

Abstract
Alzheimer’s disease (AD) is the most common form of dementia and the sixth leading cause of death in the United States. Plaques composed of aggregated amyloid-beta protein (Aβ) accumulate between the neural cells in the brain and are associated with dementia and cellular death. Many strategies have been investigated to prevent Aβ self-assembly into disease-associated β-sheet amyloid aggregates; however, a promising therapeutic has not yet been identified. In this study, a peptoid-based mimic of the peptide KLVFF (residues 16–20 of Aβ) was tested for its ability to modulate Aβ aggregation. Peptoid JPT1 includes chiral, aromatic side chains to induce formation of a stable helical secondary structure that allows for greater interaction between the aromatic side chains and the cross β-sheet of Aβ. JPT1 was found to modulate Aβ40 aggregation, specifically decreasing lag time to β-sheet aggregate formation as well as the total number of fibrillar, β-sheet structured aggregates formed. These results suggest that peptoids may be able to limit the formation of Aβ aggregates that are associated with AD.
Keywords: Alzheimer’s disease, peptoid, amyloid-beta, KLVFF, inhibitor, aggregates
Alzheimer’s disease (AD) is the most prevalent form of dementia worldwide and is a leading cause of death among the elderly population.1 AD is a neurodegenerative disorder that is classified by the buildup of aggregated amyloid-beta protein (Aβ) as plaques in the brain, which are associated with cellular death and dementia. Aβ is generated as a result of proteolytic cleavage of the amyloid precursor protein and is typically 40 or 42 amino acid residues long (Aβ40 and Aβ42, respectively).1 Though the specific mechanism that causes AD is not clear, the deposition of aggregated Aβ in the brain and the formation of tau tangles within neuronal cells are closely associated with disease progression.1 Aβ monomer typically has a random coil secondary structure, but in the case of AD it forms fibrils with a cross-β-sheet pattern that ultimately deposit as plaques around the neural cells.2 Amyloid plaques were originally thought to be directly involved in cellular death; however, recent studies suggest that smaller soluble oligomeric aggregates are the toxic species.3−5
While there is currently no cure for AD, many groups are working to develop small molecules and peptides that inhibit Aβ aggregation. One such group of peptides is based upon KLVFF (residues 16–20 of Aβ), which was first identified by Tjernberg and co-workers and serves as the key recognition element that allows for aggregation due to hydrophobic interactions.6,7 Variants of the peptide KLVFF bind to Aβ and inhibit aggregation in vitro.7−12 However, as with all peptides, KLVFF variants are rapidly degraded in vivo and have low bioavailability.13
In order to increase bioavailability, we are utilizing poly-N-substituted glycines (peptoids) to mimic the peptide KLVFF. Peptoids are a novel class of peptidomimetics that are resistant to protease degradation.14 Peptoids are similar in structure to peptides, but with the side chain appended to the amide group rather than the α-carbon.15 This change in location of the side chain group leads to a lack of backbone chirality and hydrogen bond donors, both of which are required for secondary structure formation in peptides.15−17 However, the inclusion of chiral side chains in the peptoid sequence leads to the formation of helical secondary structure.18−23 Peptoids that include chiral, aromatic side chains form polyproline type-I-like helices with as little as five monomers.20 Peptoid helices have a periodicity of three monomers per turn and exhibit a helical pitch of ∼6 Å.20 Because peptoid helices are stabilized by steric interactions rather than hydrogen bonds, they are stable under extreme conditions including 8 M urea at 70 °C.23 The bioavailability of peptoids is favorable compared to peptides due to the change in backbone structure, which results in proteolytic stability and improved promise as a therapeutic.14 Furthermore, peptoids can be synthesized using automated, solid-phase equipment via a two-step process.15 Because the side chain group is added through the addition of a free amine, there are a multitude of available chemistries that can be included.15,24
Peptoids have only recently gained momentum for use as therapeutics and detection agents in protein misfolding diseases. The peptoid ligand HQP09 specifically binds to expanded polyglutamine proteins to inhibit aggregation of Htt-N-53Q in vitro and shows promise for the treatment of Huntington’s disease (HD) by exerting Ca2+ stabilizing and neuroprotective effects in HD mice.25 The peptoid IAM1 and its dimer have been shown to selectively bind to both Aβ40 and Aβ42, as well as to inhibit Aβ aggregation.26 Peptoids have also been used to create an AD blood test that senses antibodies produced specifically in AD patients.27 Peptoids are ideal therapeutic candidates for protein misfolding diseases, such as AD, due to increased bioavailability compared to peptides14,24 and improved membrane permeability compared to small molecules.14,28,29 In the present study, a novel peptoid-based mimic of the peptide KLVFF is reported to modulate Aβ40 aggregation.
Results and Discussion
Peptoid Sequence, Rationale, and Characterization
The pentapeptide KLVFF is known to be the minimum sequence required to bind to Aβ40.6 The peptide KLVFF was later shown to stereospecifically bind to the homologous sequence in Aβ via hydrophobic and electrostatic interactions.7 As a result, a five residue peptide with a proline point substitution (LPFFD, iAβ5) was designed to inhibit Aβ aggregation by interfering with Aβ self-assembly.30,31 However, iAβ5 is susceptible to proteolytic degradation and has low bioavailability.13,32,33 In order to reduce proteolytic degradation and increase blood-brain permeability, the N-terminus of the peptide was acetylated and the C-terminus was modified with an amide group (iAβ5p).33 While this chemical modification reduced the extent of proteolytic degradation, the ability of the peptide to inhibit Aβ aggregation was also decreased.32
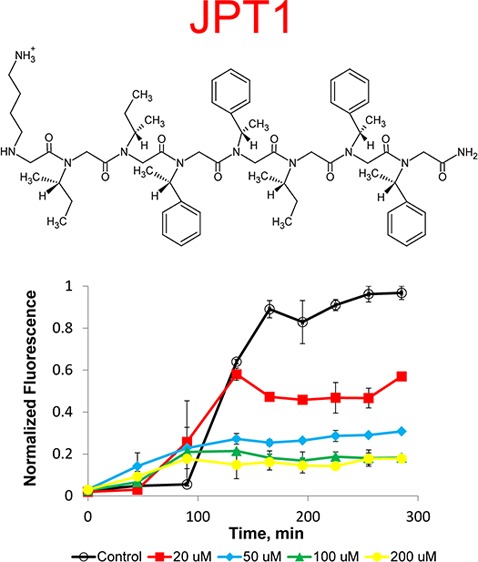
Here, we propose to use a protease-resistant, peptoid-based mimic of KLVFF to modulate the aggregation of Aβ. The peptoid JPT1 (Figure 1A) was designed to have a sequence similar to that of the peptide KLVFF. The sequence of JPT1 is KIIFFIFF, in terms of corresponding amino acids. This peptoid was designed to exhibit helical secondary structure such that the aromatic F groups align on two faces of the helix with ∼6 Å spacing between. The spacing between the F groups on the face of the helix corresponds to the spacing between backbone carbons i and i+2 in a β-sheet and may facilitate π–π stacking between the aromatic groups within the peptoid and Aβ aggregates. In order to induce helical secondary structure in the peptoid, the aliphatic L and V groups have been replaced with the chiral I-like group and the aromatic F groups have been replaced with a chiral F-like group. An additional helical turn, composed of chiral I-like and F-like groups, was added at the C-terminus to create a more robust helical structure and to allow for more potential interactions between the aromatic side chains and Aβ aggregates.
Figure 1.

Structure and characterization of peptoid JPT1. (A) Chemical structure of JPT1. (B) Circular dichroism spectra for JPT1. The spectra depict a polyproline type-I-like helical secondary structure.
The secondary structure of JPT1 was determined via circular dichroism (CD). The CD spectra for JPT1 (Figure 1B) in both methanol and water resemble that of a peptide α-helix. This corresponds to a polyproline type-I-like helix for peptoids.20 Specifically, the CD spectra exhibited a characteristic maximum near 190 nm and two minima near 205 and 220 nm.18,20−22 In methanol, the increased intensity of the peak near 220 nm as compared to the peak near 205 nm indicates that the helical secondary structure is more stable.19 In water, JPT1 continues to depict a polyproline type-I-like helical structure, though the helical structure is more loosely formed than in methanol.
Aggregation Studies
Thioflavin T (ThT) binds to β-sheet-rich structures to yield a shifted and enhanced fluorescent signal. As a result, it can be used to selectively detect β-sheet aggregates of Aβ. Since inhibitors often also work by binding to Aβ, they have the potential to disrupt the binding of ThT to aggregate structure, which can render false positive results. Thus, it is important to first evaluate whether peptoid JPT1 competes for binding sites with ThT. A competitive ThT binding assay was performed by adding 12.5 μM JPT1 to a solution of preformed 2.5 μM Aβ40 fibrils and incubating for 15 min. These concentrations are representative of those within diluted samples used for ThT measurements. ThT fluorescence results show no significant competitive binding for JPT1 (Figure 2), confirming that ThT detection of Aβ aggregates within the aggregation reaction will yield reliable results. Likewise, no ThT signal was observed when JPT1 was incubated alone, eliminating the possibility of aberrant interactions between the peptoid and ThT (Supporting Information).
Figure 2.

JPT1 does not compete competitively with ThT for binding of fibrillar Aβ. 2.5 μM Aβ40 fibrils were incubated for 15 min with 10 μM ThT in the presence and absence of 12.5 μM JPT1, and ThT fluorescence was measured following gentle mixing. Parameters are expressed as mean ± SEM, n = 3.
Aggregation assays were used to assess the ability of the peptoid JPT1 to modulate Aβ40 aggregation.34 Assays were performed with 20 μM purified Aβ40 monomer in the absence (control) and presence of JPT1 at concentrations of 20, 50, 100, and 200 μM. Aggregation was initiated via agitation on an orbital shaker. ThT fluorescence measurements (excitation at 440 ± 10 nm and emission at 490 ± 10 nm) were taken every 30–45 min to monitor aggregate formation. In the absence of peptoid, Aβ40 aggregation exhibited a lag time, the time at which ThT fluorescence begins to increase, followed by a rapid growth, and ending in a plateau, the point where the ThT fluorescence no longer increases and aggregates are in equilibrium with monomer (Figure 3). In the presence of peptoid JPT1, the lag time is shortened, indicating a more rapid formation of aggregates, and the plateau intensity is reduced, indicating the formation of fewer β-sheet aggregates at equilibrium (Figure 3). To quantify these effects, a lag extension is calculated as the ratio of the lag time to that of the control and a plateau reduction is calculated as the percentage decrease in the plateau fluorescence as compared to the control. A lag extension less than one was observed for all concentrations of JPT1, indicating that Aβ40 aggregates are forming more quickly in the presence of the peptoid (Figure 3). Further, a dose-dependence was observed up to 100 μM, where ThT fluorescence was increased at the first data point taken. A dose dependence was also observed for reduction of the equilibrium plateau, with inhibition increasing from 46.5 ± 4.9% to 81.2 ± 4.4% as the peptoid concentration is increased from 20 to 100 μM JPT1 (Figure 3). Higher concentrations exhibited similar inhibition to 100 μM JPT1, indicating that saturation of the inhibitory effect was achieved.
Figure 3.

ThT analysis shows that peptoid JPT1 modulates Aβ40 aggregation. JPT1 decreases lag time to Aβ40 aggregation and decreases the quantity of β-sheet aggregates formed. JPT1 was added to 20 μM Aβ40 monomer at concentrations of 0 (control), 20, 50, 100, and 200 μM, and the presence of fibrillar aggregates was detected by ThT fluorescence. Normalized fluorescence values are calculated as a percentage of the control plateau. Lag extension, or the fold change in lag time, and percent inhibition, or percentage decrease in the plateau, are shown below the graph. Parameters are expressed as mean ± SEM, n = 2. *p < 0.05; ***p < 0.0001.
Dot Blot Analysis
Dot blot analyses were performed in conjunction with ThT fluorescence measurements to confirm the presence of the fibrillar species of Aβ40, as detected by a conformation-specific antibody. Figure 4A shows the dot blot analysis for the time course of 20 μM Aβ40 aggregation in the absence (control) and presence of peptoid JPT1 at 100 μM. The dots were first probed with sequence-specific antibody 6E10, which recognizes residues 1–16 of Aβ, as a positive control to indicate that Aβ is present.35 The amino terminus has been implicated in playing a role in conformational changes during oligomerization.35,36 The ability of 6E10 to recognize Aβ in the presence of peptoid JPT1 is dramatically decreased compared to the control (Figure 4A). This difference may be due in part to a masking of the amino terminus due to conformation changes, a result that has been observed in other studies.37,38 Replicate dots were probed with the conformation-specific antibody OC, which binds fibrillar structures, to detect the presence of fibrillar Aβ aggregates.39 Dot blot analysis with the antibody OC confirms fibril formation of Aβ40 in the control beginning at 165 min, just slightly later than that observed via ThT fluorescence. In the presence of 100 μM JPT1, fibrils were observed at 45 min resulting in a lag extension of <0.31, also consistent with the decreased lag time observed using ThT fluorescence. Quantification of the dot blot assays showed that in the presence of JPT1 there is a plateau reduction of 63.7 ± 9.2% (Figure 4B). Although this is slightly lower than the inhibition detected by ThT, this difference is likely due to differing sensitivities between the assays. This data confirms that JPT1 is able to decrease lag time for Aβ aggregation as well as reduce the amount of fibrillar Aβ aggregates formed.
Figure 4.
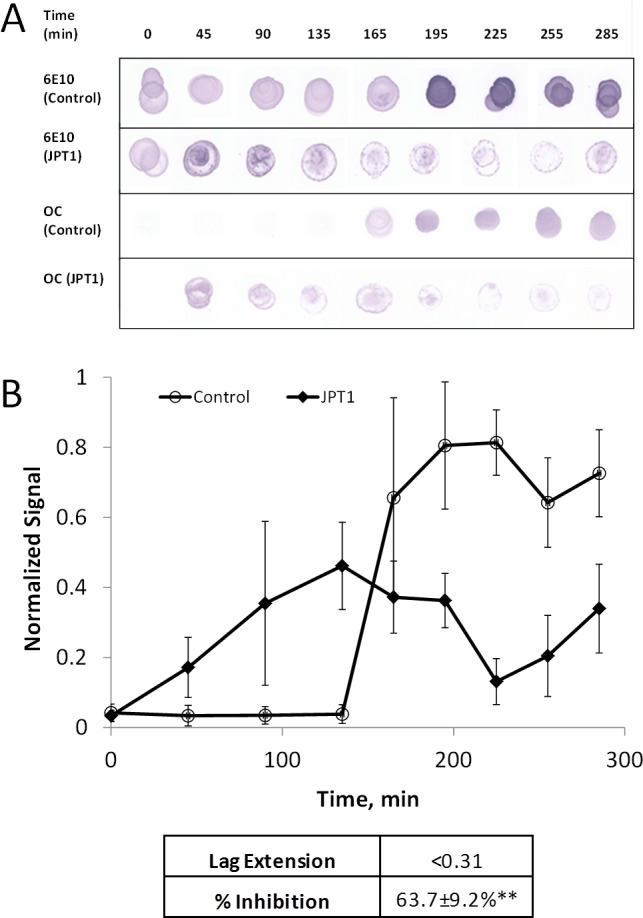
Dot blot analysis shows that peptoid JPT1 modulates Aβ40 aggregation. JPT1 decreases lag time to Aβ40 aggregation and decreases the quantity of aggregates formed. JPT1 was added to 20 μM Aβ40 monomer at a concentration of 0 (control) or 100 μM, and the presence of fibrillar structure was detected by dot blot with antibody OC. Detection with antibody 6E10 was performed as a parallel control for detection of total protein. (A) Dot blot results. (B) Quantification of antibody OC. Normalized values are calculated as percentage of the control plateau. Parameters are expressed as mean ± SEM, n = 3. **p < 0.005.
Conclusions
In this study, for the first time, a novel peptoid mimic of the peptide KLVFF was shown to modulate Aβ40 aggregation. Specifically, addition of the peptoid JPT1 to Aβ40 monomer results in a decreased time to aggregate formation and a reduction in the amount of fibrillar structured aggregates formed at equilibrium. Both effects were found to be dose dependent, with little change when the concentration of JPT1 was increased beyond 100 μM.
Experimental evidence suggests that the Aβ hydrophobic core plays a pivotal role in aggregation. Thus, the peptoid JPT1 was designed for optimal interaction with the hydrophobic regions of the Aβ cross β-sheet via incorporation of aromatic groups with ∼6 Å spacing on the peptoid helix to match that within the β-sheet backbone. The observed promotion of Aβ40 aggregation by JPT1 may occur along a different pathway that bypasses the formation of toxic soluble oligomers, as observed with “fibrillar seeds.”40 These “fibrillar seeds” propagated fibril formation and at the same time significantly inhibited total aggregate levels, similar to the peptoid JPT1.
Previous studies have shown that variants of the peptide KLVFF can act to inhibit or accelerate Aβ aggregation.10−12,26 However, little progress toward a viable therapeutic has been made with these peptides due to issues with proteolytic degradation in vivo.6,7,11 One variant of the peptide KLVFF (iAβ5p) was chemically modified to reduce the extent of proteolytic degradation. However, the ability of the peptide to inhibit Aβ aggregation was also decreased.32,33
Since peptoids can mimic peptide structure, and sometimes activity, but are not susceptible to proteolytic degradation, they are ideal candidates for treatment of neurological diseases that are associated with misfolded proteins.14,24,41−43 Luo et al. used a combinatorial library to identify peptoids that bind to Aβ40 and Aβ42.26 The peptoid IAM1 was shown to inhibit the aggregation of both Aβ40 and Aβ42 by roughly one-half at a molar ratio of 100:1 and 20:1 (peptoid to Aβ), respectively. The dimer of the peptoid, (IAM1)2, was shown to be more effective in inhibiting Aβ42 aggregation, with complete inhibition at a 10:1 molar ratio (peptoid to Aβ42). However, it did not inhibit Aβ40 aggregation effectively at any ratio tested.
Our studies with peptoid JPT1 have shown ∼84% inhibition of β-sheet containing aggregates during Aβ40 aggregation at a ratio of 5:1 (peptoid to Aβ40). Our results indicate that the peptoid JPT1 holds promise as a therapeutic agent for AD. In comparison of the efficacy of peptoid JPT1 to the peptide KLVFF, at a concentration equimolar with Aβ monomer, peptoid JPT1 inhibits fibrillar structured aggregates at ∼46%, while at this same stoichiometry peptide KLVFF either has no effect or slightly enhances the presence of β-sheet containing structures.44,45 Further studies will focus on modifications to the sequence that allow for greater than 80% reduction of Aβ aggregation at equimolar concentration or lower. In addition, the size and morphology of the aggregates formed in the presence of peptoid will be characterized and the effect that peptoid-induced changes in Aβ aggregation have upon the physiological activity of aggregates will be assessed.
Methods
Materials
(N)-(S)-sec-Butylamine (I-like group), (S)-(−)-α-methylbenzylamine (F-like group), piperidine, bovine serum albumin (BSA), and ThT were purchased from Sigma-Aldrich (St. Louis, MO). tert-Butyl N-(4-aminobutyl) carbamate (K-like group) was purchased from CNH Technologies, Inc. (Wobrun, MA). MBHA rink amide resin was purchased from NovaBiochem (Gibbstown, NJ). Aβ40 was purchased from Anaspec, Inc. (Fremont, CA). Primary antibody OC was purchased from EMD Millipore (Billerica, MA). Primary antibody 6E10, alkaline phosphatase-conjugated anti-mouse IgG, and alkaline phosphatase-conjugated anti-rabbit IgG were purchased from Thermo Scientific (Rockford, IL). All other reagents used during synthesis, purification, and sample preparation were purchased from VWR. All chemicals were used without further modification unless otherwise indicated.
Peptoid Synthesis and Purification
Peptoids were synthesized via a two-step process using an Applied Biosystems 433A automated peptide synthesizer (Carlsband, CA) that was refurbished from a 431A synthesizer.15 Rink amide resin was swelled with dimethylformamide (DMF), and the Fmoc protecting group was removed using 20% piperidine in DMF. The secondary amine was acylated by adding 1.2 M bromoacetic acid in DMF and diisopropylcarbodiimide at a ratio of 5.3:1 and vortexing for 60 min. Side chain amines were added to the resin via an SN2 reaction mechanism. The peptoid was removed from the resin by bathing it in a cleavage cocktail consisting of 95% trifluoroacetic acid (TFA), 2.5% triisoproylsilane, and 2.5% water for 5 min. The acid was removed using a Heidolph Laborota 4001 rotating evaporator (Elk Grove Village, IL), and the peptoid was diluted to a concentration of ∼3 mg mL–1 in a 35:65 solution of acetonitrile–water. Peptoids were purified using a Waters Delta 600 preparative high performance liquid chromatography unit (HPLC; Milford, MA) with a Duragel G C18 150 × 20 mm column (Peeke Scientific, Novato, CA). Gradients were run at ∼1% per minute with 5–95% solvent B in A (solvent A: water, 5% acetonitrile, 0.1% TFA; solvent B: acetonitrile, 5% water, 0.1% TFA). Peptoids were confirmed to be >98% pure via analytical HPLC (Waters 2695 Separations Module) equipped with a Duragel G C18 150 × 2.1 mm column (Peeke Scientific) using a linear gradient of 5 to 95% solvent D in C (solvent D: acetonitrile, 0.1% TFA; solvent C: water, 0.1% TFA) over 30 min. MALDI-TOF mass spectrometry was used to confirm that the purified peptoid mass matched the theoretical mass (Supporting Information). Purified peptoid solutions were dried to powder using a Labconco lyophilizer (Kansas City, MO) and stored at −20 °C.
Circular Dichroism
Circular dichroism (CD) spectrometry analysis was performed using a Jasco J-710 instrument (Easton, MD) at room temperature with a scanning speed of 20 nm min–1 and a path length of 0.1 cm. JPT1 was measured in pure methanol and water at a concentration of 120 μM. CD spectra is the cumulative average of 20 scans.
Preparation of Aβ Peptide Solution
Aβ40 (1 mg) was dissolved in 0.5 mL of 50 mM NaOH for 5 min prior to separation of aggregates from monomer via size exclusion chromatography on a Superdex 75 10/300 GL column (GE Heathcare, Pittsburgh, PA). Size exclusion chromatography was performed using an AKTA FPLC in a running buffer of 40 mM Tris-HCl (pH 8.0) and at a flow of 0.5 mL min–1 and 25 °C. The column was pretreated with 2 mg mL–1 BSA to optimize yield by reducing nonspecific interactions between Aβ and the column matrix. Fractions were eluted in 0.5 mL increments and immediately analyzed via an ND-1000 Nanodrop spectrophotometer to determine fraction concentration (277 nm, ε = 1450 M–1 cm–1).
Binding Competition Assay
Assays to assess binding competition between JPT1 and ThT were performed by incubating Aβ40 fibrils in the presence and absence of JPT1. Fibrils were formed prior to the assay by agitating purified 50 μM Aβ40 monomer in 40 mM Tris-HCl (pH 8.0) at 800 rpm on an orbital shaker at 25 °C for 20 h. Fibrils were then separated via centrifugation at 15 000 rpm for 15 min until a visible pellet was observed. The fibril pellet was removed from solution and resuspended in 40 mM Tris-HCl (pH 8.0) at a concentration of 50 μM. Competition assay solutions were prepared by gently mixing 0 (control) or 2.5 μM Aβ40 with 12.5 μM JPT1, 10 μM ThT, 1.25% (v/v) dimethyl sulfoxide (DMSO), and 40 mM Tris-HCl (pH 8.0). Solutions were allowed to incubate for 15 min to ensure binding. Fluorescence measurements were then taken on a Shimazdu RF-150 mini fluorometer (excitation at 440 nm and emission at 490 nm) with an average scan time of 4 s.
Aβ40 Monomer Aggregation Assay
Aggregation assays were performed with 20 μM purified Aβ40 in 40 mM Tris-HCl (pH 8.0) and 150 mM NaCl. Peptoid JPT1 was dissolved in DMSO and added at 0, 20, 50, 100, or 200 μM such that the final DMSO concentration was 1.25% (v/v). Assays were performed at 25 °C under agitation on an orbital shaker at 800 rpm. For ThT measurements, an aliquot was taken every 45 min and combined with ThT at a 5:1 molar ratio (ThT to Aβ monomer unit). Samples were well mixed for 8 s and then scanned with a Shimazdu RF-150 mini fluorometer (excitation at 440 ± 10 nm and emission at 490 ± 10 nm) with an average scan time of 4 s. ThT fluorescence with background (ThT alone) subtraction was normalized for the fluorescence detected at equilibrium for the control reaction and plotted against time. Lag time was noted for both the control and peptoid JPT1 as the time point before a significant increase in fluoresce was observed (5% of the plateau). Lag extension was calculated as the ratio of the lag time for peptoid JPT1 to that of the control. Equilibrium plateau was determined for both the control and peptoid JPT1 and percent inhibition was calculated for all peptoid JPT1 concentrations as the percentage decrease of the equilibrium relative to the control.
Dot Blot
In parallel with ThT fluorescence, aggregation assays were also assessed with dot blot analysis. Every 30 to 45 min, samples were spotted onto 0.1 μm nitrocellulose membrane (GE Healthcare, Pittsburgh, PA) and immediately blocked with 5% skim milk in Tris buffered saline containing 0.2% Tween20 (TBS-T) at 4 °C overnight. Membranes were washed three times with TBS-T and then incubated with either Aβ1–16 specific 6E10 antibody (1:2000 dilution) or Aβ fibril specific OC antibody (1:5000 dilution) for 1 h at room temperature with gentle agitation. Membranes were washed three times with TBS-T. Protein detection for bound 6E10 and OC antibodies was performed by binding alkaline phosphatase-conjugated anti-mouse IgG (1:2000 dilution) or alkaline phosphatase-conjugated anti-rabbit IgG (1:3000 dilution), respectively, for 1 h at room temperature with gentle agitation. Dot blots were developed using nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate in TBS-T/MgCl2 until precipitate from the reaction was no longer forming. The developing reaction was halted by a 10% acetic acid solution. Dot blots were immediately imaged using a Canon CanoScan 9000F (Tokyo, Japan) instrument. Controls performed by dotting with JPT1 alone confirmed an absence of signal for the peptoid (Supporting Information). Images were analyzed with ImageJ software using the gel lane analysis tool. Dot density was normalized to the density detected at equilibrium for the control and plotted against time. Lag extension and percent inhibition were assessed as described for ThT fluorescence.
Statistical Analysis
Data were analyzed using Prism 5 software (GraphPad Software Inc., La Jolla, CA). Differences in lag extension and percent inhibition were assessed using a one-way analysis of variance (ANOVA) or t test for ThT fluorescence and dot blot analysis, respectively. For ANOVA, Dunnett’s post-test was used to identify groups with means significantly different from control.
Acknowledgments
The authors would like to thank Dr. Srinivas Jayanthi and Dr. Rohana Liyanage for valuable training. The authors would like to thank Dr. Bob Beitle for use of and consultation regarding FPLC and fluorometer, Dr. Suresh Kumar for use of and consultation regarding FPLC and CD, and the Arkansas Statewide Mass Spectrometry Facility.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid-beta protein
- Aβ40
amyloid-beta 1–40
- Aβ42
amyloid-beta 1–42
- HD
Huntington’s disease
- CD
circular dichorism
- ThT
thioflavin T
- DMF
dimethylformamide
- TFA
trifluoroacetic acid
- HPLC
high pressure liquid chromatography
- DMSO
dimethyl sulfoxide
Supporting Information Available
Additional figures and methods as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ K.A.M.: Department of Molecular Physiology and Biophysics, Baylor College of Medicine, One Baylor Plaza BCM335 Houston, Texas 77030.
Author Contributions
J.P.T, T.L-R., K.A.M., M.A.M., and S.L.S. designed the study. J.P.T, T.L-R., K.A.M., L.R., and O.B. performed the experiments and analyses. J.P.T, T.L-R., M.A.M., and S.L.S. wrote the manuscript. M.A.M. and S.L.S. supervised the project.
This publication was made possible by Grant Number P30 GM103450-03 from the National Institute of General Medical Sciences, a component of the National Institutes of Health (NIH) (S.L.S.), Arkansas Bioscience Institute (S.L.S.), NIH Grant Number P20 RR-016461 from the National Center for Research Resources (M.A.M.), and NSF/EPSCoR Grant Number EPS-0447660 (M.A.M.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hardy J.; Selkoe D. J. (2002) The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Etienne M. A.; Edwin N. J.; Aucoin J. P.; Russo P. S.; McCarley R. L.; Hammer R. P. (2007) Beta-Amyloid Protein Aggregation. Methods Mol. Biol. 386, 203–225. [DOI] [PubMed] [Google Scholar]
- Caughey B.; Lansbury P. T. (2003) Protofibrils, Pores, Fibrils, and Neurodegeneration: Separating the Responsible Protein Aggregates from the Innocent Bystanders. Annu. Rev. Neurosci. 26, 267–298. [DOI] [PubMed] [Google Scholar]
- Glabe C. G. (2006) Common Mechanisms of Amyloid Oligomer Pathogenesis in Degenerative Disease. Neurobiol. Aging 27, 570–575. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. (2009) Amyloid Beta-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 284, 4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjernberg L. O.; Näslund J.; Lindqvist F.; Johansson J.; Karlström A. R.; Thyberg J.; Terenius L.; Nordstedt C. (1996) Arrest of Beta-Amyloid Fibril Formation by a Pentapeptide Ligand. J. Biol. Chem. 271, 8545–8548. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Lilliehöök C.; Callaway D. J.; Näslund J.; Hahne S.; Thyberg J.; Terenius L.; Nordstedt C. (1997) Controlling Amyloid Beta-Peptide Fibril Formation with Protease-Stable Ligands. J. Biol. Chem. 272, 12601–12605. [DOI] [PubMed] [Google Scholar]
- Esler W. P.; Stimson E. R.; Ghilardi J. R.; Lu Y. A.; Felix A. M.; Vinters H. V.; Mantyh P. W.; Lee J. P.; Maggio J. E. (1996) Point Substitution in the Central Hydrophobic Cluster of a Human Beta-Amyloid Congener Disrupts Peptide Folding and Abolishes Plaque Competence. Biochemistry 35, 13914–13921. [DOI] [PubMed] [Google Scholar]
- Lowe T. L.; Strzelec A.; Kiessling L. L.; Murphy R. M. (2001) Structure-Function Relationships for Inhibitors of Beta-Amyloid Toxicity Containing the Recognition Sequence KLVFF. Biochemistry 40, 7882–7889. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Nakamura K.; Akikusa S.; Okada T.; Kodaka M.; Konakahara T.; Okuno H. (2002) Inhibitors of Fibril Formation and Cytotoxicity of Beta-Amyloid Peptide Composed of KLVFF Recognition Element and Flexible Hydrophilic Disrupting Element. Biochem. Biophys. Res. Commun. 290, 121–124. [DOI] [PubMed] [Google Scholar]
- Moss M. A.; Nichols M. R.; Reed D. K. I. M.; Hoh J. A. N. H.; Rosenberry T. L. (2003) The Peptide KLVFF-K 6 Promotes β -Amyloid (1–40) Protofibril Growth by Association but Does Not Alter Protofibril Effects on Diphenyltetrazolium Bromide (MTT). Mol. Pharmacol. 64, 1160–1168. [DOI] [PubMed] [Google Scholar]
- Hamley I. W.; Castelletto V.; Moulton C. M.; Rodríguez-Pérez J.; Squires A. M.; Eralp T.; Held G.; Hicks M. R.; Rodger A. (2010) Alignment of a Model Amyloid Peptide Fragment in Bulk and at a Solid Surface. J. Phys. Chem. B 114, 8244–8254. [DOI] [PubMed] [Google Scholar]
- Aileen Funke S.; Willbold D. (2012) Peptides for Therapy and Diagnosis of Alzheimer’s Disease. Curr. Pharm. Des. 18, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S. M.; Simon R. J.; Ng S.; Zuckermann R. N.; Kerr J. M.; Moos W. H. (1995) Comparison of the Proteolytic Susceptibilities of Homologous l-Amino Acid, d-Amino Acid, and N-Substituted Glycine Peptide and Peptoid Oligomers. Drug Dev. Res. 35, 20–32. [Google Scholar]
- Zuckermann R. N.; Kerr J. M.; Kent B. H.; Moos W. H. (1992) Efficient Method for the Preparation of Peptoids [Oligo(N-Substituted Glycines)] by Submonomer Solid-Phase Synthesis. J. Am. Chem. Soc. 114, 10646–10647. [Google Scholar]
- Kirshenbaum K.; Zuckermann R. N.; Dill K. A. (1999) Designing Polymers That Mimic Biomolecules. Curr. Opin. Struct. Biol. 9, 530–535. [DOI] [PubMed] [Google Scholar]
- Barron A. E.; Zuckermann R. N. (1999) Bioinspired Polymeric Materials: In-between Proteins and Plastics. Curr. Opin. Chem. Biol. 3, 681–687. [DOI] [PubMed] [Google Scholar]
- Armand P.; Kirshenbaum K.; Falicov A.; Dunbrack R. L.; Dill K. A.; Zuckermann R. N.; Cohen F. E. (1997) Chiral N-Substituted Glycines Can Form Stable Helical Conformations. Folding Des. 2, 369–75. [DOI] [PubMed] [Google Scholar]
- Armand P.; Kirshenbaum K.; Goldsmith R. A.; Farr-Jones S.; Barron A. E.; Truong K. T. V; Dill K. A.; Mierke D. F.; Cohen F. E.; Zuckermann R. N.; Bradley E. K. (1998) NMR Determination of the Major Solution Conformation of a Peptoid Pentamer with Chiral Side Chains. Proc. Natl. Acad. Sci. U.S.A. 95, 4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirshenbaum K.; Barron A. E.; Goldsmith R. A.; Armand P.; Bradley E. K.; Truong K. T. V; Dill K. A.; Cohen F. E.; Zuckermann R. N. (1998) Sequence-Specific Polypeptoids: A Diverse Family of Heteropolymers with Stable Secondary Structure. Proc. Natl. Acad. Sci. U.S.A. 95, 4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. W.; Sanborn T. J.; Zuckermann R. N.; Barron A. E. (2001) Peptoid Oligomers with Alpha-Chiral, Aromatic Side Chains: Effects of Chain Length on Secondary Structure. J. Am. Chem. Soc. 123, 2958–2963. [DOI] [PubMed] [Google Scholar]
- Wu C. W.; Sanborn T. J.; Huang K.; Zuckermann R. N.; Barron A. E. (2001) Peptoid Oligomers with Alpha-Chiral, Aromatic Side Chains: Sequence Requirements for the Formation of Stable Peptoid Helices. J. Am. Chem. Soc. 123, 6778–6784. [DOI] [PubMed] [Google Scholar]
- Sanborn T. J.; Wu C. W.; Zuckermann R. N.; Barron A. E. (2002) Extreme Stability of Helices Formed by Water-Soluble Poly-N-Substituted Glycines (polypeptoids) with Alpha-Chiral Side Chains. Biopolymers 63, 12–20. [DOI] [PubMed] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K. (1992) Peptoids: A Modular Approach to Drug Discovery. Proc. Natl. Acad. Sci. U.S.A. 89, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Wu J.; Luo Y.; Liang X.; Supnet C.; Kim M. W.; Lotz P.; Yang G.; Muchowski P. J.; Kodadek T.; Bezprozvanny I. (2012) Expanded Polyglutamine-Binding Peptoid as a Novel Therapeutic Agent for Treatment of Huntington’s Disease. J. Biol. Chem. 18, 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Vali S.; Sun S.; Chen X.; Liang X.; Drozhzhina T.; Popugaeva E.; Bezprozvanny I. (2013) Aβ42-Binding Peptoids as Amyloid Aggregation Inhibitors and Detection Ligands. ACS Chem. Neurosci. 4, 952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C. M.; Yam A. Y.; Wang X.; Magdangal E.; Salisbury C.; Peretz D.; Zuckermann R. N.; Connolly M. D.; Hansson O.; Minthon L.; Zetterberg H.; Blennow K.; Fedynyshyn J. P.; Allauzen S. (2010) Aβ40 Oligomers Identified as a Potential Biomarker for the Diagnosis of Alzheimer’s Disease. PLoS One 5, e15725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckermann R. N.; Kodadek T. (2009) Peptoids as Potential Therapeutics. Curr. Opin. Mol. Ther. 11, 299–307. [PubMed] [Google Scholar]
- Cho S.; Choi J.; Kim A.; Lee Y.; Kwon Y.-U. (2010) Efficient Solid-Phase Synthesis of a Series of Cyclic and Linear Peptoid-Dexamethasone Conjugates for the Cell Permeability Studies. J. Comb. Chem. 12, 321–326. [DOI] [PubMed] [Google Scholar]
- Soto C.; Sigurdsson E. M.; Morelli L.; Kumar R. A.; Castaño E. M.; Frangione B. (1998) Beta-Sheet Breaker Peptides Inhibit Fibrillogenesis in a Rat Brain Model of Amyloidosis: Implications for Alzheimer’s Therapy. Nat. Med. 4, 822–826. [DOI] [PubMed] [Google Scholar]
- Sigurdsson E. M.; Permanne B.; Soto C.; Wisniewski T.; Frangione B. (2000) In Vivo Reversal of Amyloid-Beta Lesions in Rat Brain. J. Neuropathol. Exp. Neurol. 59, 11–17. [DOI] [PubMed] [Google Scholar]
- Adessi C.; Frossard M.-J.; Boissard C.; Fraga S.; Bieler S.; Ruckle T.; Vilbois F.; Robinson S. M.; Mutter M.; Banks W. A.; Soto C. (2003) Pharmacological Profiles of Peptide Drug Candidates for the Treatment of Alzheimer’s Disease. J. Biol. Chem. 278, 13905–13911. [DOI] [PubMed] [Google Scholar]
- Poduslo J.; Curran G.; Kumar A.; Frangione B.; Soto C. (1999) Beta-Sheet Breaker Peptie Inhibitor of Alzheimer’s Amyloidogenesis with Increased Blood-Brain Barrier Permeability and Resistance to Proteolytic Degradation in Plasma. J. Neurobiol. 39, 371–382. [PubMed] [Google Scholar]
- Hudson S. a; Ecroyd H.; Kee T. W.; Carver J. a. (2009) The Thioflavin T Fluorescence Assay for Amyloid Fibril Detection Can Be Biased by the Presence of Exogenous Compounds. FEBS J. 276, 5960–5972. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Overman M. J.; Cotman C. W. (1995) Amino-Terminal Deletions Enhance β-Amyloid Peptides in Vitro. J. Biol. Chem. 270, 23895–23898. [DOI] [PubMed] [Google Scholar]
- Rosenman D. J.; Connors C. R.; Chen W.; Wang C.; García A. E. (2013) Aβ Monomers Transiently Sample Oligomer and Fibril-like Configurations: Ensemble Characterization Using a Combined MD/NMR Approach. J. Mol. Biol. 425, 3338–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H. E.; Qi W.; Choi H. M.; Fernandez E. J.; Kwon I. (2011) A Safe, Blood-Brain Barrier Permeable Triphenylmethane Dye Inhibits Amyloid-B Neurotoxicity by Generating Nontoxic Aggregates. ACS Chem. Neurosci. 2, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. A.; Wong H. E.; Kwon I. (2013) Different Fates of Alzheimer’s Disease Amyloid-B Fibrils Remodeled by Biocompatible Small Molecules. Biomacromolecules 14, 264–274. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Sarsoza F.; Saing T.; Cotman C. W.; Necula M.; Margol L.; Wu J.; Breydo L.; Thompson J. L.; Rasool S.; Gurlo T.; Butler P.; Glabe C. G. (2007) Fibril Specific, Conformation Dependent Antibodies Recognize a Generic Epitope Common to Amyloid Fibrils and Fibrillar Oligomers That Is Absent in Prefibrillar Oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Liu Q.; Sun X.; Yu J.; Zhao D.; Yu Y.; Luo J.; Hu J.; Yu Z.; Zhao Y.; Li Y. (2013) Fibrillar Seeds Alleviate Amyloid-B Cytotoxicity by Omitting Formation of Higher-Molecular-Weight Oligomers. Biochem. Biophys. Res. Commun. 439, 321–326. [DOI] [PubMed] [Google Scholar]
- Yam A. Y.; Wang X.; Gao C. M.; Connolly M. D.; Zuckermann R. N.; Bleu T.; Hall J.; Fedynyshyn J. P.; Allauzen S.; Peretz D.; Salisbury C. M. (2011) A Universal Method for Detection of Amyloidogenic Misfolded Proteins. Biochemistry 50, 4322–4329. [DOI] [PubMed] [Google Scholar]
- Patch J. A., Kirshenbaum K., Seurynck S. L., Zuckermann R. N., and Barron A. E. (2004) Versatile Oligo(N-Substituted) Glycines: The Many Roles of Peptoids in Drug Discovery. In Pseudo-Peptides in Drug Development (Nielsen P.E., Ed.), pp 1–31, Wiley-VCH, Weinheim. [Google Scholar]
- Ross T. M.; Zuckermann R. N.; Reinhard C.; Frey W. H. (2008) Intranasal Administration Delivers Peptoids to the Rat Central Nervous System. Neurosci. Lett. 439, 30–33. [DOI] [PubMed] [Google Scholar]
- Findeis M. a; Musso G. M.; Arico-Muendel C. C.; Benjamin H. W.; Hundal a M.; Lee J. J.; Chin J.; Kelley M.; Wakefield J.; Hayward N. J.; Molineaux S. M. (1999) Modified-Peptide Inhibitors of Amyloid Beta-Peptide Polymerization. Biochemistry 38, 6791–800. [DOI] [PubMed] [Google Scholar]
- Austen B. M.; Paleologou K. E.; Ali S. A. E.; Qureshi M. M.; Allsop D.; El-agnaf O. M. A. (2008) Designing Peptide Inhibitors for Oligomerization and Toxicity of Alzheimer’s. Biochemistry 47, 1984–1992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.