouse brain.
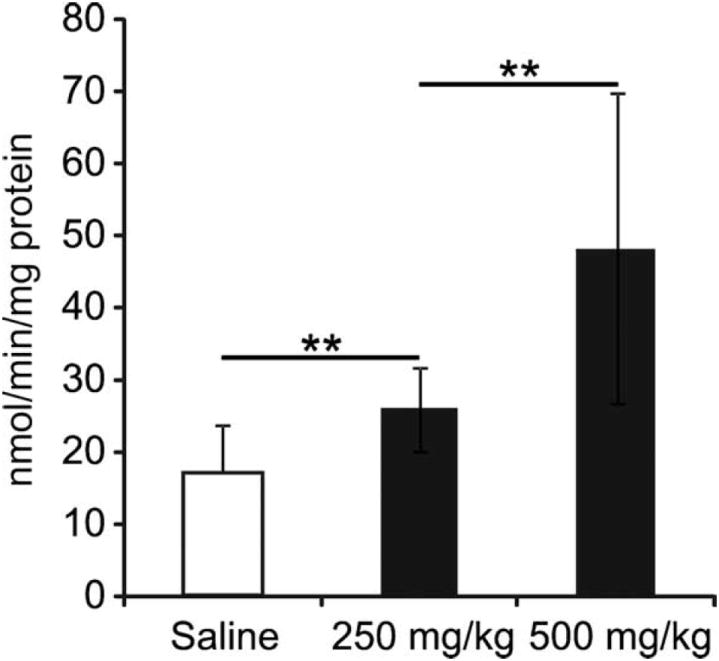
PDHC activity was measured in brain mitochondrial extracts of mice treated with phenylbutyrate [250 mg/kg/day (n=10) or 500 mg/kg/day (n=8)] and in saline controls (n=16). **p<0.01.
We also evaluated the amounts of Pdhc subunits, Pdp, and Pdk in brain RNA to determine whether increased transcription of PDHC, PDHA2 or its regulatory enzymes could contribute to elevated enzyme activity. The amounts of mRNA of Pdha1, Pdha2, Pdhb, Dld, Dlat, Pdk1, Pdk2, Pdk3, Pdk4, Pdp1, and Pdp2 were unchanged in brains of mice treated with phenylbutyrate (250 or 500 mg/kg/day) compared to untreated animals (supplementary Fig. 5). Similarly, the same mRNAs were unchanged in muscles of mice treated with 500 mg/kg/day compared to saline-treated controls (supplementary Fig. 6). These results suggest that the primary effect of phenylbutyrate is on enzyme activity and PDK-mediated phosphorylation of E1α and is not mediated by increased RNA expression of the genes encoding PDHC or its regulatory enzymes in both brain and muscle.
Phenylbutyrate inhibits E1α phosphorylation by PDK inhibition
Because of the previously shown effect of phenylbutyrate on BDK (20), we assayed for the ability of PDK to inactivate E1 in the presence and absence of phenylbutyrate. At saturating concentrations of E1α (4.2 μM), only slight PDK inhibition was observed (7%; 195 or 210 nmol/min per mg of enzyme with or without the inhibitor, respectively), but at non-saturating concentrations of E1α, phenylbutyrate prevented inactivation of E1 in the presence of PDK (Fig. 4A), by acting as a competitive inhibitor with a Ki (inhibition constant) of 0.33±0.08 mM (Fig. 4B). These results suggest that phenylbutyrate protects PDHC from inactivation by competing for binding of E1α to PDK. To confirm that phenylbutyrate binds to PDK, we carried out a docking simulation which revealed two putative binding sites of phenylbutyrate to PDK2: one site was found near the adenosine triphosphate (ATP) lid (Fig. 4C and 4D) with a binding energy of -4.0 kcal/mol and theoretical Ki of 1.2 mM and another site corresponded to the site of binding of Pfz3, a PDK inhibitor (Fig. 4E and 4F), with a binding energy of -5.06 kcal/mol and a theoretical Ki of 0.197 mM.
Fig. 4. PDK inhibition by phenylbutyrate.
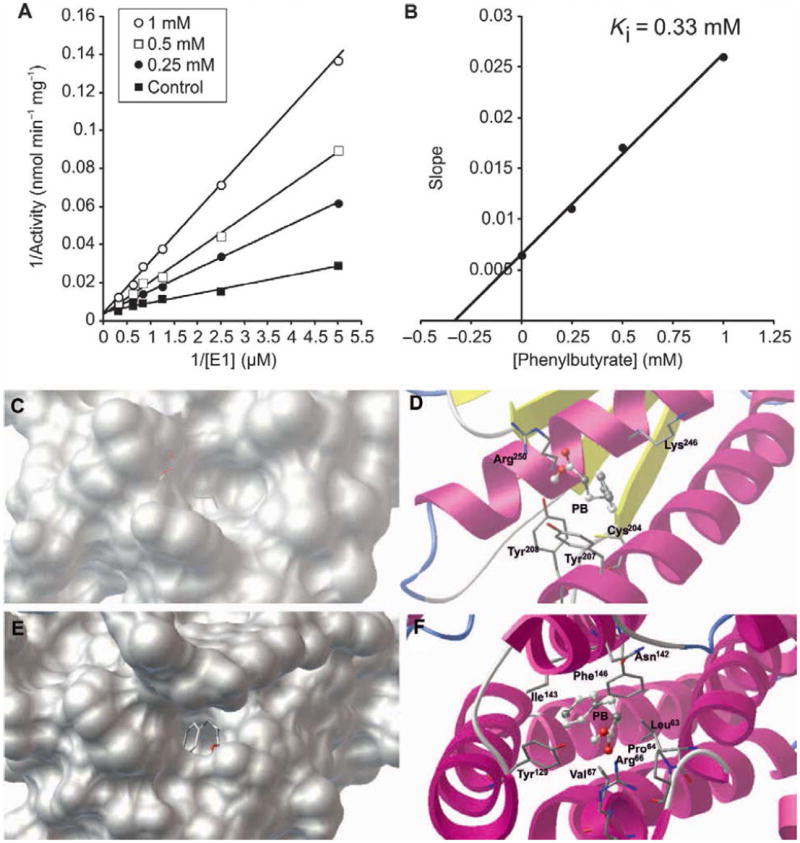
A. Lineweaver-Burk plot of WT recombinant E1α in the absence (■) or in the presence of 0.25 mM (●), 0.5 mM (□), and 1 mM (○) of phenylbutyrate. B. Replot of slopes measured from (A) against the concentration of phenylbutyrate. The intersection of the line with the x axis gives the value of Ki. C and E. Phenylbutyrate (PB, stick representation) in the two identified pockets on the protein surface of PDK2. D and F. Specific interactions of phenylbutyrate with amino acid residues (stick representation) at the binding sites. Van der Waals interaction spheres of the amino acid residues (stick representation) in contact with the inhibitor have been removed for clarity.
Phenylbutyrate increases PDHC enzyme activity in PDHC deficient fibroblasts
To evaluate phenylbutyrate’s potential for enhancing PDHC enzyme activity in PDHC deficiency, we investigated its effects in fibroblasts from patients with PDHC deficiency due to mutations in PDHA1 or PDHX genes (Table 1). Most mutations resulted in detection of mutated protein by Western blot analysis in fibroblasts (supplementary Fig. 7). PDHC-deficient cells were incubated with phenylbutyrate under optimal conditions (1 mM for 24 hours), and mitochondrial extracts were prepared to measure enzyme activity. Phenylbutyrate increased PDHC enzyme activity in 8 of 12 cell lines with PDHA1 defects and in 1 of 2 cell lines with PDHX mutations (Fig. 5 and Table 1). To correlate genotype with drug response, we mapped phenylbutyrate-responsive and non-responsive amino acid substitutions on the three-dimensional (3D) structure of the E1α protein (Fig. 6). Mutations resulting in phenylbutyrate-mediated increase of PDHC activity (p.V138M; p.P221T, p.N135S; p.W214C; p.R234G) were localized closer to the catalytic site of the enzyme than were mutations that were not associated with a drug-induced increase of PDHC activity (p.R349H; p.M181V). Because the sites of phosphorylation that regulate PDHC activity are located near the active site of the enzyme, our findings are consistent with the notion that phenylbutyrate acts to mitigate PDK-dependent E1α phosphorylation.
Table 1.
PDHC-deficient fibroblasts.
| Patient no | Gender | Enzyme activity (% of normal control) | Molecular defectˆ | ||
|---|---|---|---|---|---|
| Affected gene | Allele 1 | Allele 2 | |||
| 1 | Female | 33 | PDHA1 | p.V138M | wild-type |
| 2 | Male | 24 | PDHA1 | p.N135S | - |
| 3 | Male | 44 | PDHA1 | p.R349H | - |
| 4 | Female | 65 | PDHA1 | p.R275* | wild-type |
| 5 | Female | 46 | PDHA1 | c.904-928inv25 | wild-type |
| 6 | Male | 15 | PDHA1 | p.M181V | - |
| 7 | Female | 38 | PDHA1 | p.Y214C | wild-type |
| 8 | Female | 44 | PDHA1 | c.1063-1068del5 | wild-type |
| 9 | Male | 25 | PDHA1 | c.1159AAGTdup | - |
| 10 | Male | 25 | PDHA1 | c.1159AAGTdup | - |
| 11 | Male | 45 | PDHA1 | p.R234G | - |
| 12 | Male | 70 | PDHA1 | p.P221T | - |
| 13# | Female | 46 | N.D. | N.D. | N.D. |
| 14 | Male | 11 | PDHX | p.Q219* | c.1122_1125del |
| 15 | Male | 12 | PDHX | 10 kb del (ex 5-6) | 10 kb del (ex 5-6) |
Nucleotides are reported according to National Center for Biotechnology (NCBI) gene sequence (NM_000284.3 for PDHA1 and NM_003477.2 for PDHX). The amino acid positions of the mutations are reported considering the mature NCBI protein sequence (NP_000275.1 for PDHA1 and NP_003468.2 for PDHX) after signal peptide cleavage.
This patient was found to have PDH deficiency due to abnormal ubiquitination and proteasome degradation of E1β encoded by PDHB1 gene, as previously described (66). N.D.= not detected.
Fig. 5. PDHC activity in PDHC-deficient cells.
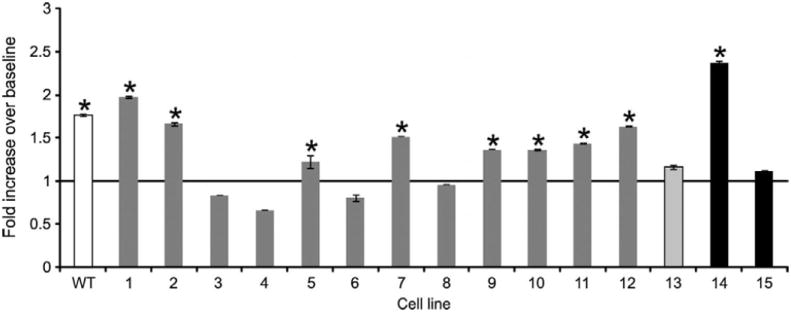
PDHC activity is shown for WT control (white bar) and PDHC-deficient cells incubated with phenylbutyrate. The molecular defects of the corresponding patient cell lines are shown in Table 1. Patients 1-12 (dark gray bars) have mutations in PDHA1, patient 13 (light gray bar) has PDHC deficiency due to abnormal ubiquitination and degradation of E1β (66), and patients 14 and 15 (black bars) have mutations in PDHX. *p<0.05.
Fig. 6. Position of responsive and nonresponsive PDHA1 mutations.
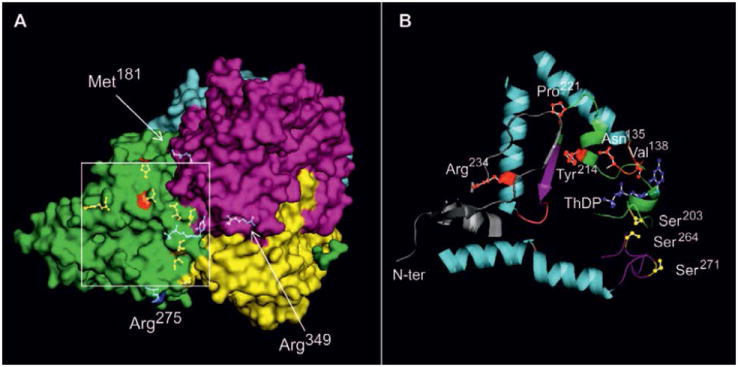
A. Picture of heterotetrameric human E1 [Protein Data Bank (PDB: 3EXE)] with the four chains highlighted (α, green; β, cyan; α′, magenta; β′, yellow), and the positions (arrows) of non-responsive mutations Met181-α and Arg349-α shown as ball and stick colored in blue. The positions of Met181-α and Arg349-α are part of the α subunit, and are underneath the β subunit. The square indicates the region with phenylbutyrate-responsive mutations, and is enlarged in (B). B. Orientation is the same as in (A). The following elements are shown: Asn135-α, Val138-α, Pro221-α, Tyr214-α, and Arg234-α (red), phosphorylation sites Ser203-α, Ser264-α, Ser271-α (yellow), and coenzyme ThDP (blue). The ribbon of the N-terminus (N-ter) of the E1α protein (residues 1 to 35) is depicted in gray; the structural elements containing Pro221-α, Tyr214-α, and Ser203-α are shown in cyan. The loop containing Ser264-α and Ser271-α is shown in magenta. The loop containing the residues Asn135-α and Val138-α is shown in green.
Phenylbutyrate corrects the morphological and biochemical phenotype of noam631 zebrafish
Targeted disruption of the E1α and E3 subunits in mice results in embryonic lethality (26-27); thus, these animals have limited utility for investigation of novel therapies. In contrast, the no optokinetic response a (noa) mutant zebrafish is available as a model of PDHC deficiency (28-29). The noam631 zebrafish shows expanded melanophores (28), absence of feeding behavior, lethargy, and premature death (30). These mutant fish also show elevations of lactate and pyruvate. The noam631 zebrafish mutant harbors a missense mutation at codon 644 in the DLAT gene, encoding the E2 subunit of PDHC (31).
Wild-type zebrafish have the ability to adjust melanin pigment levels in response to light or dark background. A marked phenotype of noa mutants is a dark pigmentation due to expanded melanophores (28), a trait common to zebrafish vision mutants (32), presumably due to poor detection of light. When noam631 mutants were treated with phenylbutyrate, a decrease in melanophore expansion was observed (Fig. 7A). Untreated mutants lay lethargically and floated on their side or back. In contrast, phenylbutyrate-treated noam631 larvae showed active locomotion behavior (Fig. 7B).
Fig. 7. Effect of phenylbutyrate on zebrafish noam631 mutants.
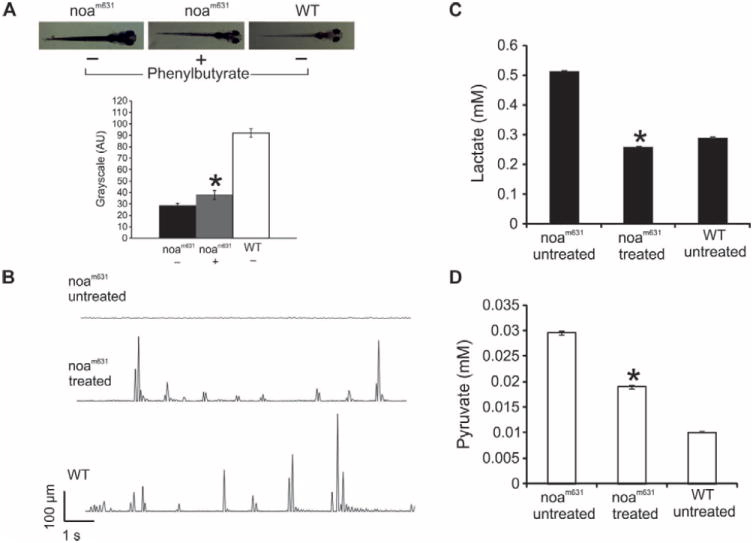
A. Pigmentation phenotype of untreated noam631, treated mutant noam631 and untreated wild-type larvae at 8 days after fertilization (n=14 per group). The bar graph shows quantification of zebrafish pigmentation, calculated with ImageJ software reporting mean gray value for a given area. The binary representation assumes that 0 is black and the maximum value (255 at 8 bit) is white. B. Movements of the larvae are plotted as the distance travelled by the larvae relative to time in 48-well plate, as previously described (31). C and D. Lactate and pyruvate concentrations in untreated or treated mutant noam631 larvae and in wild-type larvae (n=6 per group). *: p<0.05.
Elevated levels of lactate and pyruvate are biochemical hallmarks of PDHC deficiency (3). We determined the concentrations of these metabolites in homogenates of phenylbutyrate-treated mutants and controls. In the phenylbutyrate-treated fish, we observed a 2-fold reduction in lactate (Fig. 7C) and a 1.6-fold reduction in pyruvate compared to untreated mutants (Fig. 7D). These data suggest that phenylbutyrate reduces the detrimental lactic acidosis in the zebrafish PDH-deficient model.
Phenylbutyrate prevents lactic acidosis induced by partial hepatectomy
The liver is the principal organ responsible for the clearance of blood lactate (33-35). Partial hepatectomy has been experimentally used in animal models to induce systemic lactic acidosis (36). We performed partial hepatectomy in mice that were treated with phenylbutyrate (500 mg/kg/day, divided into three daily doses) from the day before the partial hepatectomy and up to 72 hours after the intervention and found that phenylbutyrate prevented blood accumulation of lactate in hepatectomized mice (Fig. 8).
Fig. 8. Effect of phenylbutyrate on blood lactate on hepatectomized mice.
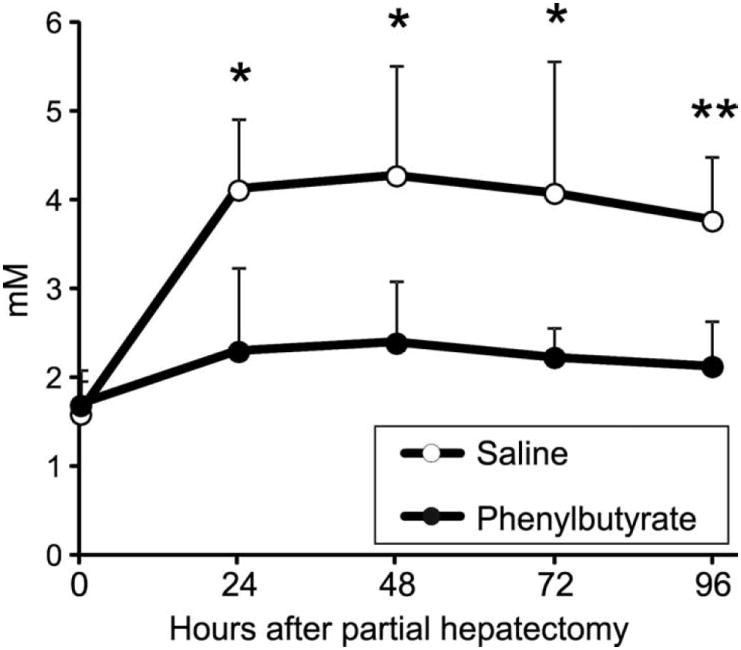
Blood lactate concentration at specified time intervals after partial hepatectomy in mice treated with saline or phenylbutyrate (n=5 per group). *: p<0.05; **: p<0.01.
Discussion
There is currently no proven treatment for patients with PDHC deficiency. Through a combination of studies in cells, mice, and in the zebrafish disease model, we demonstrate that phenylbutyrate may be a potential treatment for patients with PDHC deficiency. The results of this study show that phenylbutyrate enhances the activity of PDHC throughout the body, including the brain, which is one of the most affected tissues in PDHC deficiency. Given that phenylbutyrate is a drug already approved for use in humans, and its safety profile is well-known (37), these findings have the potential to be rapidly translated to clinical trials for patients with PDHC deficiency.
By means of recombinant enzymes, we showed that phenylbutyrate directly inhibits the PDK2, thus providing a molecular explanation for the effect of phenylbutyrate on PDHC activity. We also predicted that the binding of phenylbutyrate to PDK2 would occur near the ATP lid in proximity of the Tyr208-α, thereby reducing PDK2 activity (38). This site and another binding site found only on PDK2, have not been previously recognized as inhibitor binding sites of PDK. When a nucleotide binds to the ATP-binding pocket, the ordered ATP lid must be reorganized to accommodate the phosphate group of the nucleotide. The inherent flexibility of the ATP lid similarly allows binding of the inhibitor that interferes with the ATP-ADP cycle. In BDK, the ATP lid is largely disordered, even when nucleotide is bound (39), thus supporting the hypothesis that full ordering requires both nucleotide binding and specific protein-protein interactions. For BDK and PDKs, such interactions are likely provided by their corresponding E1 substrates and/or lipoyl-bearing domains of the E2 core (40-42). The conformational changes induced by phenylbutyrate binding to PDK could interfere with E1α binding, thus supporting the competitive inhibition.
PDHC enzyme activity was increased by phenylbutyrate in fibroblasts from some of the patients with impaired PDHC activity caused by various molecular defects. Phenylbutyrate-responsive mutations are all located near the active site of E1. Crystal structure of the heterotetrameric (α2β2) E1 protein shows that active sites 1 and 2 are located in the conserved phosphorylation loop A (Ph-loop A, residues from 259-α to 282-α), which forms the wall of the E1α active-site channel and anchors the ThDP to the active site (43). Site 3 is located in the short phosphorylation loop B segment (Ph-loop B; residues from 198-α to 205-α) and provides coordination with a Mg2+ ion chelated by the diphosphate group of ThDP. Phosphorylation of Ser264-α (site 1) prevents the cofactor ThDP-induced ordering of the Ph-loops A and B that carry the three phosphorylation sites. The disordering of these loops is caused by a steric hindrance between the phosphorylation group at site 1 and the nearby Ser266-α, which abolishes a hydrogen-binding network essential for maintaining the loop conformations. The unstructured loops impair binding of lipoyl domains of the PDHC core to E1α, negating the reductive acetylation step which disrupts substrate channeling by PDHC and leads to inactivation of the catalytic machinery (44). It is likely that phenylbutyrate-responsive PDHC-deficient cells harbor mutations that are located at the active site and quench PDHC activity by mimicking or potentiating the disordering effect of phosphorylation. Inhibition of phosphorylation by phenylbutyrate relieves the inhibitory effect of such mutations. For example, Val138-α from the E1α chain is located in the loop 136 to 140. The p.V138M mutation (patient 1) may result in steric hindrance with ThDP that hampers ThDP reorganization of loops around the active site, thus affecting PDHC activity. A similar effect can be envisaged for the p.N135S mutation (patient 2) that may push ThDP against Val138-α. Pro221-α is responsible for the turn determining the orientation of region 198 to 205, bearing the Ser203-α phosphorylation site. Therefore, the p.P221T mutation (patient 12) may affect the structural organization of the active site. Tyr214-α is inside the loop containing Ser203-α and binds by hydrophobic interactions the N-terminal region of E1α (residues 13 and 15) as well as the residues Gly222-α-Arg224-α close to Pro221-α. Therefore, the replacement of Tyr214-α with a Cys residue (patient 7) can account for the reduced enzyme activity. The p.R234G mutation (patient 11) is located inside the α-helix 230-245 and contributes to anchoring the helix to other portions of the N-terminus (Val25-α, Thr26-α, Tyr35-α); in this case it is difficult to rationalize the positive effect of phenylbutyrate on PDHC activity because this residue is more distant from the active site. The residues Met181-α and Arg349-α, which are mutated in phenylbutyrate non-responsive cells from patients 6 and 3, respectively, are distant from the active site and from the phosphorylation sites. It is conceivable that the inactivating effect of the mutations affecting these residues is due to a mechanism different from phosphorylation/dephosphorylation, and not susceptible to phenylbutyrate. Arg349-α forms a salt bridge with Glu281-β and a hydrogen bond with Pro267-α. These interactions can be changed by replacing arginine with histidine (patient 3), therefore producing structural changes in the oligomeric structure of the enzyme. Likewise, Met181-α forms hydrophobic interactions with a cluster of hydrophobic residues from chain β. Mutation to valine (patient 6) could drastically weaken this interaction that anchors α-helix 59-72 from chain β to chain α. Mutation p.E275*, which deletes the C-terminus of E1α has either the same effect as mutation 349 or more disruptive effects.
Most (84%) of patients with PDHC deficiency have mutations in the PDHA1 gene (3) and thus are potential candidates for phenylbutyrate therapy. It remains to be investigated whether patients carrying molecular defects in genes encoding the other components of the complex are also candidates for this treatment. Given the mode of action of phenylbutyrate we have elucidated, PDHC deficiency due to PDP1 defects is also highly likely to be responsive.
Lactic acidosis is a major manifestation of mitochondrial diseases caused by pathogenic mutant mtDNA. Secondary lactic acidosis has detrimental effects on mitochondrial disease phenotype, and inhibition of chronic lactic acidosis appears to provide clinical benefit. In a mouse model of mitochondrial disease resulting in a defect of mitochondrial respiration and lactic acidosis, inhibition of lactate production by DCA improved mitochondrial biogenesis and survival (45). Therefore, applications of phenylbutyrate could be extended to a broader range of mitochondrial cytopathies. Moreover, excessive lactic acid production occurs in many non-genetic conditions associated with blood vessel occlusion, asphyxia, liver disease, and other pathological conditions. For example, lactate, as a product of glycolysis of blood-derived glucose, accumulates in brain during ischemic stroke (46-47) and results in tissue acidosis (46-49) which is detrimental for cell survival (50-51). Furthermore, elevation of blood glucose concentration raises lactate and acidosis in ischemic brain areas and increases infarct size in animals (52-53) and humans (54). The reduction of blood lactate in hepatectomized mice (Fig. 8) suggests that phenylbutyrate may be effective for the treatment of lactic acidosis in a broad range of diseases, such as heart ischemia, septic shock, and stroke (55-56). Interestingly, there have been already reports of a protective effect of phenylbutyrate against cerebral ischemic insults (57-58), which could be explained, at least in part, by the effect of phenylbutyrate on PDHC phosphorylation reported in the present study.
In conclusion, we show that phenylbutyrate inhibits PDK-mediated phosphorylation, increases PDHC activity, improves the phenotype of the zebrafish model of PDHC deficiency, and reduces systemic lactic acidosis. These results may pave the way towards a clinical trial of phenylbutyrate for PDHC deficiency and possibly other mitochondrial diseases, and several non-genetic diseases with lactic acidosis.
Materials and Methods
Cell studies
The effect of phenylbutyrate on PDHC activity was investigated in synchronized wild-type human fibroblasts (BA1054 and BA1020) cultured in Dulbecco’s modified Eagle’s medium (DMEM) and 1% fetal bovine serum in the presence or absence of 0.25, 0.5, or 1 mM of phenylbutyrate (Amonaps, Swedish Orphan International Lab) for 2, 6, 24, or 48 hours. PDHC-deficient cells were treated with 1 mM phenylbutyrate for 24 hours. After drug treatment, cells were lysed in cold lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and 0.1% SDS] in the presence of protease inhibitor cocktail (Sigma). Total proteins quantified by the Bradford method were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membrane. Primary rabbit polyclonal anti-PDH-E1α (pSer264-α) (Calbiochem AP1062) and primary monoclonal anti-PDH-E1α (MitoSciences MSP02) were diluted 1:500 and 1:250, respectively. Horseradish Peroxidase (HRP)-conjugated secondary antibodies [ECL rabbit IgG, HRP-linked whole antibody from donkey (GE Healthcare) and ECL anti-mouse IgG, HRP-linked species-specific whole antibody from sheep (GE Healthcare NA931)] were diluted in 5% milk in tris-buffered saline Tween-20 (TBST). PDHC-deficient fibroblasts were analyzed for expression of proteins of the complex with primary monoclonal MitoProfile PDH Western Blot antibodies (anti-PDH-E1α, anti-PDH-E1β, anti-PDH-E2, and anti-E3BP) (MitoSciences MSP02-150). Protein bands were visualized with a chemiluminescence detection system (Pierce). Band intensities were quantified with Quantity One-4.6.7 Basic (1-D Analysis Software) (Biorad Laboratories).
Mouse studies
Mouse studies were approved by the Italian Ministry of Health. Sodium phenylbutyrate or saline was given orally to C57B6 mice (Charles River Laboratories) by gavage at the doses of 250 and 500 mg/kg/day divided into three administrations for three consecutive days (at least n=5 mice per group). After three days of treatment, the animals were sacrificed and various organs (brain, liver, and muscle) were collected after cardiac perfusion with phosphate buffered saline (PBS). For crude mitochondria purification, fresh tissue was placed in cold 20 mM Hepes, 5 mM EDTA Na2, 0.25 M sucrose, 1mM phenylmethylsulfonyl fluoride, 1mM phenanthroline (pH 7.4). Tissues were minced, washed in buffer, and the homogenate centrifuged for 15 min at 1000 g at 4°C to remove unbroken cells and nuclei. Supernatants were centrifuged at 10,000 g at 4°C for 15 min. For purification of crude mitochondria from muscles, fresh tissues were harvested and placed in 20 mM Hepes, 1 mM EDTA Na2, 100 mM KCl, 0.3% bovine serum albumin (BSA), 2 mM 2-mercaptoethanol (pH 7.4). Tissues were minced, washed in buffer, and the homogenate centrifuged for 10 min at 800 g at 4°C to remove unbroken cells and nuclei. Supernatants were combined and centrifuged at 10,000 g for 10 min at 4°C. The pellets were resuspended in MSEM buffer [220 mM mannitol, 70 mM sucrose, 1 mM EDTA Na2, 5 mM Mops (pH 7.4)] and centrifuged at 20,000 g for 10 min at 4°C. Mitochondrial extracts from brains, muscles, and livers were stored at -80°C.
Serum phenylbutyrate and phenylacetate concentrations were determined by tandem mass spectrometry according to a previously reported method (25).
Partial hepatectomy in 12 week-old C57BL/6 male mice was performed according to standard procedure (59). The animals were randomly separated into partial hepatectomy group with saline and partial hepatectomy group with phenylbutyrate (500 mg/kg/day, divided into three daily administrations). Saline or phenylbutyrate were administered by gavage from the day before the partial hepatectomy and continued up to 72 hours after the intervention (n=5 per group). Blood samples were collected at baseline, 24 hours, 48 hours, 72 hours, and 96 hours post-hepatectomy. Blood samples were collected from a small incision with a scalpel blade on the lateral tail vein in mice anesthetized with isofluorane; 10 μl of whole blood was immediately placed in 100 μl of ice cold 1.2 M HClO4 and centrifuged at 1,500 g for 10 min. Supernatants were collected and stored at -80°C until analysis. Lactate determinations were performed by lactate dehydrogenase assay (Sigma) adding lactate dehydrogenase and NAD+ (nicotinamide adenine dinucleotide) to the supernatants. After 15 min of incubation at 37°C, absorbance at 340 nm was measured to determine lactate concentrations. Each lactate measurement was performed in duplicate.
Enzyme assays
For PDH enzyme assays, cells were harvested with trypsin solution and centrifuged. After centrifugation, the pellets were washed twice with PBS. The cell pellet was resuspended in 2 ml Buffer A [Mops, 20 mM KOH (pH 7.4), 250 mM sucrose] and 0.2 mg of digitonin for every ml of Buffer A. The solution was mixed and kept on ice for 5 min and centrifuged at 5000 × g for 3 min. The supernatant was mixed with 3 ml Buffer B [Mops, KOH 20 mM (pH 7.4), sucrose 250 mM, EDTA Na4 1mM], kept on ice for 5 min, then centrifuged at 10,000 × g for 3 min. The supernatant was discarded and pellets were resuspended in 0.5 ml of 20 mM K-phosphate buffer (pH 7.4), then frozen in liquid nitrogen and thawed at 37°C three times. Pellets were stored at -80°C and used for enzyme assays. PDHC enzyme activity was measured in mitochondrial extracts as described previously (60). Briefly, the assay mixture contained 30 mM Hepes-KOH, 10 mM β-mercaptoethanol, 1 mM CoASH, 0.1 mM NAD [nicotinamide adenine dinucleotide (oxidized form)], 0.3 mM thiamine pyrophosphate, 0.02% Triton, 10 mM MgCl2, and 1mM CaCl2. The enzyme reaction was initiated with 20 mM of 14C-Na Pyruvate (Perkin Elmer) and incubated for 10 min at 37°C. Reaction was terminated with 250 μl of 1N HCl on ice and left at 37°C for 60 min. The 14CO2 released from the reaction was captured onto filter paper and transferred in scintillation liquid overnight; the amount of 14C was counted by a β-counter (Beckman LS6500). PDHC enzyme activity was expressed as nanomoles per minute per milligram of protein.
Western blots
Pellets of crude mitochondria were re-homogenized in cold lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, and 0.1% SDS] in the presence of protease inhibitors (Sigma). Total proteins quantified by the Bradford method were resolved by SDS-PAGE and transferred onto PVDF membrane.
Western blot analyses with 10 μg of mitochondria were performed with PhosphoDetect anti-PDH-E1α (pSer264-α; Calbiochem AP1062), PhosphoDetect anti-PDH-E1α (pSer203-α; Calbiochem AP1063), PhosphoDetect anti-PDH-E1α (pSer271; Calbiochem AP1064), anti-cytochrome c oxidase IV (COXIV; Cell Signaling Technology Inc.), anti-PDH-E1α (Abcam ab110416) and HRP-conjugated secondary antibodies (GE Healthcare) diluted in 5% milk in TBST and in 1% BSA in TBST. Bands were visualized with a chemiluminescence detection system (Pierce).
Real time PCR
Fibroblasts and mouse tissues were homogenized with 1 ml of TRIzol (Qiagen Inc.) with TissueLyser (Qiagen Inc.) for 2 min at 20 Hz, and total RNA from brains was extracted with RNeasy Lipid Tissue Mini Kit (Qiagen Inc.). Concentration and purity of total RNA were assessed spectrophotometrically at 260 and 280 nm. Agarose gel electrophoresis was performed to confirm presence of 18S ribosomal RNA (rRNA) and 28S rRNA. First-strand complementary DNA (cDNA) was synthesized from total RNA with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Sequences of primers for human genes PDHA1, PDHA2, PDHB, DLAT, DDLD, PDP1, PDP2, PDHX, PDK1, PDK2, PDK3, PDK4 and β-actin are shown as supplementary Table 2. Sequences of primers for mouse genes Pdha1, Pdha2, Pdhβ, Pdhx, Pdk1, Pdk2, Pdk3, Pdk4, Pdp1, Pdp2, Dlat, Dld, CoxIV, and β2-microglobulin are shown as supplementary Table 3. Quantitative real-time PCR was performed with SYBR Green in Light Cycler 480 multi-well plates (Roche).
The real time reaction mixture contained 2 μl of a 1:20 dilution of cDNA, 12.5 μl of Light Cycler 480 SYBR Green I Master Mix (Roche) and 10.5 μl of gene-specific primers, in a final reaction volume of 25 μl. The cycling conditions were as follows: pre-incubation at 95°C for 5 min, 45 cycles of amplification and quantification at 95°C for 10 s, 60°C for 10 s, and 72°C for 15 s, melting curve program at 95°C for 5 s, 65°C for 1 min and 97°C for continuous fluorescence measurement and finally a cooling step at 40°C for 20 min. Calculations were made by 2-ΔΔCT method, where CT is defined as the threshold cycle for target amplification. The data are presented as fold changes in target gene expression in cells or tissues normalized to the internal control gene (β-actin or β2-microglobulin) and relative to the untreated tissue or cells. For the untreated or saline-treated control samples, ΔΔCT equals zero, and because 20 equals one, the fold change in gene expression relative to the untreated control equals one, by definition. For treated samples, evaluation of 2-ΔΔCT indicates the fold change in gene expression relative to the untreated or saline-treated control.
Pyruvate dehydrogenase kinase activity assay
Recombinant human PDK2 (SRP5248, Sigma-Aldrich), PDHA1 (30 to 390) (SRP5238, Sigma-Aldrich), and pyruvate dehydrogenase E2 peptide (ab73826, Abcam), were used for these studies.
PDK activity was measured in duplicate as the initial rate of incorporation of [32P]-phosphate into E1 with 0.2 mM [γ-32P]ATP (150 to 500 cpm/pmol) at 30°C (61-63). The assay used 0.02 μg of PDK2 and measured incorporation after 45 s of reaction time. The assay was conducted with a buffer A which had a final pH of 7.4 and the following composition: 113 mM Hepes-Tris (pH 7.4), 60 mM KCl, 30 mM K-Hepes, 2 mM MgCl2, and 0.2 mM EDTA. The assay mixture in these studies also contained 2 mM dithiothreitol. Concentrated protein components (8, 4, 3, 2, 1, and 0.5 μg of E1 and 7.6, 3.8, 2.8, 1.9, 0.95, and 0.48 μg of E2) were preincubated together for 60 min at 4°C in the buffer in which they were prepared and then were added to reaction mixtures for 2 min at 30°C prior to initiation of PDK2 activity. Phenylbutyrate was added at the concentration of 0.25, 0.5, or 1mM and then the proteins were incubated in buffer A at 22°C for 60 s, and 0.2 mM of [γ-32P]ATP was added to a final volume of 50 μl. The reaction was terminated by adding 2 mM of ATP, and labeled E1 was separated from unbound ATP by loading 35 μl of the mixture onto G-25 Sephadex gel filtration columns (10×0.45 mm). The reaction mixtures eluted at the void volume (around 200 μl) were applied to dry paper (Whatman 3MM, 22 mm) previously soaked in 10% (w/v) trichloroacetic acid. The assay was completed by reading incorporated cpm, in vials containing liquid scintillation cocktail in a Beckman LS6500 multi-purpose scintillation counter. The data were analyzed with GraFit Data Analysis Software version 5.0.
Docking of phenylbutyrate on PDK
Docking of phenylbutyrate was performed with the Autodock 4.2 force field and the Lamarckian Genetic algorithm in the Autodock molecular modeling package (Abagyan, R., Orry, A., Raush, E. & Totrov, M. ICM Manual v.3.0, MolSoft LLC, 2011). The initial PDK model was generated in Autodock by building hydrogen atoms for the crystal structure of human PDK2 (PDB chain ID 2BU7) and adding Gasteiger charges. An initial conformation of the ligand phenylbutyrate was generated by Cartesian optimization of the ligand model in GROMOS87 force field (PRODRG at http://davapc1.bioch.dundee.ac.uk/prodrg/submit2.html). All side chains and the backbone of the protein were kept rigid as in the crystal structure. Docking was performed first by placing the ligand in a random position by centering the grid on the macromolecule and setting the grid with a 1 Å spacing on the entire protein; after the identification of the best binding sites, a further analysis was performed by starting with the ligand in the binding pockets and setting the grid with a 0.375 Å spacing. The global energy of the complex was calculated as a sum of van der Waals, electrostatic, hydrogen-bonding and torsion stress terms. In repeated runs, conformation clustering of 10 dockings within a root mean squared deviation of 0.5 Å to the overall best energy pose of the ligand allowed to identify at least five conformations with distinct binding energy above -5 kcal/mol. Control of docking procedure was obtained by docking the compound dichloracetate on the kinase structure (PDB chain ID 2Q8H) after ligand and potassium ion removal. Pictures were drawn with the tools available under the Autodock 4.2 suite.
Structural analysis of mutations
Structural analysis of PDHA mutations was performed with the three-dimensional structure of PDHC component (E1) of human PDHC available at the RCBS Protein Data Bank (PDB chain ID 3EXE). Coordinates were loaded in the Swiss PDB Viewer software (Swiss Institute of Bioinformatics), and analyses of both localization and mutation of these residues were performed in their structural context with the tools available under the Swiss PDB Viewer software. 3D structures of the proteins were drawn with PyMol software (DeLano Scientific LLC).
Zebrafish studies
The noa strain carrying the m631 allele was obtained from the Zebrafish International Resource Center (ZIRC), University of Oregon, and maintained as described (64). The noam631 mutants were treated from 5 to 8 days post-fertilization (dpf) with 0.25 mM phenylbutyrate. For genotyping, DNA was extracted from whole or partial larvae with 40 μl of extraction buffer [10 mM Tris-HCl (pH 8.2), 10 mM EDTA, 200 mM NaCl, 0.5% SDS, proteinase K (200 μg/ml)] and incubated at 50°C overnight. One hundred μl of 95% ethanol was added to the mix, after which it was placed on ice for 20 to 30 min, and centrifuged for 10 min. The supernatant was removed, 200 μl of 70% ethanol was added, and samples were centrifuged for 2 min. DNA was resuspended in 20 μl TE and used for genotyping with PCR conditions available at ZIRC (http://zebrafish.org/zirc/fish/pdf/pcr/m631.pdf).
To compare the size of melanophores between wild-type, noam631 phenylbutyrate-treated and noam631 untreated zebrafish, we analyzed the mean gray value of larvae in the region of the otic vesicle on photographs taken at 8 days after fertilization. Images were converted to 8-bit grayscale and then processed with the auto threshold function in ImageJ software [version 1.46, National Institutes of Health (NIH)] according to previous published method (65). After application of the auto threshold function, a selection was created by highlighting pixels, and the mean gray value for the selected area was reported. The images were taken with a Leica IC80 HD camera. Group averages were calculated for all areas where melanophores were measured, and standard deviations were calculated.
Locomotion behavior was videotaped and digitized, and the position of each larva in the 48-well plate was determined frame-by-frame with NIH image and the “MTrack2” plug-in that tracks particles and reports their coordinates at each time-point after the adjustment of the threshold. The distance travelled by the larvae in each of 300 frames and the time (10 s) were plotted on a graph after the conversion of pixels to μm in “R software”, an integrated suite of software facilities for data manipulation, calculation and graphical display. The genotype of each fish was confirmed by PCR after melanophore and behavioural assays.
Quantification of lactate and pyruvate in zebrafish
Lactate and pyruvate levels were quantified by standard spectrophotometric analysis (Sigma). The heads of three noam631 mutants and three wild-type larvae at 8 dpf were cut into two pieces for DNA extraction and lactate/pyruvate determination, respectively. One piece of each larva was tested for genotype and the other halves were pooled and homogenized in ice-cold 1.2 M HClO4. Proteins and insoluble materials were pelleted, exogenous lactate dehydrogenase (Sigma) and 5 mM NAD+ (for lactate measurements) or 83 μM NADH (reduced form of NAD+) (for pyruvate measurements) were added, and the change in absorbance at 340 nm was measured after 1 hour incubation at 24°C. Each measurement was performed in duplicate.
Statistical analyses
Data are expressed as mean values ± SD in the bar and line plots. Statistical significance was computed using the Student’s 2 tail test. A p-value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank A. Carissimo from Telethon Institute of Genetics and Medicine Bioinformatic Core for assistance with quantification of zebrafish melanophores and movement
Funding: This work was supported by grants of the United Mitochondrial Disease Foundation (to N.B.-P), the Italian Telethon Foundation (P37TELC to N.B.-P. and GPP10005 to M.Z.), the European Research Council (IEMTx to N.B.-P.), and FIRB-MERIT RBNE08LN4P grant (to G.M.).
Footnotes
Author contributions: R. F. performed all in vitro and in vivo experiments and all assays included in the study; G.M. performed docking studies, protein structure analysis, and supervised PDK inhibition assay; E.L. helped with PDHC enzyme activity; E. N. performed partial hepatectomy in mice and helped with the animal procedures; M.I.F. and P.S. helped with zebrafish studies; B. L. participated to the study design; P.W.S. and M.Z. provided PDHC deficient cell lines; N. B-P. led and supervised the project through all stages.
Competing interests: the authors declare that they have no competing interests.
References and notes
- 1.Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes. Faseb J. 1990;4:3224. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- 2.Robinson BH. Lactic acidemia and mitochondrial disease. Mol Genet Metab. 2006;89:3. doi: 10.1016/j.ymgme.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 3.Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;105:34. doi: 10.1016/j.ymgme.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson BH, Chun K, Mackay N, Otulakowski G, Petrova-Benedict R, Willard H. Isolated and combined deficiencies of the alpha-keto acid dehydrogenase complexes. Ann N Y Acad Sci. 1989;573:337. doi: 10.1111/j.1749-6632.1989.tb15009.x. [DOI] [PubMed] [Google Scholar]
- 5.Cederbaum SD, Blass JP, Minkoff N, Brown WJ, Cotton ME, Harris SH. Sensitivity to carbohydrate in a patient with familial intermittent lactic acidosis and pyruvate dehydrogenase deficiency. Pediatr Res. 1976;10:713. doi: 10.1203/00006450-197608000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Falk RE, Cederbaum SD, Blass JP, Gibson GE, Kark RA, Carrel RE. Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics. 1976;58:713. [PubMed] [Google Scholar]
- 7.Wexler ID, Hemalatha SG, McConnell J, Buist NR, Dahl HH, Berry SA, Cederbaum SD, Patel MS, Kerr DS. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology. 1997;49:1655. doi: 10.1212/wnl.49.6.1655. [DOI] [PubMed] [Google Scholar]
- 8.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329(Pt 1):191. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koga Y, Povalko N, Katayama K, Kakimoto N, Matsuishi T, Naito E, Tanaka M. Beneficial effect of pyruvate therapy on Leigh syndrome due to a novel mutation in PDH E1alpha gene. Brain Dev. 2012;34:87. doi: 10.1016/j.braindev.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka M, Nishigaki Y, Fuku N, Ibi T, Sahashi K, Koga Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases. Mitochondrion. 2007;7:399. doi: 10.1016/j.mito.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Komaki H, Nishigaki Y, Fuku N, Hosoya H, Murayama K, Ohtake A, Goto Y, Wakamoto H, Koga Y, Tanaka M. Pyruvate therapy for Leigh syndrome due to cytochrome c oxidase deficiency. Biochim Biophys Acta. 2010;1800:313. doi: 10.1016/j.bbagen.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121:e1223. doi: 10.1542/peds.2007-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 14.Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 15.Linn TC, Pettit FH, Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci U S A. 1969;62:234. doi: 10.1073/pnas.62.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korotchkina LG, Patel MS. Mutagenesis studies of the phosphorylation sites of recombinant human pyruvate dehydrogenase. Site-specific regulation. J Biol Chem. 1995;270:14297. doi: 10.1074/jbc.270.24.14297. [DOI] [PubMed] [Google Scholar]
- 17.Korotchkina LG, Patel MS. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J Biol Chem. 2001;276:37223. doi: 10.1074/jbc.M103069200. [DOI] [PubMed] [Google Scholar]
- 18.Teague WM, Pettit FH, Wu TL, Silberman SR, Reed LJ. Purification and properties of pyruvate dehydrogenase phosphatase from bovine heart and kidney. Biochemistry. 1982;21:5585. doi: 10.1021/bi00265a031. [DOI] [PubMed] [Google Scholar]
- 19.Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM. Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J Biol Chem. 1998;273:17680. doi: 10.1074/jbc.273.28.17680. [DOI] [PubMed] [Google Scholar]
- 20.Brunetti-Pierri N, Lanpher B, Erez A, Ananieva EA, Islam M, Marini JC, Sun Q, Yu C, Hegde M, Li J, Wynn RM, Chuang DT, Hutson S, Lee B. Phenylbutyrate therapy for maple syrup urine disease. Hum Mol Genet. 2011;20:631. doi: 10.1093/hmg/ddq507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roche TE, Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol Life Sci. 2007;64:830. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 23.Pinheiro A, Faustino I, Silva MJ, Silva J, Sa R, Sousa M, Barros A, de Almeida IT, Rivera I. Human testis-specific PDHA2 gene: methylation status of a CpG island in the open reading frame correlates with transcriptional activity. Mol Genet Metab. 2010;99:425. doi: 10.1016/j.ymgme.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Gilbert J, Baker SD, Bowling MK, Grochow L, Figg WD, Zabelina Y, Donehower RC, Carducci MA. A phase I dose escalation and bioavailability study of oral sodium phenylbutyrate in patients with refractory solid tumor malignancies. Clin Cancer Res. 2001;7:2292. [PubMed] [Google Scholar]
- 25.Yu X, Thompson MM, Shi D, Tuchman M. Quantification of benzoic, phenylacetic, and phenylbutyric acids from filter-paper blood spots by gas chromatography--mass spectrometry with stable isotope dilution. Clin Chem. 2001;47:351. [PubMed] [Google Scholar]
- 26.Johnson MT, Yang HS, Magnuson T, Patel MS. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A. 1997;94:14512. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson MT, Mahmood S, Hyatt SL, Yang HS, Soloway PD, Hanson RW, Patel MS. Inactivation of the murine pyruvate dehydrogenase (Pdha1) gene and its effect on early embryonic development. Mol Genet Metab. 2001;74:293. doi: 10.1006/mgme.2001.3249. [DOI] [PubMed] [Google Scholar]
- 28.Brockerhoff SE, Hurley JB, Janssen-Bienhold U, Neuhauss SC, Driever W, Dowling JE. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc Natl Acad Sci U S A. 1995;92:10545. doi: 10.1073/pnas.92.23.10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brockerhoff SE, Dowling JE, Hurley JB. Zebrafish retinal mutants. Vision Res. 1998;38:1335. doi: 10.1016/s0042-6989(97)00227-7. [DOI] [PubMed] [Google Scholar]
- 30.Taylor MR, Van Epps HA, Kennedy MJ, Saari JC, Hurley JB, Brockerhoff SE. Biochemical analysis of phototransduction and visual cycle in zebrafish larvae. Methods Enzymol. 2000;316:536. doi: 10.1016/s0076-6879(00)16748-5. [DOI] [PubMed] [Google Scholar]
- 31.Taylor MR, Hurley JB, Van Epps HA, Brockerhoff SE. A zebrafish model for pyruvate dehydrogenase deficiency: rescue of neurological dysfunction and embryonic lethality using a ketogenic diet. Proc Natl Acad Sci U S A. 2004;101:4584. doi: 10.1073/pnas.0307074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuhauss SC, Biehlmaier O, Seeliger MW, Das T, Kohler K, Harris WA, Baier H. Genetic disorders of vision revealed by a behavioral screen of 400 essential loci in zebrafish. J Neurosci. 1999;19:8603. doi: 10.1523/JNEUROSCI.19-19-08603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiolero R, Tappy L, Gillet M, Revelly JP, Roth H, Cayeux C, Schneiter P, Leverve X. Effect of major hepatectomy on glucose and lactate metabolism. Ann Surg. 1999;229:505. doi: 10.1097/00000658-199904000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchalter SE, Crain MR, Kreisberg R. Regulation of lactate metabolism in vivo. Diabetes Metab Rev. 1989;5:379. doi: 10.1002/dmr.5610050405. [DOI] [PubMed] [Google Scholar]
- 35.Kreisberg RA. Lactate homeostasis and lactic acidosis. Ann Intern Med. 1980;92:227. doi: 10.7326/0003-4819-92-2-227. [DOI] [PubMed] [Google Scholar]
- 36.Park R, Arieff AI. Treatment of lactic acidosis with dichloroacetate in dogs. J Clin Invest. 1982;70:853. doi: 10.1172/JCI110682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, McCarter RJ, Krischer JP, Batshaw ML. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hitosugi T, Fan J, Chung TW, Lythgoe K, Wang X, Xie J, Ge Q, Gu TL, Polakiewicz RD, Roesel JL, Chen GZ, Boggon TJ, Lonial S, Fu H, Khuri FR, Kang S, Chen J. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell. 2011;44:864. doi: 10.1016/j.molcel.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Machius M, Chuang JL, Wynn RM, Tomchick DR, Chuang DT. Structure of rat BCKD kinase: nucleotide-induced domain communication in a mitochondrial protein kinase. Proc Natl Acad Sci U S A. 2001;98:11218. doi: 10.1073/pnas.201220098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davie JR, Wynn RM, Meng M, Huang YS, Aalund G, Chuang DT, Lau KS. Expression and characterization of branched-chain alpha-ketoacid dehydrogenase kinase from the rat. Is it a histidine-protein kinase? J Biol Chem. 1995;270:19861. doi: 10.1074/jbc.270.34.19861. [DOI] [PubMed] [Google Scholar]
- 41.Wynn RM, Chuang JL, Cote CD, Chuang DT. Tetrameric assembly and conservation in the ATP-binding domain of rat branched-chain alpha-ketoacid dehydrogenase kinase. J Biol Chem. 2000;275:30512. doi: 10.1074/jbc.M005075200. [DOI] [PubMed] [Google Scholar]
- 42.Ono K, Radke GA, Roche TE, Rahmatullah M. Partial activation of the pyruvate dehydrogenase kinase by the lipoyl domain region of E2 and interchange of the kinase between lipoyl domain regions. J Biol Chem. 1993;268:26135. [PubMed] [Google Scholar]
- 43.Ciszak EM, Korotchkina LG, Dominiak PM, Sidhu S, Patel MS. Structural basis for flip-flop action of thiamin pyrophosphate-dependent enzymes revealed by human pyruvate dehydrogenase. J Biol Chem. 2003;278:21240. doi: 10.1074/jbc.M300339200. [DOI] [PubMed] [Google Scholar]
- 44.Kato M, Wynn RM, Chuang JL, Tso SC, Machius M, Li J, Chuang DT. Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: role of disordered phosphorylation loops. Structure. 2008;16:1849. doi: 10.1016/j.str.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ogasawara E, Nakada K, Hayashi J. Lactic acidemia in the pathogenesis of mice carrying mitochondrial DNA with a deletion. Hum Mol Genet. 2010;19:3179. doi: 10.1093/hmg/ddq228. [DOI] [PubMed] [Google Scholar]
- 46.Wagner KR, Kleinholz M, de Courten-Myers GM, Myers RE. Hyperglycemic versus normoglycemic stroke: topography of brain metabolites, intracellular pH, and infarct size. J Cereb Blood Flow Metab. 1992;12:213. doi: 10.1038/jcbfm.1992.31. [DOI] [PubMed] [Google Scholar]
- 47.Folbergrova J, Zhao Q, Katsura K, Siesjo BK. N-tert-butyl-alpha-phenylnitrone improves recovery of brain energy state in rats following transient focal ischemia. Proc Natl Acad Sci U S A. 1995;92:5057. doi: 10.1073/pnas.92.11.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nedergaard M, Kraig RP, Tanabe J, Pulsinelli WA. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am J Physiol. 1991;260:R581. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Widmer H, Abiko H, Faden AI, James TL, Weinstein PR. Effects of hyperglycemia on the time course of changes in energy metabolism and pH during global cerebral ischemia and reperfusion in rats: correlation of 1H and 31P NMR spectroscopy with fatty acid and excitatory amino acid levels. J Cereb Blood Flow Metab. 1992;12:456. doi: 10.1038/jcbfm.1992.63. [DOI] [PubMed] [Google Scholar]
- 50.Kraig RP, Petito CK, Plum F, Pulsinelli WA. Hydrogen ions kill brain at concentrations reached in ischemia. J Cereb Blood Flow Metab. 1987;7:379. doi: 10.1038/jcbfm.1987.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siesjo BK, Katsura K, Kristian T. Acidosis-related damage. Adv Neurol. 1996;71:209. [PubMed] [Google Scholar]
- 52.Li PA, Shamloo M, Katsura K, Smith ML, Siesjo BK. Critical values for plasma glucose in aggravating ischaemic brain damage: correlation to extracellular pH. Neurobiol Dis. 1995;2:97. doi: 10.1006/nbdi.1995.0010. [DOI] [PubMed] [Google Scholar]
- 53.Anderson RE, Tan WK, Martin HS, Meyer FB. Effects of glucose and PaO2 modulation on cortical intracellular acidosis, NADH redox state, and infarction in the ischemic penumbra. Stroke. 1999;30:160. doi: 10.1161/01.str.30.1.160. [DOI] [PubMed] [Google Scholar]
- 54.Parsons MW, Barber PA, Desmond PM, Baird TA, Darby DG, Byrnes G, Tress BM, Davis SM. Acute hyperglycemia adversely affects stroke outcome: a magnetic resonance imaging and spectroscopy study. Ann Neurol. 2002;52:20. doi: 10.1002/ana.10241. [DOI] [PubMed] [Google Scholar]
- 55.Preiser JC, Moulart D, Vincent JL. Dichloroacetate administration in the treatment of endotoxin shock. Circ Shock. 1990;30:221. [PubMed] [Google Scholar]
- 56.Ayala P, Montenegro J, Vivar R, Letelier A, Urroz PA, Copaja M, Pivet D, Humeres C, Troncoso R, Vicencio JM, Lavandero S, Diaz-Araya G. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol. 2012;92:97. doi: 10.1016/j.yexmp.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 57.Srinivasan K, Sharma SS. Sodium phenylbutyrate ameliorates focal cerebral ischemic/reperfusion injury associated with comorbid type 2 diabetes by reducing endoplasmic reticulum stress and DNA fragmentation. Behav Brain Res. 2011;225:110. doi: 10.1016/j.bbr.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 58.Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol. 2004;66:899. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- 59.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 60.DeVivo DC, Haymond MW, Obert KA, Nelson JS, Pagliara AS. Defective activation of the pyruvate dehydrogenase complex in subacute necrotizing encephalomyelopathy (Leigh disease) Ann Neurol. 1979;6:483. doi: 10.1002/ana.410060605. [DOI] [PubMed] [Google Scholar]
- 61.Baker JC, Yan X, Peng T, Kasten S, Roche TE. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J Biol Chem. 2000;275:15773. doi: 10.1074/jbc.M909488199. [DOI] [PubMed] [Google Scholar]
- 62.Rahmatullah M, Roche TE. Modification of bovine kidney pyruvate dehydrogenase kinase activity by CoA esters and their mechanism of action. J Biol Chem. 1985;260:10146. [PubMed] [Google Scholar]
- 63.Ravindran S, Radke GA, Guest JR, Roche TE. Lipoyl domain-based mechanism for the integrated feedback control of the pyruvate dehydrogenase complex by enhancement of pyruvate dehydrogenase kinase activity. J Biol Chem. 1996;271:653. doi: 10.1074/jbc.271.2.653. [DOI] [PubMed] [Google Scholar]
- 64.Westerfield M, editor. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) 5. University of Oregon Press; Eugene: 2007. [Google Scholar]
- 65.Van Otterloo E, Li W, Bonde G, Day KM, Hsu MY, Cornell RA. Differentiation of zebrafish melanophores depends on transcription factors AP2 alpha and AP2 epsilon. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han Z, Zhong L, Srivastava A, Stacpoole PW. Pyruvate dehydrogenase complex deficiency caused by ubiquitination and proteasome-mediated degradation of the E1 subunit. J Biol Chem. 2008;283:237. doi: 10.1074/jbc.M704748200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.