

Abstract
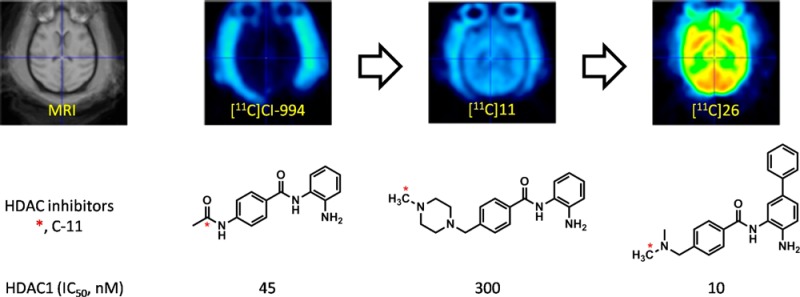
Recent studies have revealed that several histone deacetylase (HDAC) inhibitors, which are used to study/treat brain diseases, show low blood-brain barrier (BBB) penetration. In addition to low HDAC potency and selectivity observed, poor brain penetrance may account for the high doses needed to achieve therapeutic efficacy. Here we report the development and evaluation of highly potent and blood-brain barrier permeable HDAC inhibitors for CNS applications based on an image-guided approach involving the parallel synthesis and radiolabeling of a series of compounds based on the benzamide HDAC inhibitor, MS-275 as a template. BBB penetration was optimized by rapid carbon-11 labeling and PET imaging in the baboon model and using the imaging derived data on BBB penetration from each compound to feed back into the design process. A total of 17 compounds were evaluated, revealing molecules with both high binding affinity and BBB permeability. A key element conferring BBB penetration in this benzamide series was a basic benzylic amine. These derivatives exhibited 1–100 nM inhibitory activity against recombinant human HDAC1 and HDAC2. Three of the carbon-11 labeled aminomethyl benzamide derivatives showed high BBB penetration (∼0.015%ID/cc) and regional binding heterogeneity in the brain (high in thalamus and cerebellum). Taken together this approach has afforded a strategy and a predictive model for developing highly potent and BBB permeable HDAC inhibitors for CNS applications and for the discovery of novel candidate molecules for small molecule probes and drugs.
Keywords: Histone deacetylase, positron emission tomography, blood-brain barrier permeability, benzamides
Epigenetic regulation of gene expression via enzymatic modification of DNA and histone proteins is implicated in development,1 inflammation,2 heart disease,3 cancer,4 and neuropsychiatric disorders5,6 including depression, Alzheimer’s disease, and substance use disorders. Two common and influential epigenetic transformations are DNA methylation and chromatin modifications, which provide a mechanism for the transmission of environmentally cued information to subsequent generation of cells thereby producing phenotypic diversity without changing DNA sequence.
Histone acetyl transferase (HAT) and histone deacetylase (HDAC) are histone modifying enzymes that regulate gene expression by catalyzing the addition and removal of acetyl groups from the lysine groups of histone proteins, which in association with DNA form chromatin. HDAC removes an acetyl group of histone proteins (by hydrolysis of acetamido group of ε-carbon of lysine residues), inducing a tight charge–charge interaction between DNA and histone proteins, thereby rendering the DNA inaccessible to transcription factor binding and repressing gene expression. In general, histone acetylation is associated with activation of gene expression exposing DNA to transcription factors whereas deacetylation is linked to gene repression by condensing the chromatin rendering DNA less available for transcription.
There are four classes of HDAC (I, II, III, IV) and 11 isoforms. Class I HDACs (1, 2, 3, 8 subtype) are usually found in the nucleus, while class II HDAC (4, 5, 7, 9 subtype) and class IV (HDAC 11 subtype) occur in both cytoplasm and nucleus. Except for Class III HDAC, all HDACs contain a Zn ion in the substrate binding pocket where the hydrolysis of the acetamide bond occurs. Class I HDAC inhibitors (HDACi) have been developed as potential treatment of cancers. Suberoylanilide hydroxamic acid (1, SAHA, Zolinza, Figure 1A) was approved as the first drug targeting pan HDAC for cutaneous T-cell lymphoma treatment.7 In addition, SAHA has been clinically tested for diverse types of cancers including breast cancer8 and gliomas9 when used in combination with conventional cancer drugs. Recently, a benzamide type of HDACi, MS-275 (6, Entinostat, Figure 1A) has also been under many clinical trials, notably, showing its clinical efficacy for the treatment of estrogen-positive breast cancer.10
Figure 1.

Known HDAC inhibitor drugs (A) and the flowchart for image-guided systematic approach (B).
Though mostly investigated for the treatment of cancer, an increasing numbers of studies are evaluating HDACi for central nervous system (CNS) diseases, such as schizophrenia, neurodegenerative disorders, seizures, depression, and addiction.11−15 One potential advantage of HDAC drugs is the potential for reversing abnormal cell transcription, rather than treating downstream translational endophenotypes. The potential therapeutic benefits that HDACi might confer in CNS disorders stimulated us to investigate the brain uptake of a number of well-known HDAC inhibitors as potential templates for the development of highly potent blood-brain barrier (BBB) permeable HDAC inhibitors for CNS applications. In our initial studies, we used carbon-11 labeled versions of the known HDAC inhibitor drugs, butyric acid, valproic acid, and 4-phenylbutyric acid, to measure their brain uptake and whole-body pharmacokinetics.16 All of these drugs have CNS applications, particularly valproic acid, which has been used for decades to treat seizure disorders. Yet all of them have very limited BBB permeability. Similarly, we found that the benzamide HDACi, MS-275 (6), has low brain uptake when administered intravenously to nonhuman primates,17 suggesting its limitation as a therapeutic agent for CNS disorders. Recently, Hanson et al.18 also demonstrated that the lack of behavioral effects of SAHA is likely to be due to poor BBB permeability, even though its therapeutic potential for CNS applications was suggested by in vitro studies. Clearly, a systematic approach to better predict BBB penetration of small molecule probes and drugs for CNS therapeutics is needed, including that for HDACi.19
Here we report a positron emission tomography (PET) image-guided synthesis, radiolabeling, and evaluation of highly potent and BBB permeable HDAC inhibitors targeting mainly HDAC isoforms 120 and 221,22 due to their CNS disorder relevance. We modeled our series on the HDAC inhibitor MS-275 (6, Figure 1A). Although MS-275 is not BBB permeable, its relatively low molecular weight (MW) and nonionic structure at physiological pH provide the opportunity to vary different parameters and the introduction of readily labelable functional groups enabled the rapid assessment of brain uptake using PET. In parallel, we assessed the in vitro potency for HDAC inhibition of the compounds with the highest brain uptake.
Results
Overview
The development of a BBB permeable HDAC inhibitor is based on an iterative plan with four components: in silico structural design and prediction, parallel synthesis and in vitro HDAC assay, radiolabeling, quantification, and in vivo imaging (Figure 1B). Briefly, as a starting point, four known highly potent HDAC drug candidates and their derivatives (7, 8, 19) including [11C]MS-275 (6)17 were labeled with C-11 and their BBB permeability was evaluated by in vivo PET imaging. This first set of data was used to generate initial guidelines for structural modification, compared with calculated log BB (BB, ratio of brain to plasma concentration of drug in steady state (Supporting Information (SI) Table 1).23 Then, based on these results, the next set of compounds were designed under the consideration of parallel synthesis, radiolabeling, and in vitro assay. In general, three radiolabeled compounds were designed at a time, creating one set for in vivo evaluation and generating a quantitative structure–property relationship (QSPR) model (SI). The accumulated set of benzamides provided determinant physicochemical properties to be modified for further optimization. We used baboon (Papio Anubis) as it is expected to be similar to human24 and allows more accurate blood analysis at multiple time points than rodent.
Table 1. Inhibition Assay of HDAC1, HDAC2, and HDAC3 (IC50, nM).

Recombinant histone deacetylase (the value in parentheses was obtained after 3 h incubation).
Chemistry: Structural Design, Synthesis, and Radiochemistry
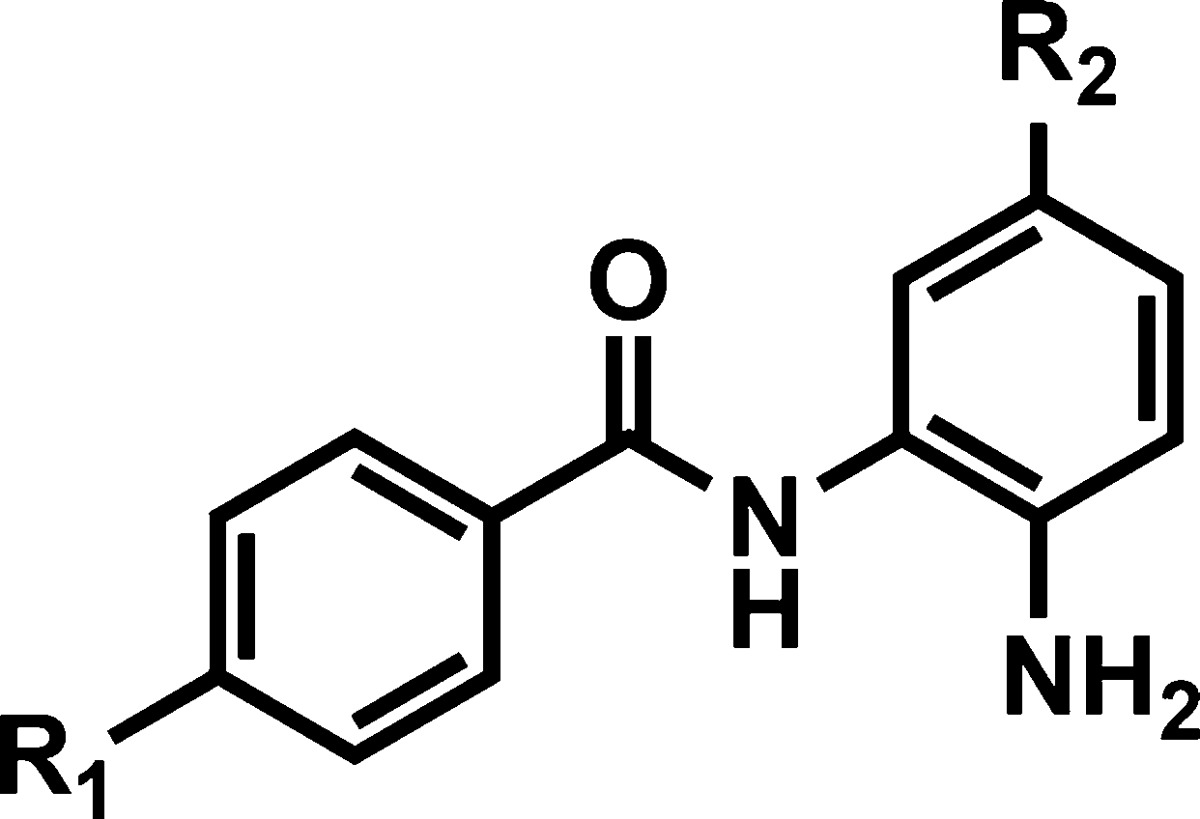
Despite the low BBB permeability of [11C]MS-275, the core benzamide structure was chosen as a template for our systematic approach varying polar surface area (PSA), charge, molecular volume (MV) (SI Table 1), and lipophilicity.25 Structurally, substituent modification of benzamides was confined at R1 of phenyl ring A and at R2 of phenyl ring B (Figures 1B and 2) because both positions have been shown to be critical for HDAC.26 Among various PET isotopes, carbon-11 was chosen in that its 20.4 min half-life made multiple PET studies possible in 1 day in the same animal. For facile and rapid labeling with carbon-11, N-methylation was adopted using [11C]methyl iodide27−29 or [11C]methyl triflate.30 In some cases, N-acetylation was also performed with [11C]acetyl chloride.31
Figure 2.

Parallel synthesis and radiolabeling of benzamides.
Initially, we chose two known benzamides,32,33 CI-994 (7, Tacedinaline) and 8, both of which have low molecular weight and sub micromolar HDAC inhibitory activity (Table 1). Carbon-11 versions of these compounds were synthesized by amidation using [11C]acetyl chloride (Figure 2B), which was prepared from [11C]CO2. The spirobicyclic compound 19 was also labeled with [11C]methyl iodide, since its nor-precursor 18 was one of the most potent HDAC1 inhibitors (IC50 = 8 nM) among the known benzamides.26 Precursor and unlabeled standards were synthesized by modification of known procedures in multistep synthesis (SI). However, none of these compounds proved to be BBB permeable in the baboon brain PET imaging study.
The first QSPR model built with these compounds (including [11C]MS-275) using the linear cross-correlation matrix revealed that PSA was the main determinant property (r2 = 0.39) for brain total distribution volume (VT, capacity of the tissue to bind the drug, SI). A set of methylpiperazines varying R1 was selected to reduce PSA in the second round. For the rapid optimization of BBB permeability, we used the parallel synthesis approach as shown in Figure 2.26 Boc-protected phenylenediamines (2 and 3) were coupled with 4-chloromethylbenzoyl choride (4) to provide various benzamides. We set R2 as a labeling position for N-methylation with [11C]methyl iodide or [11C]methyl triflate, introducing a piperazine ring and an N-mono or N-dimethyl amino group. [11C]Methyl iodide or [11C]methyl triflate was used for N-methylation of the piperazine group to give high radiochemical yield (40–60%, decay-corrected) in N,N-dimethylformamide (DMF) containing 1,2,2,6,6-pentamethylpiperidine (PMP) within 1 h of radiosynthesis from the end of cyclotron bombardment (Figure 2B). However, N-methylation of benzyl amines, which were chosen as the third set of benzamides, did not provide desired labeled product under these conditions probably due to the reduced nucleophilicity. After exploring many conditions with different combination of bases and solvents, we found that using ammonia as a base gave a moderate yield (10–30%) and sufficient labeled compound for the in vivo imaging study.
In Vitro HDAC Inhibition Assay
In vitro HDAC inhibition assay data of benzamides are shown in Table 1. The purpose of this evaluation is to compare the inhibitory potency between benzamides, based on the result of a well-characterized hydroxamte inhibitor, SAHA (1) as a reference. For HDAC1, inhibitory activity was measured after 1 and 3 h incubation with each compound. Piperazinylmethyl substitution at R1 instead of an acetamido group of CI-994 decreases potency 15-fold. Halogen substitution at R1 also decreased potency. In contrast, introduction of a phenyl group at R1 dramatically increased inhibitory activity up to the nanomolar level for HDAC1 and -2, but not -3, indicating cation−π electron interaction between the phenyl group and guanidinium cation of the arginine residue in the deep cavity inside the HDAC binding pocket.34,35 Another notable point was that the in vitro assay showed that compounds 15 and 16 displayed fast and potent inhibitory activity similar to SAHA. These properties are abolished with 4-fluoro substituent of the phenyl ring at R2. Methylated spirobicyclic compound 19 also showed similar potency to known nor-precursor compound 18.26 We also found that a relatively simple amino compound, 20 was a potent inhibitor for HDAC1 and -2. In order to reduce PSA and molecular weight to improve BBB permeability, the piperazinylmethyl group (compounds 9–17) was replaced with simple aminomethyl substituent on R1 (compounds 21–27), maintaining the C-11 labeling position. Similar to results with the piperazine derivatives, potency was recovered by adding an aromatic group at R1 resulting in a potency of 3 nM, after 3 h incubation in vitro (Table 1).
PET Study: BBB Permeability and Binding Specificity
PET studies were performed with 17 carbon-11 labeled benzamides in the baboon brain. Arterial blood was sampled over the time course of the study and selected samples were analyzed by HPLC to measure the concentration of unmetabolized parent benzamide. The % fraction of all intact carbon-11 labeled benzamides in plasma was approximately 60 ± 24% at 10 min after injection (Figure 3A). A series of benzamides with N,N-di- or N-monomethylamino methyl substituent on R2 showed significantly higher intact percent (72 ± 5%) (Figure 3A), compared with piperazinyl derivatives. MS-275 (6) and CI-994 (7), which have been used for human clinical trials, also showed relatively higher intact percent. A methylated version of spirobicylic 19 showed a very low intact fraction in the early time points and a low area under the curve (AUC), which might be consistent with fast clearance of its potent nor-compound, 18 in plasma in a prior report (Figure 3B).26
Figure 3.
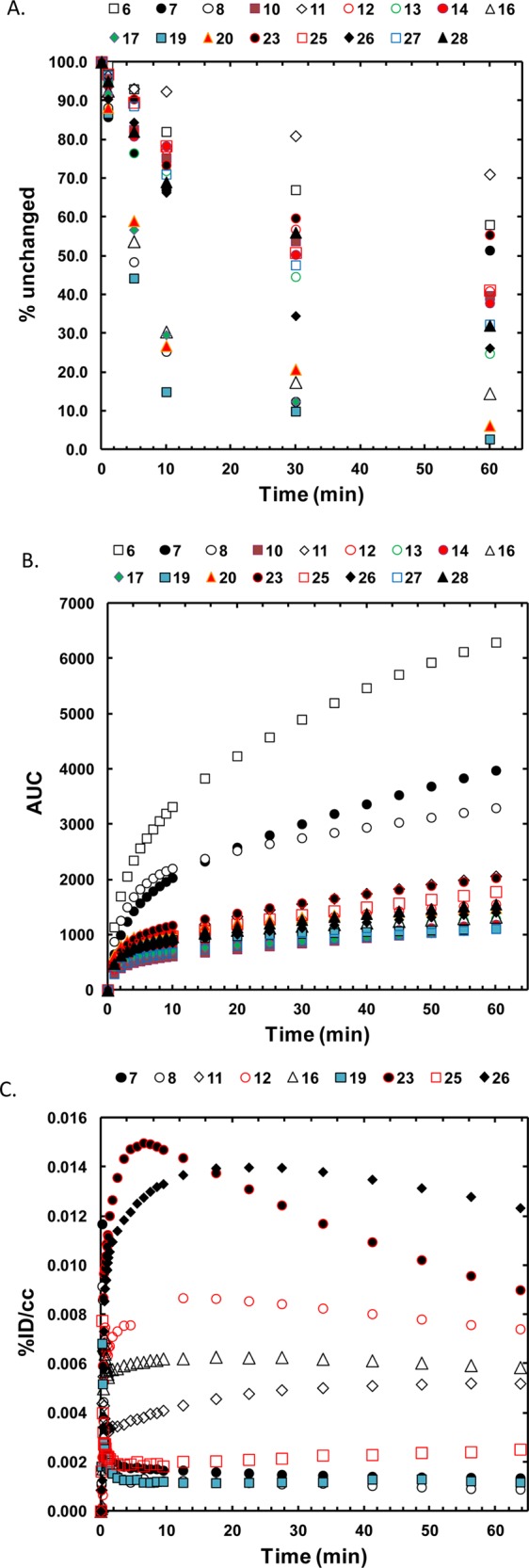
Fraction of unchanged carbon-11 labeled benzamides (A), area under the curves in baboon plasma over 60 m in after time of injection (B), and time–activity curves of global brain uptake for the selected benzamides (C).
Figure 3C shows the brain uptake (% injected dose per cc (%ID/cc)) of the selected carbon-11 labeled benzamides. The carbon-11 labeled CI-994 as well as previously reported HDAC inhibitor, 8,32 showed very low brain uptake, similar to [11C]MS-275 (6). However, the whole brain uptake of 4-methylpiperazinyl-1-methyl derivatives (10–14, 16, 17) was 4–8 times higher than [11C]MS-275 (6) at 15 min after injection. Further improvement was shown in N,N-dimethylaminomethyl derivatives (23, 26–28) which showed peak brain uptake up to 0.015%ID/cc, at 5 min post injection.
For highly potent benzamide HDAC inhibitors, the total distribution volume (VT, as determined by a one-tissue compartment model) in the brain and AUC ratio of brain versus plasma (BAUC/PAUC) were calculated (Table 2) over the 90 min scan session. VT and BAUC/PAUC were 0–17 mL/cm3 and 0.1–7.5, respectively. While both parameters of MS-275 and CI-994 were minimal, N,N-dimethylamino compounds 26 and 28 showed very high VT and BAUC/PAUC.
Table 2. Brain Total Distribution Volume and AUC Ratio of Brain to Plasma for Selected [11C]Benzamides.
| compd | log D7.4a | cLogP | PSAb | VT (mL/cm3) | BAUC/PAUC |
|---|---|---|---|---|---|
| 6, MS-275 | 1.8 | 2.5 | 119.2 | 0 | 0.14 |
| 7, CI-994 | 1.0 | 1.5 | 98.0 | 0.41 | 0.26 |
| 8 | 3.0 | 98.2 | 0.22 | 0.24 | |
| 16 | 2.2 | 3.8 | 70.2 | 8.02 | 3.54 |
| 19 | 4.7 | 5.4 | 70.1 | 4.81 | 0.93 |
| 20 | 4.8 | 70.0 | 1.35 | 0.77 | |
| 23 | 2.2 | 64.8 | 5.51 | 3.93 | |
| 25 | 4.0 | 74.8 | 2.13 | 0.90 | |
| 26 | 2.1 | 4.2 | 64.8 | 11.96 | 7.53 |
| 28 | 3.9 | 64.8 | 17.81 | 5.95 |
Measured.
Polar surface area (Å).
Considering both in vitro binding potency and BBB permeability, we chose compound 26 to evaluate binding specificity to brain HDACs. In the baseline studies, the brain distribution of [11C]26 was heterogeneous, showing highest VT in thalamus, cerebellum, and striatum. A repeated baseline study of [11C]26 on the same day in the same animal showed less than 10% of averaged variability of global brain uptake (n = 3). We examined the regional VT change of [11C]26 caused by pretreatment with unlabeled compound 26 (1 mg/kg) or the potent HDAC inhibitor, SAHA (1 mg/kg) (Table 3). The degree of VT reduction ranged 8–24%, compared with the baseline studies in various brain regions.
Table 3. Difference of Total Distribution Volumes (VT) of Test–Retest and Blocking Study (n = 3)a.
| test/retest | pretreatment with SAHA | pretreatment with 26 | |
|---|---|---|---|
| CB | 12.6 (±23.5) | –2.5 (±26.8) | –13.4 (±18.4) |
| TH | –3.9 (±14.6) | –15.4 (±11.1) | –12.9 (±27.0) |
| TEMP | 3.1 (±19.8) | –8.9 (±20.7) | –24.8 (±5.7) |
| STR | –4.5 (±21.6) | 2.5 (±36.0) | 3.8 (±48.5) |
CB, cerebellum; TH, thalamus; TEMP, temporal cortex; STR, striatum.
Discussion
We report the development of BBB permeable, highly potent HDAC inhibitors using PET in the nonhuman primate brain. Previously, almost all HDAC inhibitors tested in vivo showed lack of BBB permeability. Recently, similarly with two SAHA-based hydroxamates,36,37 we and Shuiyu et al. also reported minimal brain uptake of trichostatin A (TSA)-like hydroxamates38 and aryl hydroxamate derivative (KB631) in the baboon brain,39 respectively. Brain efflux transporters may play a critical role for these failures in that a hydroxamate drug, SAHA is known to be a P-gp substrate in in vitro assay.18 In contrast, the brain uptake of a benzamide, [11C]MS-275, was not improved by pretreatment of P-gp substrate, verapamil.17
In order to develop a predictive approach for the development of BBB permeable HDACi, we initiated a systematic strategy (that allows rapid iterative feed-back to structural modification from the initial design) using highly sensitive image-guided quantification of BBB penetration in a semi-high throughput manner. Carbon-11, a short-lived isotope of carbon (half-life: 20.4 min) that decays by positron emission, producing two highly energetic photons, can be detected external to the body barrier using PET imaging. Carbon-11 labeled compounds and nonhuman primate PET imaging provide the opportunity to assess the BBB penetration of drugs noninvasively to facilitate their translation for applications in humans.40 Due to the short half-life of carbon-11, a serial study with three different [11C]benzamides was readily achieved in the same day using the same animal. We coupled this with a QSPR model evaluating the utility of different molecular descriptors for predicting BBB uptake.
Two measures of brain uptake relative to plasma were used for the highly potent benzamide HDAC inhibitors: the total tissue distribution volume (VT) and the area under the curve (AUC) ratio of brain to plasma (BAUC/PAUC) (Table 2). VT represents the capacity of the tissue to bind the drug, whereas BAUC/PAUC represents the amount of drug in the brain over the whole time course of the experiment relative to that in plasma for the same time period. As expected, there was a strong linear correlation between VT and BAUC/PAUC (R2 = 0.81, SI Figure 1). Measures of VT and BAUC/PAUC were highest for compounds 26 and 28 in Table 2, whereas they were the lowest for CI-994 (7) and MS-275 (6). On the other hand, compound 19 has a higher VT (4.81 mL/cc) relative to BAUC/PAUC. The higher than expected VT is most likely related to the high log P and log D7.4, indicating that the higher lipophilicity results in a slower brain clearance and therefore a higher VT. Even though BAUC/PAUC for CI-994 is 0.256, which is similar to the brain/plasma ratio (B/P) reported previously (0.43),41 we point out that extrapolation of our result into steady-state B/P would be limited because these data were obtained using bolus IV administration with tracer-doses of the labeled compounds.
Considering chemical structure and based on our previous results and predicted log BB (log(B/P)), the core benzamide structure was selected and modified as a template for BBB permeability optimization. We excluded charged and high molecular weight derivatives. We found that the polar surface area (PSA)42 is a critical property for brain uptake and it must be not more than 65 for these types of benzamides (Table 2). For example, the removal of one nitrogen atom by changing N-methylpiperazine (16) to dimethylamino group (26) changed the PSA from 70.19 to 64.75 and improved BBB permeability, showing a 2-fold increase in BAUC/PAUC. Even more dramatically, the slight difference between compound 25 (monomethyl) and 26 (dimethyl) resulted in a 7-fold increase in BAUC/PAUC, which also could be explained by the number of hydrogen bonding donors (HBD). Following this reasoning, we suggest that the low brain permeability of CI-994 (7) may be attributed to its high PSA (119.16) whereas both molecular volume and PSA predicted the poor brain entry of MS-275. For all previously reported hydroxamate-based compounds, it became evident that their PSA (74–107) was not within the appropriate range.
In in vitro binding assays, the potent hydroxamic acid HDACi, SAHA, inhibits three isoforms of class I HDACs in the low nM range without any apparent selectivity. In contrast, benzamides showed selective inhibitory activity for HDAC1 and -2, which have recently been reported for their therapeutic potential in CNS disorders.20−22,43 Interestingly, some of piperazynyl derivatives (17) showed moderate HDAC2 selectivity. It also should be noted that most of the benzamides reported here showed time-dependent inhibition, indicating slower kinetics than SAHA. Chou et al. and others observed slow kinetics but long residence time for other benzamides.44,45 However, we found that some of the piperazinyl compounds (15, 16) demonstrated fast HDAC inhibitory activity (Table 1).
We also point out that recent studies showed that benzamides bind various HDAC-involving chromatin modifying complexes,46 indicating that the results of binding assays on purified HDAC may not reflect in vivo HDAC activity. Nonetheless, unlike SAHA, CI-994 was devoid of non-HDAC target binding. Our findings suggest that the development of hydroxamte and benzamide types radiotracers hold promise as potential PET ligands for measuring brain HDACs in vivo.47
Based on its high brain uptake and the in vitro IC50 results, compound 26 (IC50, 3 nM for HDAC1) was selected to evaluate its in vivo binding specificity. While in vitro autoradiography47 showed relatively high [3H]CI-994 binding density in cerebellum in the rodent brain, VT reduction of [11C]26 in the baboon cerebellum were similar to that in in thalamus and striatum, all of which showed higher reduction than cortex. There are no published reports of the regional density of HDAC in the primate brain that might have helped us interpret the regional differences we observed. The low displacement signal might be due to the slow binding kinetics of benzamides on HDAC as well as to nonspecific binding. However, since there are no BBB permeable PET radiotracers available except for [18]FAHA, compound [11C]26 would offer a good template for further development of brain permeable HDAC radiotracers. Notably, while [18]FAHA, an HDAC substrate, binds into mainly class II HDAC, benzamides are reversible HDAC I inhibitors and therefore might be useful to measure class I HDACs.
Study Limitations and Other Considerations
Some of the synthesized benzamides could be substrates of efflux transporters, and this would introduce another variable in our studies. We reported that [11C]MS-27517 showed no observable increase in brain uptake in the rodent after verapamil pretreatment. In addition, BAUC/PAUC could be affected by high brain nonspecific binding, reducing pharmacologically active free fraction in the brain.48 BBB permeability differences between species might result in different rates of brain uptake of benzamides when translated into humans. Though we focused on BBB permeability, some of the potent compounds developed in this report including piperazinyl derivatives could be good radioligand candidates for cancer imaging and therapeutics for which high BBB permeability is not necessary. Slow binding kinetics of some benzamides observed by in vitro assay may limit the visualization of target engagement in vivo over the short PET scan time.
In summary, an image-guided iterative synthesis approach yielded new highly potent and BBB permeable benzamide type HDAC inhibitors, one of which was assessed using PET. Importantly, we determined the value of the molecular descriptor PSA for predicting BBB permeability for this series. Taken together, this approach has afforded a strategy and a predictive model for developing highly potent and BBB permeable HDAC inhibitors for CNS applications which could also serve as a tool for the discovery of novel candidates for small molecule probes and for drugs.
Methods
Radiosynthesis of [11C]26
[11C]carbon dioxide produced by the 14N(p,α)11C nuclear reaction on an EBCO cyclotron (TR-19, EBCO Industry Ltd., Richmond, Canada) was converted into [11C]methyl iodide using an automatic PETtrace MeI Microlab (GE Medical Systems, Milwaukee, WI) apparatus, and then further converted to [11C]methyl triflate by passage over a AgOTf furnace at 190 ± 10 °C.30 [11C]Methyl triflate was transferred by argon flow to a V-shaped reaction vessel (5 mL) containing compound 25 (1 mg) in DMSO (300 μL) at room temperature. Upon completion of heating at 50 °C for 3 min, the reaction mixture was diluted with HPLC eluent (1 mL) and purified by reverse-phase HPLC (Column, Phenomenex Luna C18(2), 250 mm × 10 mm; flow rate, 6 mL/min; eluent, ammonium formate solution (0.1 M, water/acetonitrile = 25/75)). The desired product (retention time, 40 min) was collected and concentrated using a rotary evaporator under reduced pressure. The product was reconstituted with sterile saline (4 mL) and filtered through a sterile filter (Acrodisc 13 mm with 0.2 μm HT Tuffryn Membrane, Pall Co., Ann Arbor, MI) into a sterile, pyrogen-free vial. The total synthesis time was 75 min from the end of bombardment. Radiochemical yield was 40% (n = 16, decay corrected) and radiochemical purity was >99% (specific activity = 5–18 Ci/μmol at the end of bombardment). Radiochemical purity and identity were determined by analytical HPLC (Column, Phenomenex Luna C18, 250 mm × 4.6 mm; flow rate, 1.0 mL/min; eluent, ammonium formate solution (0.1 M, water/acetonitrile = 32.5/67.5; retention time, 11 min)).
In Vitro HDAC Inhibition Assay
Purified HDACs were incubated with 2 μM carboxyfluorescein (FAM)-labeled acetylated or trifluoroacetylated peptide substrate (Broad Substrate A and B respectively) and test compound for 60 min at room temperature, in HDAC assay buffer that contained 50 mM HEPES (pH = 7.4), 100 mM KCl, 0.01% BSA, and 0.001% Tween-20. Reactions were terminated by the addition of the known pan HDAC inhibitor LBH-589 (panobinostat) with a final concentration of 1.5 μM. Substrate and product were separated electrophoretically, and fluorescence intensity and substrate and product peaks were determined and analyzed by Labchip EZ Reader. The reactions were performed in duplicate for each sample. IC50 values were automatically calculated by Origion8 using 4 Parameter Logistic Model. The percent inhibition was plotted against the compound concentration, and the IC50 value was determined from the logistic dose–response curve fitting by Origin 8.0 software.49 Compounds were tested in duplicate in a 12-point dose curve with 3-fold serial dilution starting from 33.33 μM.
PET Studies in the Baboon
Animal studies were approved by the Brookhaven Institutional Animal Care and Use Committee. PET studies were performed in 9 female baboons (Papio anubis, 13.5–21 kg). Briefly, anesthesia was induced by intramuscular injection of ketamine hydrochloride (10 mg/kg) and then maintained with isoflurane (Forane, 1–4%), nitrous oxide (1500 mL/min), and oxygen (800 mL/min) during the PET scan. The brain was positioned in the field of view of the PET scanner (Siemen’s high-resolution HR+). Each labeled compound was injected into a radial arm vein and arterial blood was sampled through the popliteal artery from the time of injection and analyzed by HPLC. Each PET scan was performed in 3D mode for 90 min, monitoring heart rate, respiration rate, and body temperature. Arterial blood samples were collected at every 5 s for 2 min, then at 5, 10, 20, 30, 60, and 90 min after each radiotracer injection. To evaluate reproducibility and specificity of compound [11C]26, two injections in the same day in the same baboon were carried out with or without pretreatment of compound 26 (bolus, 1 mg/kg, 20 min prior to radiotracer injection) or SAHA (1 mg/kg, intravenous infusion for 30 min before the labeled compound).
Acknowledgments
We thank Michael Schueller for cyclotron operations and Donald Warner for PET operations. We also thank Tiffany St. Bernard, John Dobbs Jr., and Samuel Wilson supported by BNL summer internship program for their efforts for synthesis work. We used two computational chemistry software packages available in the Center for Molecular Modeling (http://cmm.cit.nih.gov) and the Helix Systems (http://helix.nih.gov) at the National Institutes of Health, Bethesda, MD.
Glossary
Abbreviations
- HDAC
histone deacetylase
- BBB
blood-brain barrier
- CNS
central nervous system
- PET
positron emission tomography
- HAT
histone acetyl transferase
- HDACi
histone deacetylase inhibitor
- SAHA
suberoylanilide hydroxamic acid
- MW
molecular weight
- QSPR
quantitative-property relationship
- MV
molecular volume
- PSA
polar surface area
- DMF
N,N-dimethylformamide
- PMP
1,2,2,6,6-pentamethylpiperidine
- AUC
area under the curve
- TSA
trichostatin A
- VT
total distribution volume
Supporting Information Available
Information on chemical syntheses and radiosynthesis of [11C]CI-994, QSPR model, and calculated values of molecular descriptors for tested compounds are available. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.S.F., J.M.H., and S.W.K designed research; Y.J.S., L.M., A.R., S.C., F.W., E.H., P.K., P.C., S.J.H., P.W., S.W.K., and J.M.H. performed research; Y.K., J.L., J.M.H., and S.W.K. analyzed data; J.L., N.D.V., J.S.F., J.M.H, and S.W.K. wrote the paper.
This work was supported by National Institutes of Health grant 1R01DA030321 (Y.J.S., J.M.H., S.W.K.), National Institute of Alcohol Abuse and Alcoholism Intramural Program (S.W.K., L.M., N.D.V.), and the Deutscher Akademischer Austauschdienst (DAAD), Bonn, Germany (P.W.). This study was carried out in part at Brookhaven National Laboratory under contract DE-AC02-98CH10886 with the U.S. Department of Energy and with infrastructure support from its Office of Biological and Environmental Research.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bernal A. J.; Jirtle R. L. (2010) Epigenomic Disruption: The Effects of Early Developmental Exposures. Birth Defects Res., Part A 88, 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H.; Hamon M.; Cossart P. (2012) Epigenetics and Bacterial Infections. Cold Spring Harbor Perspect. Med. 2, x. 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar G.; Adris P.; Bhargava N.; Chen H.; Raghow R. (2012) Pan-Histone Deacetylase Inhibitors Regulate Signaling Pathways Involved in Proliferative and Pro-Inflammatory Mechanisms in H9c2 Cells. BMC Genomics 13, 709–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. (2007) Cancer Epigenomics: DNA Methylomes and Histone-Modification Maps. Nat. Rev. Genet. 8, 286–298. [DOI] [PubMed] [Google Scholar]
- Tsankova N.; Renthal W.; Kumar A.; Nestler E. J. (2007) Epigenetic Regulation in Psychiatric Disorders. Nat. Rev. Neurosci. 8, 355–367. [DOI] [PubMed] [Google Scholar]
- Hyman S. E. (2012) Target Practice: Hdac Inhibitors for Schizophrenia. Nat. Neurosci. 15, 1180–1181. [DOI] [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. (2007) FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 12, 1247–1252. [DOI] [PubMed] [Google Scholar]
- Ramaswamy B.; Fiskus W.; Cohen B.; Pellegrino C.; Hershman D. L.; Chuang E.; Luu T.; Somlo G.; Goetz M.; Swaby R.; Shapiro C. L.; Stearns V.; Christos P.; Espinoza-Delgado I.; Bhalla K.; Sparano J. A. (2012) Phase I-II Study of Vorinostat Plus Paclitaxel and Bevacizumab in Metastatic Breast Cancer: Evidence for Vorinostat-Induced Tubulin Acetylation and Hsp90 Inhibition in Vivo. Breast Cancer Res. Treat. 132, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. Q.; Puduvalli V. K.; Reid J. M.; Kuhn J. G.; Lamborn K. R.; Cloughesy T. F.; Chang S. M.; Drappatz J.; Yung W. K. A.; Gilbert M. R.; Robins H. I.; Lieberman F. S.; Lassman A. B.; McGovern R. M.; Xu J. H.; Desideri S.; Ye X. B.; Ames M. M.; Espinoza-Delgado I.; Prados M. D.; Wen P. Y. (2012) Phase I Study of Vorinostat in Combination with Temozolomide in Patients with High-Grade Gliomas: North American Brain Tumor Consortium Study 04–03. Clin. Cancer Res. 18, 6032–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley D. A.; Ismail-Khan R. R.; Melichar B.; Lichinitser M.; Munster P. N.; Klein P. M.; Cruickshank S.; Miller K. D.; Lee M. J.; Trepel J. B. (2013) Randomized Phase II, Double-Blind, Placebo-Controlled Study of Exemestane with or without Entinostat in Postmenopausal Women with Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer Progressing on Treatment with a Nonsteroidal Aromatase Inhibitor. J. Clin. Oncol. 31, 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore M.; Miller C. A.; Fass D. M.; Hennig K. M.; Haggarty S. J.; Sweatt J. D.; Rumbaugh G. (2010) Inhibitors of Class 1 Histone Deacetylases Reverse Contextual Memory Deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacol. 35, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden S. A.; Christoffel D. J.; Heshmati M.; Hodes G. E.; Magida J.; Davis K.; Cahill M. E.; Dias C.; Ribeiro E.; Ables J. L.; Kennedy P. J.; Robison A. J.; Gonzalez-Maeso J.; Neve R. L.; Turecki G.; Ghose S.; Tamminga C. A.; Russo S. J. (2013) Epigenetic Regulation of Rac1 Induces Synaptic Remodeling in Stress Disorders and Depression. Nat. Med. 19, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T.; Zukin R. S. (2008) Epigenetic Targets of Hdac Inhibition in Neurodegenerative and Psychiatric Disorders. Curr. Opin. Pharmacol. 8, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A. G.; Thompson L. M. (2008) Therapeutic Application of Histone Deacetylase Inhibitors for Central Nervous System Disorders. Nat. Rev. Drug Discovery 7, 854–868. [DOI] [PubMed] [Google Scholar]
- Price S.; Dyke H. J. (2007) Histone Deacetylase Inhibitors: An Analysis of Recent Patenting Activity. Expert Opin. Ther. Pat. 17, 745–765. [Google Scholar]
- Kim S. W.; Hooker J. M.; Otto N.; Win K.; Muench L.; Shea C.; Carter P.; King P.; Reid A. E.; Volkow N. D.; Fowler J. S. (2013) Whole-Body Pharmacokinetics of Hdac Inhibitor Drugs, Butyric Acid, Valproic Acid and 4-Phenylbutyric Acid Measured with Carbon-11 Labeled Analogs by Pet. Nucl. Med. Biol. 40, 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooker J. M.; Kim S. W.; Alexoff D.; Xu Y. W.; Shea C.; Reid A.; Volkow N.; Fowler J. S. (2010) Histone Deacetylase Inhibitor MS-275 Exhibits Poor Brain Penetration: Pharmacokinetic Studies of [11C]MS-275 Using Positron Emission Tomography. ACS Chem. Neurosci. 1, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J. E.; La H.; Plise E.; Chen Y. H.; Ding X.; Hanania T.; Sabath E. V.; Alexandrov V.; Brunner D.; Leahy E.; Steiner P.; Liu L.; Scearce-Levie K.; Zhou Q. (2013) SAHA Enhances Synaptic Function and Plasticity in Vitro but Has Limited Brain Availability in Vivo and Does Not Impact Cognition. PLoS One 8, e69964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L. (2011) Organic Synthesis toward Small-Molecule Probes and Drugs. Proc. Natl. Acad. Sci. U.S.A. 108, 6699–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y.; Shen S.; Dietz K.; He Y.; Howell O.; Reynolds R.; Casaccia P. (2010) HDAC1 Nuclear Export Induced by Pathological Conditions Is Essential for the Onset of Axonal Damage. Nat. Neurosci. 13, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J. S.; Haggarty S. J.; Giacometti E.; Dannenberg J. H.; Joseph N.; Gao J.; Nieland T. J.; Zhou Y.; Wang X.; Mazitschek R.; Bradner J. E.; DePinho R. A.; Jaenisch R.; Tsai L. H. (2009) HDAC2 Negatively Regulates Memory Formation and Synaptic Plasticity. Nature 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J.; Rei D.; Guan J. S.; Wang W. Y.; Seo J.; Hennig K. M.; Nieland T. J.; Fass D. M.; Kao P. F.; Kahn M.; Su S. C.; Samiei A.; Joseph N.; Haggarty S. J.; Delalle I.; Tsai L. H. (2012) An Epigenetic Blockade of Cognitive Functions in the Neurodegenerating Brain. Nature 483, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortagere S.; Chekmarev D.; Welsh W. J.; Ekins S. (2008) New Predictive Models for Blood-Brain Barrier Permeability of Drug-Like Molecules. Pharm. Res. 25, 1836–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen S.; Lindhe O.; Palner M.; Kornum B. R.; Rahman O.; Langstrom B.; Knudsen G. M.; Hammarlund-Udenaes M. (2009) Species Differences in Blood-Brain Barrier Transport of Three Positron Emission Tomography Radioligands with Emphasis on P-glycoprotein Transport. Drug Metab. Dispos. 37, 635–643. [DOI] [PubMed] [Google Scholar]
- Fan Y.; Unwalla R.; Denny R. A.; Di L.; Kerns E. H.; Diller D. J.; Humblet C. (2010) Insights for Predicting Blood-Brain Barrier Penetration of Cns Targeted Molecules Using Qspr Approaches. J. Chem. Inf. Model. 50, 1123–1133. [DOI] [PubMed] [Google Scholar]
- Kattar S. D.; Surdi L. M.; Zabierek A.; Methot J. L.; Middleton R. E.; Hughes B.; Szewczak A. A.; Dahlberg W. K.; Kral A. M.; Ozerova N.; Fleming J. C.; Wang H.; Secrist P.; Harsch A.; Hamill J. E.; Cruz J. C.; Kenific C. M.; Chenard M.; Miller T. A.; Berk S. C.; Tempest P. (2009) Parallel Medicinal Chemistry Approaches to Selective HDAC1/HDAC2 Inhibitor (SHI-1:2) Optimization. Bioorg. Med. Chem. Lett. 19, 1168–1172. [DOI] [PubMed] [Google Scholar]
- Marazano C.; Maziere M.; Berger G.; Comar D. (1977) Synthesis of Methyl Iodide-11C and Formaldehyde-11C. Int. J. Radiat. Appl. Instrum. A 28, 49–52. [DOI] [PubMed] [Google Scholar]
- Link J. M.; Krohn K. A.; Clark J. C. (1997) Production of [11C]Ch3I by Single Pass Reaction of [11C]Ch4 with I2. Nucl. Med. Biol. 24, 93–97. [DOI] [PubMed] [Google Scholar]
- Larsen P.; Ulin J.; Dahlstrøm K.; Jensen M. (1997) Synthesis of [11C]Iodomethane by Iodination of [11C]methane. Appl. Radiat. Isot. 48, 153–157. [Google Scholar]
- Jewett D. M. (1992) A Simple Synthesis of [11C]Methyl Triflate. Int. J. Radiat. Appl. Instrum. A 43, 1383–1385. [DOI] [PubMed] [Google Scholar]
- Le Bars D.; Luthra S. K.; Pike V. W.; Duc C. L. (1987) The Preparation of a Carbon-11 Labelled Neurohormone--[11C]Melatonin. Int. J. Radiat. Appl. Instrum. A 38, 1073–1077. [DOI] [PubMed] [Google Scholar]
- Methot J. L.; Chakravarty P. K.; Chenard M.; Close J.; Cruz J. C.; Dahlberg W. K.; Fleming J.; Hamblett C. L.; Hamill J. E.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C. M.; Kral A. M.; Meinke P. T.; Middleton R. E.; Ozerova N.; Sloman D. L.; Stanton M. G.; Szewczak A. A.; Tyagarajan S.; Witter D. J.; Secrist J. P.; Miller T. A. (2008) Exploration of the Internal Cavity of Histone Deacetylase (HDAC) with Selective HDAC1/HDAC2 Inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 18, 973–978. [DOI] [PubMed] [Google Scholar]
- el-Beltagi H. M.; Martens A. C.; Lelieveld P.; Haroun E. A.; Hagenbeek A. (1993) Acetyldinaline: A New Oral Cytostatic Drug with Impressive Differential Activity against Leukemic Cells and Normal Stem Cells--Preclinical Studies in a Relevant Rat Model for Human Acute Myelocytic Leukemia. Cancer Res. 53, 3008–3014. [PubMed] [Google Scholar]
- Finnin M. S.; Donigian J. R.; Cohen A.; Richon V. M.; Rifkind R. A.; Marks P. A.; Breslow R.; Pavletich N. P. (1999) Structures of a Histone Deacetylase Homologue Bound to the Tsa and Saha Inhibitors. Nature 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Wang D. F.; Wiest O.; Helquist P.; Lan-Hargest H. Y.; Wiech N. L. (2004) On the Function of the 14 a Long Internal Cavity of Histone Deacetylase-Like Protein: Implications for the Design of Histone Deacetylase Inhibitors. J. Med. Chem. 47, 3409–3417. [DOI] [PubMed] [Google Scholar]
- Hendricks J. A.; Keliher E. J.; Marinelli B.; Reiner T.; Weissleder R.; Mazitschek R. (2011) In Vivo Pet Imaging of Histone Deacetylases by F-18-Suberoylanilide Hydroxamic Acid (F-18-Saha). J. Med. Chem. 54, 5576–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeglis B. M.; Pillarsetty N.; Divilov V.; Blasberg R. A.; Lewis J. S. (2011) The Synthesis and Evaluation of N-1-(4-(2-[F-18]-Fluoroethyl)Phenyl)-N-8-Hydroxyoctanediamide ([F-18]-Fesaha), a Pet Radiotracer Designed for the Delineation of Histone Deacetylase Expression in Cancer. Nucl. Med. Biol. 38, 683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y. J.; Muench L.; Reid A.; Chen J.; Kang Y.; Hooker J. M.; Volkow N. D.; Fowler J. S.; Kim S. W. (2013) Radionuclide Labeling and Evaluation of Candidate Radioligands for Pet Imaging of Histone Deacetylase in the Brain. Bioorg. Med. Chem. Lett. 23, 6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuiyu L.; Zhang Y.; Kalin J.; Liow J.-S.; Gladding R.; L; Innis R. B.; Koziokowski A. P.; Pike V. W. (2013) Synthesis and Evaluation of [Methyl -11c]Kb631 – a Candidate Radioligand for Histone Deacetylase Isozyme 6 (Hdac6). J. Labelled Compd. Radiopharm. 56, S319. [Google Scholar]
- Hooker J. M.; Xu Y.; Schiffer W.; Shea C.; Carter P.; Fowler J. S. (2008) Pharmacokinetics of the Potent Hallucinogen, Salvinorin a in Primates Parallels the Rapid Onset and Short Duration of Effects in Humans. NeuroImage 41, 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L.; Blaney S. M.; Dauser R.; Nuchtern J. G.; Durfee J.; McGuffey L.; Berg S. L. (2000) Pharmacokinetics and Cerebrospinal Fluid Penetration of CI-994 (n-acetyldinaline) in the Nonhuman Primate.. Clin. Cancer Res. 6(3), 994–997. [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. (2000) Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 43, 3714–3717. [DOI] [PubMed] [Google Scholar]
- Schroeder A.; Lewis M. C.; Fass D. M.; Wagner F. F.; Zhang Y. L.; Hennig K. M.; Gale J.; Zhao W. N.; Reis S.; Barker D. D.; Berry-Scott E.; Kim S. W.; Clore E. L.; Hooker J. M.; Holson E. B.; Haggarty S. J.; Petryshen T. L. (2013) A Selective Hdac 1/2 Inhibitor Modulates Chromatin and Gene Expression in Brain and Alters Mouse Behavior in Two Mood-Related Tests. PLoS One 8, e71323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C. J.; Herman D.; Gottesfeld J. M. (2008) Pimelic Diphenylamide 106 Is a Slow, Tight-Binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer B. E.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C. M.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. (2013) Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A. M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dumpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. (2011) Chemoproteomics Profiling of Hdac Inhibitors Reveals Selective Targeting of Hdac Complexes. Nat. Biotechnol. 29, 255–265. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang Y.-L.; Hennig K.; Gale J. P.; Hong Y.; Cha A.; Riley M.; Wagner F.; Haggarty S. J.; Holson E.; Hooker J. (2013) Class I HDAC Imaging Using [3H]CI-994 Autoradiography. Epigenetics 8, 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L.; Rong H. J.; Feng B. (2013) Demystifying Brain Penetration in Central Nervous System Drug Discovery. J. Med. Chem. 56, 2–12. [DOI] [PubMed] [Google Scholar]
- Katragadda M.; Magotti P.; Sfyroera G.; Lambris J. D. (2006) Hydrophobic Effect and Hydrogen Bonds Account for the Improved Activity of a Complement Inhibitor, Compstatin. J. Med. Chem. 49, 4616–4622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.