

Abstract
In this Viewpoint, we summarize and discuss the recent serendipitous discovery of an astrocyte Kir4.1 potassium channel dysfunction in two mouse models of Huntington’s disease (HD). Restoration of Kir4.1 channels within astrocytes in vivo attenuated neuronal dysfunction, some aspects of motor dysfunction and increased survival time in a HD mouse model. Overall, the data show that aspects of altered neuronal excitability associated with HD may be secondary to changes in astrocyte-mediated K+ homeostasis, thereby revealing a new striatal neural microcircuit mechanism in HD, and Kir4.1 channels and astrocytes as potential therapeutic targets for drug development.
Keywords: Huntington’s disease, astrocyte, striatum, potassium channel, microcircuit, basal ganglia, Kir4.1
One major goal of applied neuroscience is to understand the mechanisms that lead to neurological and psychiatric disorders. Although tremendous progress has been made in exploring neuronal mechanisms for brain function and disease, astrocytes, which represent about half the cells in the human brain, are less well explored. Astrocytes are known to serve various important roles within neural circuits, but the possibility that they may also contribute to, or perhaps even drive disease mechanisms, remains incompletely explored. Investigating astrocytes in brain diseases is increasingly warranted because broad analyses show that recent neuroscience drug development is correlated with failure rather than with success.1 This realization, together with the growing health burden of neurological and psychiatric diseases, calls for alternative approaches to past efforts that have focused almost exclusively on cell autonomous neuronal mechanisms. Simply stated, there is a need to (i) understand how astrocytes contribute to brain diseases and (ii) target astrocytes to determine if desirable effects can be produced within neural circuits of mouse models of brain diseases with predictive value for humans. Huntington’s disease (HD) presents itself as an exemplar disease to explore roles for astrocytes because it is associated with a range of psychiatric and neurological dysfunctions, but is caused by a single known molecular defect. Here, we present a summary of our efforts to explore astrocyte mechanisms in HD.
HD is characterized by progressive motor, cognitive, and psychiatric disturbances associated with neuronal dysfunction and atrophy of the striatum and other brain regions. There are currently no effective treatments for HD. HD is caused by an expanded chain of polyglutamines on the N-terminal region of the huntingtin protein (HTT) leading to intracellular accumulation and aggregation of mutant huntingtin. Several studies suggest that astrocytes may be involved in HD. Brains from HD patients and from mouse models of HD show accumulation of mHTT in striatal astrocytes, and expression of mHTT in astrocytes leads to age-dependent HD-like pathology and premature mortality.2 However, an open question centers on if/how astrocytes contribute to HD pathology or disease mechanisms. We therefore used two HD mouse models (the R6/2 and Q175 models) to assess astrocyte contributions to HD pathophysiology.3
We started by assessing astrogliosis in HD mouse models, because astrogliosis often accompanies brain disorders. Concomitant with the onset of symptoms, significantly more astrocytes displayed mHTT inclusions and exhibited significant reductions in important functional proteins. One of these proteins was Kir4.1. In contrast, there were no major phenotypic changes associated with astrocyte reactivity. These findings suggest that mHTT is associated with early disruption of the expression of important astrocyte functional proteins (e.g., Kir4.1), which alters astrocyte function without triggering astrogliosis. Congruent with other studies of mouse models and human HD, we found progressively increasing astrogliosis at later disease stages that exhibit overt neurodegeneration.
Kir4.1 channels are weakly inwardly rectifying potassium channels that critically regulate extracellular K+ levels and are expressed in the CNS primarily by astrocytes. Astrocyte electrophysiological properties have not been reported in mouse models of HD, although it is well established that striatal medium spiny neurons (MSNs) are depolarized by up to 12 mV in HD mouse models and display lower K+ currents. Broadly in accord with the past MSN data, we found that striatal astrocytes were depolarized by 5 mV. Our experiments show that the underlying cause can be attributed to a loss of Kir4.1 function and expression within mHTT expressing striatal astrocytes. Accordingly, the deficits in resting membrane potential were rescued by delivery of Kir4.1-GFP channels using adeno associated viruses (AAVs) and an astrocyte-specific promoter. Taken together, our studies provide evidence for a loss of striatal Kir4.1 responses and thus provide the first direct evidence for astrocyte dysfunction in conventional mouse models of HD.
Based on past studies, the loss of Kir4.1 currents in striatal astrocytes predicts reduced spatial K+ buffering, which in the simplest interpretation would lead to higher ambient K+ levels in HD mouse models. We tested for this and found that the extracellular K+ concentration was doubled in R6/2 mice. This prompted us to explore the impact of increased K+ on the properties of MSNs in WT mice. To our surprise, we found that these changes reproduced the elevated excitability features of MSNs described in a variety of HD mouse models. These data, summarized in Figure 1, provide strong evidence that subtle increases in extracellular K+ secondary to astrocyte Kir4.1 loss can produce significant effects on the properties of MSNs and can phenocopy MSN properties reported in HD mouse models. To further explore the relationship between astrocyte Kir4.1 channels and HD-like phenotypes in R6/2 mice, we delivered Kir4.1-GFP channels to striatal astrocytes using AAVs and found that one motor symptom (stride length and width) was attenuated by this approach. We also found that MSN membrane properties were partly recovered by astrocyte expression of Kir4.1-GFP, strongly supporting the notion that some HD-like phenotypes derive from neuronal dysfunction that derives in part from astrocyte disturbances. It is highly unlikely that all motor phenotypes can be recovered by any single striatal-specific intervention, because R6/2 mice have multiple abnormalities that affect distinct aspects of cortical and basal ganglia function, as well as other neural systems. Also, mutant HTT has multiple effects within cells.
Figure 1.
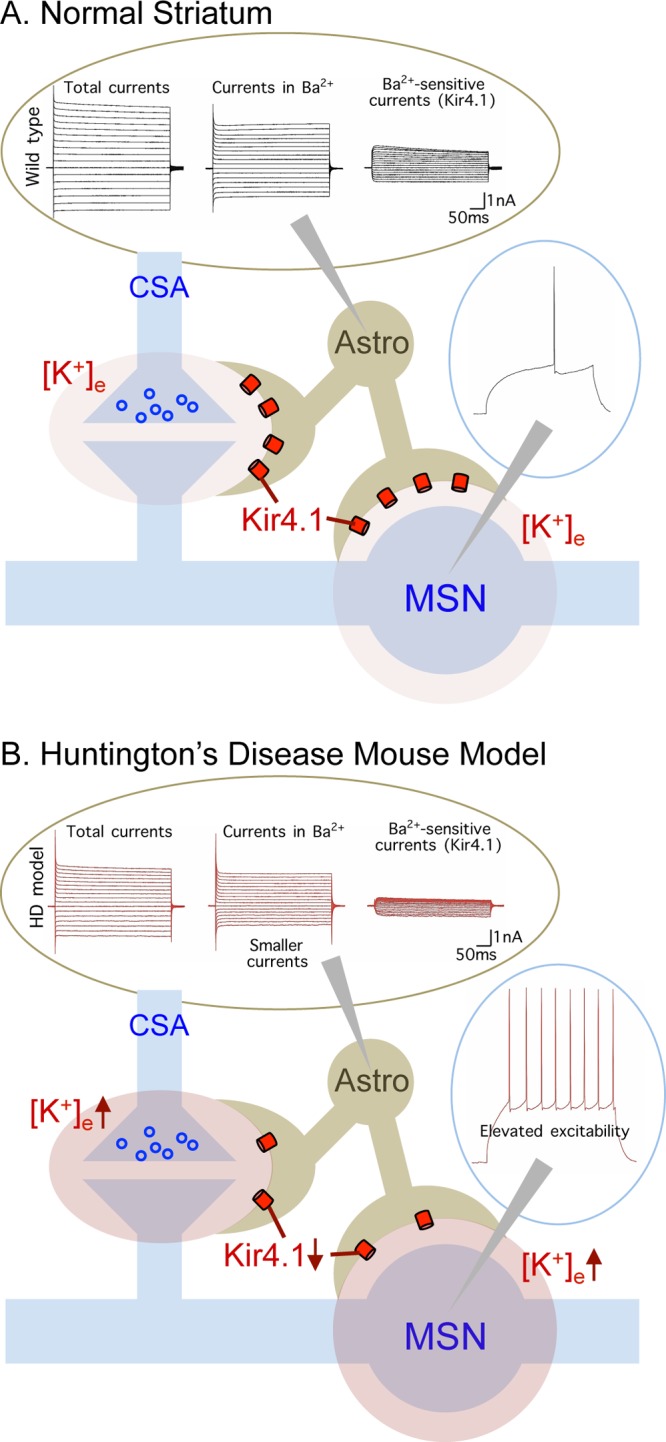
Schematic of changes in astrocyte physiology and their impact on neuronal physiology in a mouse model of HD. Abbreviations: CSA = cortical striatal afferents, MSN = medium spiny neuron, Astro = astrocyte. Ba2+ ions are used to block Kir4.1. Full details are found in Tong et al.,3 and a summary of the key findings is provided in Box 1.
To date, research efforts have been focused almost exclusively on identifying neuronal cell autonomous mechanisms to account for changes in MSN properties in HD models. Our findings, summarized in Box 1, provide evidence for the hypothesis that key aspects of altered MSN excitability in HD are secondary to disturbance of astrocyte maintenance of extracellular K+. In this view, a non cell autonomously mediated loss of K+ homeostasis has secondary consequences for MSNs such as through glutamate uptake via transporters, which use K+ as a counter transported ion. Chronic disturbances in K+ and glutamate homeostasis could contribute to excitotoxic neurodegeneration as well as to functional disturbances. By providing the first direct evidence that astrocyte physiology is altered in two conventional mouse models of HD before detectable evidence of reactive astrogliosis, our data reveal Kir4.1 channels and astrocytes as new potential therapeutic targets. Such approaches need not necessarily target Kir4.1 channels themselves, but may seek to utilize astrocyte biology to control Kir4.1 delivery to the membrane. The precise cellular functions of HTT are not known, and it is not clear how mHTT impacts on Kir4.1. Interestingly, however, transcriptome profiling of astrocyte responses to inflammatory mediators revealed HTT at the center of one of the top three most significantly altered gene networks.4 This intriguing finding warrants further dissection.
Box 1. Main Findings on Astrocyte Roles in Mouse Models of HD.
-
•
Striatal astrocytes from HD model mice contain mHTT nuclear inclusions when the mice are symptomatic. This happens before detectable evidence of astrogliosis.
-
•
Striatal astrocytes from R6/2 mice display depolarized membrane potentials and lower membrane conductances when the mice are symptomatic.
-
•
Striatal astrocyte electrophysiological defects are accounted for by lower function and expression of Kir4.1 channels, and are recovered by selective AAV2/5 mediated delivery of Kir4.1 to astrocytes.
-
•
Extracellular K+ buffering is reduced in the brains of HD mouse models as evidenced by significantly elevated extracellular K+ levels in the striatum of R6/2 mice.
-
•
Increasing K+ concentrations (like those measured in vivo in R6/2 mice) in brain slices from healthy WT mice reproduced some of the features of altered MSN excitability observed in HD mice.
-
•
Expression of Kir4.1-GFP in astrocytes rescued some neuronal deficits observed in R6/2 mice, attenuated a HD-like motor deficit and prolonged survival in R6/2 mice.
-
•
Astrocyte Kir.4.1 channels and other astrocyte molecular mechanisms may represent valuable targets for therapeutic development.
We propose that experiments similar to ours exploring astrocyte physiology are now warranted in the context of other neurodegenerative diseases such as spinocerebellar ataxias, Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Interestingly, astrocytes display a distinct repertoire of molecules as compared to neurons.5 Hence, it is opportune to determine if astrocyte specific molecular processes and pathways can be exploited by applied pharmacologists and medicinal chemists to produce desirable effects, either directly or indirectly, on neural circuits in brain disease. If so, astrocyte specific strategies may significantly expand the existing pharmacopeia to treat brain disorders.
Supported by the CHDI Foundation (B.S.K. and M.V.S.) and partly by the NIH (NS060677, MH104069 to B.S.K.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Ringel M.; Tollman P.; Hersch G.; Schulze U. (2013) Does size matter in R&D productivity? If not, what does?. Nat. Rev. Drug Discovery 12, 901–902. [DOI] [PubMed] [Google Scholar]
- Bradford J.; Shin J. Y.; Roberts M.; Wang C. E.; Li X. J.; Li S. (2009) Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. U.S.A. 106, 22480–22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X.; Ao Y.; Faas G. C.; Nwaobi S. E.; Xu J.; Haustein M. D.; Anderson M. A.; Mody I.; Olsen M. L.; Sofroniew M. V.; Khakh B. S. (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 17, 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby M. E.; Coppola G.; Ao Y.; Geschwind D. H.; Khakh B. S.; Sofroniew M. V. (2012) Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J. Neurosci. 32, 14489–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy J. D.; Emery B.; Kaushal A.; Foo L. C.; Zamanian J. L.; Christopherson K. S.; Xing Y.; Lubischer J. L.; Krieg P. A.; Krupenko S. A.; Thompson W. J.; Barres B. A. (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]