

Abstract
There are no disease-modifying drugs for any old age associated neurodegenerative disease or stroke. This is at least in part due to the failure of drug developers to recognize that the vast majority of neurodegenerative diseases arise from a confluence of multiple toxic insults that accumulate during normal aging and interact with genetic and environmental risk factors. Thus, it is unlikely that the current single target approach based upon rare dominant mutations or even a few preselected targets is going to yield useful drugs for these conditions. Therefore, the identification of drug candidates for neurodegeneration should be based upon their efficacy in phenotypic screening assays that reflect the biology of the aging brain, not a single, preselected target. It is argued here that this approach to drug discovery is the most likely to produce safe and effective drugs for neurodegenerative diseases.
Keywords: Phenotypic screening, Alzheimer’s, neurodegeneration, drug discovery, medicinal chemistry, cell culture, aging
There are currently no drugs or therapies that prevent the progression of any old age associated neurodegenerative condition. There are likely many reasons for this failure by both the pharmaceutical industry (henceforth called pharma) and academic medicine. Paramount among these reasons is the innate complexity of the pathological conditions within a patient’s brain and the high level of disease heterogeneity among the patient population. However, we believe that another reason for the high rate of failure is the current reductionist approach of pharma to these complex diseases in which the focus is on preselected molecular targets1 and its disregard of the fact that over half of the chemical scaffolds in the modern pharmacy are related to natural products that were initially identified via phenotypic screens.2
With the advent of molecular cloning, combinatorial chemistry, and robotic screening of vast synthetic chemical libraries against preselected molecular targets, there has been a shift by pharma from functional biology-based phenotypic screening using living disease models to artificial assays that measure molecular interactions.3 Unfortunately, this has resulted in a dramatic decline in new drug discovery over the past decade4 and a complete drought in the area of neurodegeneration. Over the last 30 years, only one new chemical scaffold came from combinatorial chemistry, the antitumor multikinase inhibitor sorafenib (Nexavar 1).5
The cost of drug development has also become inversely proportional to the number of new medicines created. It is therefore worth examining how the methods used for drug discovery have changed. Of the multiple approaches, only the two major ones will be discussed, phenotypic screening and the single target paradigm. Initially, material was purified from plants of therapeutic value based upon traditional medicines of different cultures. This was followed by the use of multiple phenotypic screening assays to identify most of the medicines that are in the clinic today. In contrast, the single target approach is usually based upon the use of a preselected molecule that is thought to be involved in the disease and has yielded relatively few drugs. In the following paragraphs, we will outline the apparent fallacies associated with the current approach to drug discovery and state why the return to phenotypic screening may be the only viable way forward for the treatment of neurodegenerative diseases.
Phenotypic Screening
Phenotypic screening is defined as drug screening in cellular or animal disease models to identify compounds that modify the phenotype in such a way as to generate a positive outcome relative to the disease. Examples are digoxin from foxglove in 1785, morphine from poppies in 1806, and aspirin from salicylic acid in white willow bark in 1897.6 This led to the use of both animal and microbial phenotypic screening for the isolation of most antibiotics and many other compounds that are used in the clinic today. Importantly, the compounds derived by phenotypic screening from natural products led to the identification of the Na+-K+-ATP pump (digoxin), opiate receptors (morphine), cyclooxygenases (aspirin), transpeptidase (penicillin), and many other enzymes, receptors, and transporters. A succession of follow-on drugs based upon the initial compound structures are currently in use. An approach of phenotypic drug screening will be described, and the focus will be on cell culture screening assays and the use of chemically pure small organic synthetic or natural product molecules. But first it is worth briefly outlining why pharma had abandoned phenotype screening and as a result has been unable to produce a viable drug for neurodegenerative diseases.
New Technology
While all of the early drugs were discovered by phenotypic screening, the past three decades have given rise to new technologies for making large chemical libraries (combinatorial chemistry) and high throughput screening (HTS) (robotics) that have since dominated the pharmaceutical industry. In this approach, a hypothesis is created founded upon the notion that a specific protein can be disease modifying. It requires picking both a protein target and the best binder as determined by either HTS screening or “rational design” based upon the three-dimensional structure of the target’s binding pocket. The protein is then purified and chemical libraries are screened against this single target. To date there about 1000 United States Food and Drug Administration (FDA) approved small molecule drugs, but there are only slightly over 200 defined individual molecular targets. Most of these targets were discovered well after the therapeutic introduction of the drug with which they interact.7 Therefore, picking viable drug targets is not easy.
Given the innate complexity of the toxicities associated with central nervous system (CNS) diseases and stroke, which include multiple accumulated stresses (aging, life style, and environment) as well as genetic risk factors with varying degrees of penetrance, it may be unwise to assume that the activity of a single molecule is responsible for the condition or that a single target drug can halt the entire process.
There are two apparent reasons that pharma chose to invest so heavily in the single target approach and high throughput combinatorial chemistry. First, this paradigm was introduced in the 1990s and was at the time considered innovative and did meet with some initial success outside of the area of neurodegeneration.8 Second, it is much more efficient and less labor intensive than phenotypic screening and it is believed that it will yield drugs with fewer off-target effects than those derived from phenotypic screening. The latter argument is curious given that the majority of drug scaffolds are derived from phenotypic screens that initially reduce many toxicity issues. In addition, technical problems with the modern HTS screening assays can eliminate many of the best lead compounds that are perfectly screenable in biological assays. The problems with the HTS/single target approach include the following:
(1) The robotic equipment itself presents a critical technical limitation because CNS drugs are hydrophobic and tend to stick to the plastic equipment, which is also hydrophobic.
(2) Polyphenolics and related natural products, perhaps the most promising family of lead compounds,6 interfere with many assays, generating nonspecific “hits”. For this reason and erroneous assumptions about their pharmacology, they are shunned by pharma and actively excluded from HTS libraries using theoretical filters.
(3) Once a lead compound is identified by HTS assays, it then needs to be determined if it is compatible with living cells. Therefore, its ability to get into cells must be assessed, along with its toxicity and chemical stability in a cellular environment. In contrast to the single target/HTS approach, any drug candidate that is successfully identified by phenotypic screening in living cells by definition passes most of these initial filters. For the reasons outlined below, the logical source of the drug candidates is still plant secondary metabolites.
Nature of Molecular Targets
It is dogmatically argued by pharma that blocking a single pharmaceutical target with a high affinity drug is the best approach to identifying an effective drug candidate, because, in theory, it minimizes undesirable side effects. This is simply not true for multiple reasons. The drug target is likely to have additional biological functions required for normal functions, thereby leading to toxicities. A recent example is the γ-secretase inhibitor semagacestat which failed in clinical trials because γ-secretase is required for the cleavage of another substrate (Notch 1), which if blocked leads to skin cancer.9 As pointed out by Richard Elliott, “we are simply not very good at picking drug targets.”10 Furthermore, many times the “dirtiest” drugs (those with the most off-target effects) work best. A good example is the antiarrhythmic compound amiodarone.11 Indeed, many of the best drugs in their class have multiple activities required for their function, sometimes that are unrelated to the target at the time of FDA approval.12 In addition, several approved anticancer drugs (sorafenib and sunitinib) were designed to inhibit multiple kinases, and most antipsychotics (for example clozapine) inhibit multiple CNS receptors and neurotransmitter uptake systems.13 Modern genetics has shown that cells have a great deal of built-in redundancy; therefore, inhibiting one enzyme many have a limited effect, but in some cases it may be lethal.
Although combinatorial chemistry helped optimize drug candidates,8 between 1981 and 2010, only one de novo combinatorial compound, sorafenib, was approved for clinical use.5 If not synthetic, what was the derivation of the approximately 1000 FDA approved small molecules over the past three decades? According to the data from Newman and Cragg,5b the majority of new FDA approved drugs between 1981 and 2010 were derived from natural product structures, and among CNS drugs over two-thirds were based upon natural products.
Natural Products as Lead Compounds
Natural products and the structural backbones of these molecules are major contributors to drug discovery and development.5 Most of these come from the original pharmacopoeia, intermediate metabolites from plants. Plant-derived secondary metabolites are relevant to drug discovery because they have a wide range of molecular targets and are therefore able to compete with substrates for multiple enzymes.14 The evolutionary selection for this type of small molecule is based upon the fact that the enormous number of plant secondary metabolites are made by a very limited repertoire of enzymes, but must be expressed in different amounts at different times. In addition, plants have an innate immune response derived from secondary metabolites which has some similarities to the human innate immune system.15
A basic concept from immunology may help explain the relevance of plant compounds to human disease therapy. The efficacy of an immune response is determined by an imprecise concept called avidity, which dictates that the strength of the interaction of antibodies with antigen molecules containing several epitopes is a multiple of the affinities of the interactants and is not simply the sum of their individual affinities. Extrapolating this concept to pharmacology, a drug that modestly interacts with multiple relevant target pathways, as many secondary metabolites are designed to do, may be more potent and less toxic in terms of therapeutic outcome than one that completely shuts down or activates a single pathway. A good example is the highly potent, single target drug donepezil (oral mouse LD50 45 mg/kg16) in comparison to a multitarget Alzheimer’s disease (AD) drug candidate, J147, selected through phenotypic screening, with an oral LD50 greater than 2000 mg/kg.17
Although the majority of FDA approved drugs are derived from chemical scaffolds related to natural products, these lead compounds have fallen out of favor with pharma for a number of reasons, some of which are valid, but many are unjustified or reflect the failure of the industry’s technology, not the compounds themselves. Valid concerns about natural products as a whole are that many are very large molecules, are hard to synthesize, and will not cross the blood-brain barrier (BBB). Therefore, some filters must be applied when screening these molecules. However, many classes of the 40 000–50 000 plant secondary metabolites tend to include smaller molecules with easier synthetic chemistry. Among these, small polyphenolic compounds have great therapeutic potential and will be used as an example.
The following are the standard arguments used by pharma. (1) Polyphenolic compounds are poor lead compounds because they nonspecifically bind and modify the activity of many proteins. (2) They lack a structure–activity relationship (SAR). (3) They simply function as general antioxidants. (4) The metabolism of the aromatic hydroxy groups prevents them from entering the brain. (5) They sometimes work in animals, but never in clinical trials. Only the first of these claims (nonspecific binding) may be correct for the technical reasons discussed above in the context of HTS against purified protein targets. However, it is clearly not valid when cell-based phenotypic screening procedures are used. The reasons for this are discussed below.
The protein-based, high throughput screens generate false positive hits with many hydrophobic small molecules such as polyphenolics because they nonspecifically bind to some protein targets. But in living cells a certain level of “stickiness” may be an advantage for a drug because it localizes the drug to the cell surface where it can interact with a cell surface receptor or enter the cell by a number of mechanisms. This may, in fact, be the reason why plants make these “sticky” polyphenolics. The same holds true for animals, for most, if not all, protein growth factors are quite basic and nonspecifically adhere to cell surfaces via heparin binding to more readily associate with their receptors. For example, when the heparin-binding site is removed from the cell surface, the receptor binding and growth factor potency decrease significantly for fibroblast growth factor.18 Furthermore, drug candidates for CNS diseases may be missed because they need to have a high cLogP (higher hydrophobicity) to cross the BBB and therefore may nonspecifically stick to proteins or to the extensive plastic hardware associated with robotic screening.
With respect to the SARs of polyphenolics, we have recently shown in the case of curcumin and flavones that their biological activities are exquisitely sensitive to ring substitutions, but that it is possible to improve their medicinal chemical properties and potency while maintaining multiple biological activities.19 Two examples with synthetic derivatives of curcumin and fisetin are discussed below. The derivatives are many-fold more potent, maintain most biological activities of the parent compound, and have much better medicinal chemical properties. Therefore, it is indeed possible to chemically modify plant polyphenolics and maintain multiple activities at the same time.
Furthermore, many polyphenolics are not direct antioxidants, and their biological activity is unrelated to antioxidant activity.19b In fact, it is impossible for the direct antioxidant properties of polyphenolics to have any relevance in animals because of the overwhelming antioxidant capabilities of endogenous antioxidant molecules in blood and tissues.20 It is now clear that polyphenols do not function in vivo as direct antioxidants.
A significant concern about the use of plant polyphenolics as lead compounds is their pharmacology. Most aromatic hydroxyl groups are either sulfated or glucuronidated in vivo, which in theory limits their biological activity, stops BBB penetration, and promotes their metabolic clearance, resulting in a poor drug. While it is true that these compounds are modified by in vivo metabolism, sulfation and glucuronidation are reversible, resulting in an equilibrium between the various derivatives. Therefore, given the long serum half-lives of many of these compounds in their modified state, it is likely that the less abundant unmodified compound could cross the BBB for target engagement. Furthermore, because sulfation is frequently used in vivo to avoid potential toxicity, some drug developers are currently using sulfated metabolites as lead compounds.21
However, the most important point is that many polyphenolics have therapeutic efficacy in animal models where they must get into the brain. A good example is curcumin, a relatively unstable polyphenolic, which still enters the brain and is used for imaging the amyloid plaques that it dissociates.22 In addition, the lead compound can usually be modified to improve its pharmacology by the removal or reduction of the problematic hydroxyl groups. Alternatively, the target for the polyphenol could be the vascular system or infections, so only gut penetrance is needed. Essentially all old age neurodegenerative diseases involve vascular pathology.23
There are thousands of plant polyphenols, all with distinct chemical properties and susceptibility to modification. Therefore, it is a mistake to lump all compounds in this class together. While there is not a large number of FDA approved drugs with two or more hydroxylated aromatic rings (a few examples are Fulyzag, Rifampin, Fenoldopam, and Tolcapone), there are many with aromatic rings containing one or more hydroxyl groups, including at least 17 catechols.24
Finally, among the clinical trials for AD drugs published on the NIH Web site, there have been no large-scale trials for any single polyphenolic compound, only two trials using mixtures of compounds derived from plants (tea and ginkgo) and two very small, short, and underpowered trials with curcumin with no clear outcome. A major reason for the small number of natural products in clinical trials is the inability to protect their intellectual property. However, dozens of trials for AD have been done based upon single targets and all have failed at disease modification.
In summary, the current aversion of the pharmaceutical industry to consider polyphenolic lead compounds derived from natural products cannot be based upon their lack of therapeutic potential, and their medicinal chemical properties, while perhaps not ideal, can be modified. Thus, drug discovery programs based upon natural products and phenotypic screening rather than a single selected “target” molecule should not be disregarded. Penicillin and a vast number of other drugs saved many lives before their molecular targets were identified and synthesized.
Phenotypic Screens Reflecting Neurodegeneration
While the single target approach may have found success in pharmacologically simple indications where, for example, a single ion channel or receptor is unambiguously associated with a disease, it has not generated any disease-modifying drugs for neurodegenerative diseases and stroke.3 Even in the current era where the vast majority of drug discovery has been based upon the single target approach, over 60% of the first in class drugs with a new mode of action were the result of phenotypic screening. Importantly, because phenotypic screening against human disease models leads to the identification of new disease-related molecular pathways and targets, this information has resulted in breakthrough follow-on drugs. Therefore, it is likely that phenotypic screening is necessary to advance drug discovery, particularly for the complex CNS diseases that are appearing more frequently in our longer lived population.
To illustrate the alternative phenotypic screening approach, we will use two examples from our own laboratories in the context of neurodegeneration. One of the AD drug candidates developed using this approach, J147, is currently in the Investigational New Drug (IND) phase of clinical development, and another, fisetin, is being put into a small clinical trial for Parkinson’s disease in La Jolla, California.
The ultimate phenotypic drug-screening paradigm would employ the end user, people; however, while this is how most of the natural product-based, first in class drugs were discovered, recruiting patients would be difficult. Laboratory animals, primarily disease models in mice, are currently used for most preclinical testing, but using them for initial screening of drug candidates is impractical because of time constraints and costs. In addition, results in animal neurodegeneration models have, to date, not translated into clinically approved, disease-modifying drugs. For example, well over 400 compounds reportedly improve behavior or pathology in transgenic AD mice,25 but no therapeutics have emerged from these results. Therefore, we believe that a more rigorous, in vitro drug candidate selection process is required.
This has been done in our laboratories by creating cell-based assays that define molecular toxicity pathways relevant to age-associated neurodegeneration, and selecting drug candidates that work in multiple assays, not just one. In this way, the screening paradigms have disease relevance, reproducibility, and reasonable throughput. The assays will be discussed first, followed by two examples in which the cell-based assays were used to identify or synthesize multiple potent compounds that are disease-modifying in several preclinical animal neurodegenerative disease models. In all of the assays, the nerve cells are caused to die by a toxic event that is associated with CNS aging or disease.
Although pro-survival molecules could prevent cancer cells from dying or even stimulate their growth, this possibility can never be eliminated from any neurodegeneration drug discovery program where cell viability is the selection criterion. An example is glucose metabolism, which is frequently enhanced at the expense of respiration (the Warburg effect) in cancer cells. Compounds that promote survival in low glucose media could have this potential. It is therefore important to assay the effects of drug candidates on cell division of normal cells such as NIH 3T3 fibroblasts, examine drug effects on mitochondrial respiration, and follow other FDA guidelines (www/fda/gov).
The following seven assays represent distinct neurotoxicity pathways related to aging and neurodegenerative disease that we have used to identify drug candidates (Table 1).19a,26 While these assays represent different aspects of old age brain pathology, the end result in all is nerve cell death. If it were assumed that the molecular pathway leading to death in each is unique, then it would clearly be impossible for SAR optimization using all of the assays. However, while components of each cell death pathway may be unique, this assumption is certainly not necessarily correct because there are potent compounds such as J147 that are effective in the low nanomolar range in all seven assays, and cell lines selected for resistance to death in one assay are not killed in other assays.27 It is therefore extremely likely that the rate limiting toxic pathway can be shared by many of the toxic insults in these assays. The prediction is that any drug candidate selected by these screens will have therapeutic efficacy in multiple animal neurodegeneration disease models, which has been demonstrated (see Conclusion). However, it is also the case that compounds developed through these assays are very sensitive to SAR constraints. For example, J147 is much more potent in all seven assays than its parent CNB-001, yet CNB-001 is an excellent 5-LOX inhibitor (EC50 ∼ 70 nM), while J147 is 10-fold less potent.28 Therefore, the use of these assays for SAR optimization requires an empirical approach. We have selected lead compounds that have some activity in all assays because we are looking for an ultimate common pathway in nerve cell death.
Table 1. Cell Culture Based Phenotypic Screens, Their Putative Drug Target Pathways, and Pathology in Old Brain.
| assay | target | old brain | |
|---|---|---|---|
| 1 | oxytosis | cystine antiporter | low GSH/high ROS |
| 2 | trophic factor withdrawal | receptor | reduced nerve support |
| 3 | BDNF signaling | downstream signaling | memory problems |
| 4 | glucose starvation | glycolysis | low energy |
| 5 | in vitro ischemia | ATP/mitochondria | stroke |
| 6 | intracellular Aβ | endoplasmic reticulum | proteotoxicity |
| 7 | extracellular Aβ | plasma membrane | ROS production |
Assays
1. Oxytosis
Because oxytosis is the primary screen in our drug discovery paradigm, it will be discussed first. In 1989, Murphy and colleagues reported that glutamate induced calcium-dependent cell death in a neuroblastoma cell line by inhibition of cystine import via the cystine/glutamate antiporter system xc–. The result is glutathione (GSH) depletion due to the lack of cystine, which in its reduced form is one of the three amino acids that comprise GSH, resulting in oxidative stress induced cell death.29 This type of cytotoxicity was named oxidative glutamate toxicity or oxytosis,30 and has been extensively studied in the hippocampal cell line HT22.31,32 Oxytosis is distinct from excitotoxicity where increased extracellular glutamate overstimulates ionotropic glutamate receptors thereby leading to a massive calcium influx and rapid nerve cell death.33 Importantly, oxytosis shares many of the physiological and morphological features of nerve cell death observed in AD and stroke and, therefore, is an excellent screen for drug candidates for neurodegenerative disease.30 Since the depletion of GSH in the brain is a common aspect of normal aging that is even greater in old age associated diseases, this model is relevant to many pathologies.34
The series of events leading to cell death by oxytosis have been well characterized (Figure 1). Following the inhibition of cystine import through system xc– by glutamate, GSH levels drop in a time-dependent manner. When the GSH levels fall below ∼20% (about 6 h after glutamate treatment), there is an exponential increase in reactive oxygen species (ROS) from complex I of the mitochondrial electron transport chain. GSH depletion also results in the activation of 12/15 lipoxygenase (12/15-LOX) whose eicosanoid products are activators of soluble guanylate cyclases, which then increase intracellular cGMP. Elevated cGMP eventually opens an uncharacterized calcium channel, resulting in a detrimental influx of calcium, the activation of proteases, and cell death. The synthesis of RNA and protein is required for the execution of the oxytosis cell death pathway.
Figure 1.
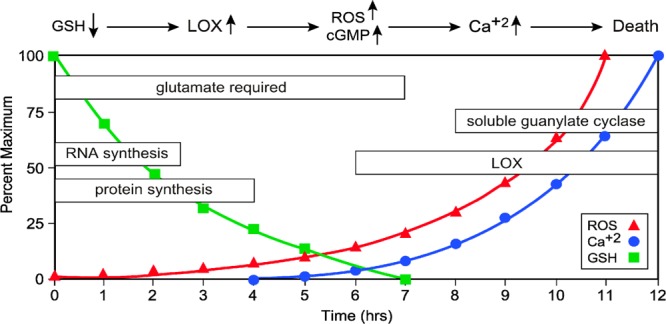
Time course of major metabolic events associated with oxytosis, a form of programmed cell death initiated by glutathione (GSH) depletion that occurs in conditions of oxidative stress and even normal aging.
Two of the major practical advantages of the HT22 model of oxytosis in the context of drug screening are its reproducibility and direct relevance to old age associated CNS diseases. Moreover, the oxidative stress induced in this model is not due to externally applied oxidants, as for example, in hydrogen peroxide toxicity, but the ROS are generated endogenously and therefore likely more physiological. The relevance of this cell death pathway to neurodegenerative disease is supported by the fact that HT22 cells selected for resistance to oxytosis are also less vulnerable to amyloid-β toxicity, endoplasmic reticulum stress brought about by the glycosylation inhibitor tunicamycin, and the overexpression of the pro-apoptotic protein Bax.27,35 Together these data highlight the pathophysiological importance of oxytosis and the potential therapeutic use of compounds that protect against this particular kind of cell death.
The oxytosis assay itself is very simple. HT22 cells or 1 day old cultures of rat E18 cortical neurons are exposed to 5 mM glutamate along with the test compound, and cell death measured 24 h later with any viability assay (for details, see Liu et al.).26a The HT22 assay is highly reproducible and has been adapted to HTS.
2. Loss of Trophic Factors
The levels of neurotrophic factors such as brain-derived neurotrophic factor (BDNF), glial derived neurotrophic factor (GDNF), fibroblast growth factor (FGF), and nerve growth factor (NGF) decline with age in the brain, and to a greater extent in AD, Parkinson’s disease, and related conditions.36 Therefore, there has been a great deal of effort to identify ways to replace these proteins in the diseased brain, but success has been limited because most proteins do not cross the BBB. A number of therapeutic strategies have been developed to circumvent this problem, including gene therapy,37 the injection of fibroblasts transfected to make growth factors,38 and, in the case of Parkinson’s disease, a surgically implanted pumping system to deliver GDNF.39 Only the latter appeared to be therapeutically effective, but was withdrawn from the clinic by its manufacturer.
Because of the heterogeneity of growth factor expression in the CNS, assays that reflect loss of trophic support are difficult to identify. Perhaps the best makes use of the fact that E18 rat embryonic nerve cells require these molecules to remain alive in cell culture when plated at low densities. At cell densities above 1 × 106/mL, they are self-supporting in tissue culture, but not at densities below this number. Once it was found that growth factors can support viability in low density culture,40 this observation evolved into a rapid screening assay for molecules that either activate receptors or their pro-survival pathways.
In an assay for trophic factor activity, E18 primary rat embryonic cortical cells are plated at 2 × 105 cells per 35 mm tissue culture dish in N2/DME/F12 medium.26a Under these conditions, the cells die within 2 days but can be rescued by combinations of neurotrophic growth factors such as BDNF and FGF.40 Cell death is prevented not only by compounds that activate neurotrophic cell-signaling pathways, but also a number of molecules with antioxidant activity.19b
3. BDNF Pathway Activation
BDNF is a neurotrophic molecule that is also involved in promoting memory. It is dramatically reduced in the brain with age and in AD, as well as in other neurological and neuropsychiatric disorders.41 The BDNF pathway has long been considered a drug target for AD.42 While BDNF will not cross the BBB, attempts have been made to make small molecules that activate the BDNF receptor and can also cross the BBB.43 However, because the function of the BDNF receptor is reduced with aging, and to an even greater extent in AD,41c,44 it is unlikely that this therapeutic approach will be effective. A much better alternative would be to activate the neuroprotective BDNF receptor signaling pathway downstream of the receptor. A robust screen for testing this potential uses cell culture conditions where the HT22 hippocampal nerve cell line dies, but can be rescued by BDNF only if the receptor is expressed by the cells. Effective drugs may activate alternative neuroprotective pathways, but protection certainly indicates an interesting drug candidate. Neurotrophic compounds are able to rescue a clone of the hippocampal nerve cell line HT22 expressing the BDNF receptor, transmembrane receptor kinase B (TrkB), from serum starvation under conditions where cells can be protected by BDNF.45 Cells lacking TrkB are used to determine if compounds that rescue the cells from serum starvation activate the receptor or act on a signaling pathway downstream of the receptor.
4. Glucose Starvation
Glucose is the brain’s major energy source and the reduction of glucose is one of the causes of nerve cell death in a variety of CNS pathologies, as well as in hypoglycemia associated with diabetes.46 Therefore, drugs that maintain viability under conditions of reduced glucose availability would be of therapeutic value. To assay for neuroprotection from this condition, PC12 cells are washed with glucose-free medium and then replated in the absence or presence of glucose and the drug candidate, and cell viability assayed 48 h later. This is an extreme form of energy depletion, but we have identified compounds that promote short-term survival in this assay.26a
5. In Vitro Ischemia
Energy metabolism and ATP levels in the brain decrease with age. The loss of mitochondrial energy metabolism and ATP levels can be mimicked using an in vitro ischemia model.47 To induce ischemia, iodoacetic acid (IAA), a well-known irreversible inhibitor of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase, is used in combination with the HT22 mouse hippocampal cell line.26c IAA has been used in a number of other studies to induce ischemia in nerve cells48 but not previously as a screen for neuroprotective compounds. Treatment of the HT22 cells with IAA shows a dose-dependent increase in cell death 20 h later with <5% survival at 20 μM. Compounds such as fisetin have been shown to maintain ATP levels in this assay.26c
6. Intracellular Amyloid Toxicity
Although extracellular toxicity of Aβ oligomers is thought to be a significant player in AD, the intracellular accumulation of Aβ may be more important. Yet it has received essentially no interest as a drug target, which we believe is a mistake. The accumulation of intracellular Aβ is likely to be a major cause of nerve cell death in AD. This conclusion is supported by a number of observations: (1) In humans, intracellular Aβ accumulation precedes plaques.49 (2) Nerve cell death occurs in mutant PS1 transgenic mice that have extensive intraneuronal Aβ, but no plaques.50 (3) There is intraneuronal Aβ accumulation well before extracellular amyloid in several different rodent AD models.51 (4) The reduction in intracellular Aβ also reduces soluble Aβ in vivo.52 (5) The removal of intracellular Aβ by apomorphine in 3XFAD mice before there are plaques improves memory and reduces AD pathology.53
For the screening against intracellular amyloid toxicity, MC65 cells are used. These cells conditionally express the C99 fragment of the amyloid precursor protein (APP) under the control of a tetracycline (tet) promoter.54 Upon the removal of tet, MC65 cells express C99, which is cleaved to Aβ by γ-secretase. Aβ polymers accumulate within the cells, leading to cell death within 4 days.26a,28,54,55 The addition of γ-secretase inhibitors blocks cell death but allows the accumulation of the nontoxic C99 protein.56
7. Extracellular Aβ Toxicity
Aβ peptide is thought to be one of the toxic entities in AD, and exogenous Aβ can kill cultured hippocampal neurons.57 There are, however, multiple problems associated with Aβ toxicity in terms of drug screening. The first is that Aβ is not very toxic, and the only cell type that Aβ reproducibly kills are cultured hippocampal neurons, which unfortunately are difficult to prepare in sufficient quantity for routine screening. Primary cortical neurons rarely die, nor does any nerve cell line. Some forms of Aβ molecular aggregates are thought to be more toxic,58 but in our hands, even using more toxic Aβ, only hippocampal neurons are completely killed.
A surrogate for cell death caused by Aβ is the MTT assay, which is normally used as a marker for bona fide cell death (cell lysis), but in the case of Aβ only gives an illusion of death. It is a valid marker for Aβ (negatively) interacting with a cell, but not ultimate cell lysis.
A consistent observation on the interaction of Aβ with nerve cells is the rapid inhibition of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction.59 MTT is a tetrazolium salt that forms a purple-colored, water-insoluble formazan upon reduction. Because only living cells can reduce MTT, MTT reduction has been developed into one of the most widely used methods for measuring cell proliferation and viability. Although it was widely assumed that MTT is reduced by active mitochondria in living cells, mitochondria are unlikely to play a significant role in cellular MTT reduction.60 Instead, MTT is taken up by cells through endocytosis and reduced by a flavin oxidase. Reduced MTT formazan accumulates in the endosome/lysosome compartment and is then transported to the cell surface through exocytosis.60 With these new insights into the mechanism of cellular MTT reduction, we subsequently demonstrated that Aβ and other cytotoxic amyloid peptides (human amylin and calcitonin) inhibit cellular MTT reduction by dramatically enhancing MTT formazan exocytosis, a phenomenon that is closely associated with the cytotoxicity of the amyloid peptides.61 Aβ and human amylin do not have sequence homology, but both of them form amyloid fibrils that are rich in β-pleated sheet conformation. These results suggest that all proteins with a β-pleated sheet structure will induce the same MTT phenomenon.
Because of the difficulties in dealing with primary hippocampal neuron culture for drug screening, the MTT assay with clonal PC12 cells can be used as a primary screen,60,61 and confirmed by hippocampal primary cultures. To determine if a compound is able to inhibit extracellular Aβ toxicity, PC12 cells are treated with the test compound and Aβ1–42 (2 μM) for 24 h followed by the MTT assay. Hippocampal neurons are treated with the test compound and 10 μM Aβ1–42, and cell death monitored 4 days later.26a
J147, A Potent Curcumin Derivative
Curcumin, the main ingredient of the Indian curry spice turmeric, is a multitarget compound that reduces inflammation, ROS production, amyloid toxicity, and excitotoxicity, and is very effective in rodent models of AD.62 However, curcumin has very low neurotrophic activity, poor bioavailability, and poor brain penetrance. To improve the neurotrophic activity and metabolic stability of curcumin, we used SAR driven iterative chemistry to improve the pharmacological properties while at the same time increasing its potency and aspects of its biological activities. Initially the highly labile diketo system of curcumin was modified to a pyrazole to make CNB-001, with improved stability and neuroprotective activity over curcumin. Systemic exploration of groups on three phenyl rings of CNB-001 revealed that the hydroxyl groups are not necessary for activity in the seven screening assays (Table 1). The addition of two methyl groups to the pyrazole attached phenyl ring led to CNB-023 with improved potency over CNB-001. However, CNB-023 is highly lipophilic (cLogP = 7.66), and compounds with high lipophilicity have multiple liabilities. To reduce the lipophilicity and identify the minimal structural requirements for activity, one of the two cinnamyl groups was removed and further optimization led to an extremely potent small molecule J147 (Figure 2). J147 is 5–10 times more potent in all of the screening assays as CNB-001 (Figure 3), while curcumin has little or no activity in any assay. J147 is not only highly potent but also it has good physicochemical properties (MW = 350, cLogP = 4.5, tPSA = 42). J147 has been studied extensively in normal aged and AD models where it has outstanding therapeutic efficacy.17
Figure 2.
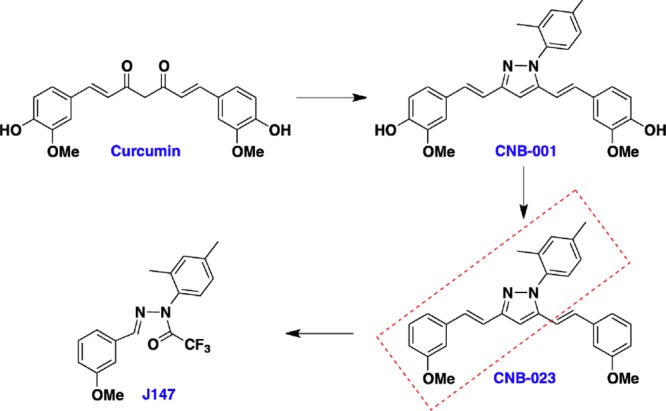
Structure activity relationship analysis results in the identification of potent small molecule J147.
Figure 3.

Biological activities of J147. J147 is active with EC50’s between 10 and 200 nM in six different assays for neurotrophic activity and neurotoxicity. Blue circles, J147; green cubes, CNB-001; red triangles, curcumin. (A) Trophic factor withdrawal: Primary cortical neurons were prepared from 18-day-old rat embryos and cultured at low cell density with or without the three compounds. Cell viability was assayed 2 days later. (B) BDNF-like activity: HT22 cells expressing the TrkB (BDNF) (yellow circles, J147) receptor or no TrkB (blue circles, J147) were placed in serum-free medium in the presence of 50 ng/mL BDNF or the indicated amounts of compounds. Cell viability was determined 2 days later. Curcumin had no activity in this assay up to 1 μM. BDNF was used at 50 ng/mL and active only in cells expressing TrkB (open bar), not in its absence (black bar). (C) Oxytosis: E18 rat cortical neurons were treated with 5 mM glutamate and different concentrations of compounds 1 day after plating when no ionotropic glutamate receptors are expressed. Cell viability was measured 24 h later. (D) Glucose starvation: PC12 cells were starved for glucose plus or minus 20 nM J147, 0.2 mM CNB-001, or 10 μM curcumin, and cell viability determined 48 h later. J147 and NGF increase cell viability in the absence of glucose, *P, 0.001 vs control. CNB-001 and curcumin are inactive at 0.2 and 10 μM, respectively (curcumin not shown). (E) Chemical ischemia: HT22 cells were treated with 20 μM iodoacetic acid for 2 h alone or in the presence of varying concentrations of J147, CNB-001, or curcumin. Percent survival was measured after 24 h. (F) Amyloid toxicity: Primary hippocampal cells were exposed to 5 μM Aβ1–42 in the presence of increasing amounts of compounds, and cell viability determined 48 h later. All data shown are mean ± SEM, n = 3 or 4. The curcumin and CNB-001 data were included for comparison with J147.
J147’s ability to enhance memory was tested against the most commonly prescribed Alzheimer’s drug, Aricept/donepezil (Pfizer), in the scopolamine-induced memory impairment model using the same cognitive behavioral assays that were used for preclinical testing of donepezil. Both compounds were comparable at rescuing short-term memory, but J147 was superior at rescuing spatial memory, and a combination of the two worked best for contextual and cued memory.17
Since J147 is a phenyl hydrazide, there was concern that it can be degraded to aromatic amines/hydrazines that are potentially carcinogenic. To explore this possibility, the metabolic stability of J147 was studied in microsomes, in mouse plasma, and in vivo. It was shown that J147 is not degraded to aromatic amines or hydrazines, that the scaffold is exceptionally stable, and that it is modified to two or three oxidative metabolites in human, mouse, rat, monkey, and dog liver microsomes. To examine the safety of these metabolites, we have synthesized all three human liver microsomal metabolites and assayed them for biological activity in the neuroprotection assays. None of these metabolites are toxic, and many of the metabolites have biological activities similar to those of J147.63
Fisetin
Over the past few years, we have shown that the flavonoid fisetin is an orally active, neuroprotective, and cognition-enhancing molecule in several animal models of CNS disorders. Fisetin has direct antioxidant activity and can maintain the intracellular levels of GSH under stress. In addition, fisetin has both neurotrophic and anti-inflammatory activity. This wide range of actions suggests that fisetin has the ability to reduce the loss of neurological function associated with multiple disorders. However, its relatively high EC50 in cell based assays (2–5 μM), low lipophilicity (cLogP 1.24), high tPSA (107), and poor bioavailability have limited fisetin for further development as a drug candidate.
The challenge was to improve the potency of fisetin in multiple neuroprotective pathways while at the same time altering its physicochemical properties to be more consistent with those of successful CNS drugs (molecular weight ≤ 400, cLogP ≤ 5, tPSA ≤ 90, HBD ≤ 3, HBA ≤ 7).64 Two different approaches were used to improve fisetin. In the first, the different hydroxyl groups were modified in a systematic manner to eliminate possible sulfate/glucuronidate metabolites. In the second approach, the flavone scaffold was changed to a quinoline, while at the same time maintaining key structural elements of fisetin.19b Utilizing our multitarget drug discovery approach, we have generated a number of derivatives with greatly enhanced activities in the neuroprotective oxytosis and in vitro ischemia assays. Three additional activities of fisetin were retained in the derivatives, including the maintenance of GSH, inhibition of bacterial lipopolysaccharide (LPS) induced microglial activation, and PC12 cell differentiation, a measure of neurotrophic activity. Flavone derivative CMS-140 and quinolone derivative CMS-121 are 600 and 400 times more potent, respectively, than fisetin in the ischemia assay (Figure 4). Thus, it is possible to maintain the multitarget qualities of a polyphenol while improving both the physiochemical and pharmacological properties of the compound.
Figure 4.
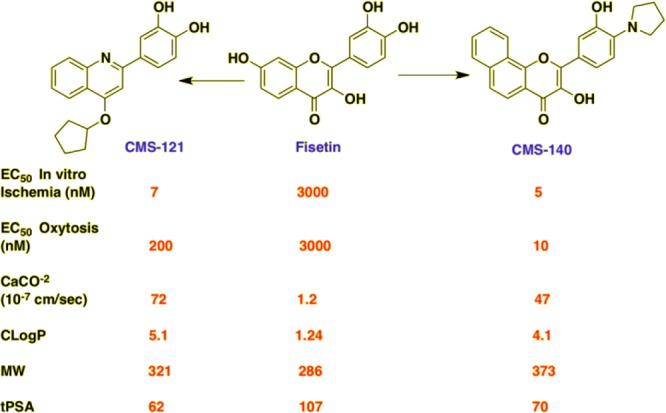
Improvement of fisetin in terms of potency (in vitro ischemia and oxytosis), medicinal chemistry properties (cLogP, MW, tPSA), and transport through CaCO-2 gut-epithelial cells.
Conclusion
The drug discovery scheme described here has been effective at identifying several highly potent families of neuroprotective molecules. These include the curcumin derivative called CNB-00126a that is effective in animal models of memory enhancement,65 stroke66 traumatic brain injury,67 lung inflammation,68 and AD.28 The more potent derivative of CNB-001, J147, is effective in memory enhancement in both normal and AD transgenic mice and neuroprotective in AD animals.19a Furthermore, it is uniquely able to reverse memory deficits in old symptomatic AD mice.17 Both compounds have neurotrophic and BDNF-like activities that are independent of the BDNF receptor.
In addition, these assays were used to discover the rare flavone fisetin that can enhance memory69 and which has therapeutic efficacy in multiple animal disease models including stroke,26c,70 Huntington’s disease,71 diabetic complications,72 and AD.73 A subset of these assays guided the synthesis of more potent fisetin derivatives,19b and has also led to the de novo isolation of a neuroprotective compound from an African plant used in traditional medicine.74
Therefore, a drug discovery paradigm based upon phenotypic screens related to multiple, age-related pathologies can yield compounds that get into cells, are likely not toxic, and target disease-related toxicity pathways. Lead compounds identified by these assays can then be improved by medicinal chemistry while maintaining activities in several screening assays, and put into animal models to advance them toward the clinic. The failure of the single target approach to drug discovery for neurodegenerative diseases makes an alternative approach necessary. We believe that it is time to use a modern version of the historically most successful pathway toward the clinic, phenotypic screening, for this purpose.
Acknowledgments
We acknowledge Jamie Simon from the Salk Institute for help preparing the graphic for the Abstract.
Glossary
Abbreviations
- Aβ
amyloid-beta
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- ATP
adenosine triphosphate
- BBB
blood-brain barrier
- BDNF
brain-derived neurotrophic factor
- cGMP
cyclic guanosine monophosphate
- CNS
central nervous system
- FDA
Food and Drug Administration
- FGF
fibroblast growth factor
- GDNF
glial derived neurotrophic factor
- GSH
glutathione
- HTS
high throughput screening
- NGF
nerve growth factor
- pharma
pharmaceutical industry
- ROS
reactive oxygen species
- SAR
structure–activity relationship
- TrkB
transmembrane receptor kinase B
Author Contributions
† M.P. and C.C. contributed equally to the manuscript. All authors meet all of the following criteria: (1) Contributing to the conception and design of the research program; (2) drafting the article or revising it critically for important intellectual content; and (3) approving the final version to be published.
These works were supported by a grant from the National Institutes of Health (NIH-R01-AG035055) and the Burns Foundation.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Pangalos M. N.; Schechter L. E.; Hurko O. (2007) Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat. Rev. Drug Discovery 6, 521–532. [DOI] [PubMed] [Google Scholar]
- Zhang H. Y.; Chen L. L.; Li X. J.; Zhang J. (2010) Evolutionary inspirations for drug discovery. Trends Pharmacol. Sci. 31, 443–448. [DOI] [PubMed] [Google Scholar]
- Swinney D. C.; Anthony J. (2011) How were new medicines discovered?. Nat. Rev. Drug Discovery 10, 507–519. [DOI] [PubMed] [Google Scholar]
- a Munos B. (2009) Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discovery 8, 959–968. [DOI] [PubMed] [Google Scholar]; b Paul S. M.; Mytelka D. S.; Dunwiddie C. T.; Persinger C. C.; Munos B. H.; Lindborg S. R.; Schacht A. L. (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discovery 9, 203–214. [DOI] [PubMed] [Google Scholar]
- a Newman D. J.; Cragg G. M.; Snader K. M. (2003) Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 66, 1022–1037. [DOI] [PubMed] [Google Scholar]; b Newman D. J.; Cragg G. M. (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishton G. M. (2008) Natural products as a robust source of new drugs and drug leads: past successes and present day issues. Am. J. Cardiol. 101, 43D–49D. [DOI] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. (2006) How many drug targets are there?. Nat. Rev. Drug Discovery 5, 993–996. [DOI] [PubMed] [Google Scholar]
- Kennedy J. P.; Williams L.; Bridges T. M.; Daniels R. N.; Weaver D.; Lindsley C. W. (2008) Application of combinatorial chemistry science on modern drug discovery. J. Comb. Chem. 10, 345–354. [DOI] [PubMed] [Google Scholar]
- Demehri S.; Turkoz A.; Kopan R. (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott R. L. (2012) Four lessons from global health drug discovery: Medicine for an ailing industry?. ACS Med. Chem. Lett. 3, 688–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden D. M. (2003) Antiarrhythmic drugs: past, present and future. J. Cardiovasc. Electrophysiol. 14, 1389–1396. [DOI] [PubMed] [Google Scholar]
- Imming P.; Sinning C.; Meyer A. (2006) Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discovery 5, 821–834. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. (2004) Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discovery 3, 353–359. [DOI] [PubMed] [Google Scholar]
- Ganesan A. (2008) The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 12, 306–317. [DOI] [PubMed] [Google Scholar]
- Nurnberger T.; Brunner F.; Kemmerling B.; Piater L. (2004) Innate immunity in plants and animals: striking similarities and obvious differences. Immunol. Rev. 198, 249–266. [DOI] [PubMed] [Google Scholar]
- Pfizer. Pfizer Material Safety Data Sheet Version 4.0 for Donepezil hydrochloride film coated tablets; www.pfizer.com/files/products/material_safety_data/PZ01067.pdf.
- Prior M.; Dargusch R.; Ehren J. L.; Chiruta C.; Schubert D. (2013) The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice. Alzheimer’s Res. Ther. 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer N. J. (2006) Insights into the role of heparan sulphate in fibroblast growth factor signalling. Biochem. Soc. Trans. 34, 442–445. [DOI] [PubMed] [Google Scholar]
- a Chen Q.; Prior M.; Dargusch R.; Roberts A.; Riek R.; Eichmann C.; Chiruta C.; Akaishi T.; Abe K.; Maher P.; Schubert D. (2011) A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease. PLoS One 6, e27865. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chiruta C.; Schubert D.; Dargusch R.; Maher P. (2012) Chemical modification of the multitarget neuroprotective compound fisetin. J. Med. Chem. 55, 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Halliwell B. (2011) Free radicals and antioxidants - quo vadis?. Trends Pharmacol. Sci. 32, 125–130. [DOI] [PubMed] [Google Scholar]; b Scalbert A.; Johnson I. T.; Saltmarsh M. (2005) Polyphenols: antioxidants and beyond. Am. J. Clin. Nutr. 81, 215S–217S. [DOI] [PubMed] [Google Scholar]
- Correia-da-Silva M.; Sousa E.; Pinto M. M. (2014) Emerging sulfated flavonoids and other polyphenols as drugs: nature as an inspiration. Med. Res. Rev. 34, 223–279. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M.; Borrelli L. A.; Rozkalne A.; Hyman B. T.; Bacskai B. J. (2007) Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 102, 1095–1104. [DOI] [PubMed] [Google Scholar]
- Toledo J. B.; Arnold S. E.; Raible K.; Brettschneider J.; Xie S. X.; Grossman M.; Monsell S. E.; Kukull W. A.; Trojanowski J. Q. (2013) Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 136, 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D. P.; Ji H. F.; Tang G. Y.; Ren W.; Zhang H. Y. (2007) How many drugs are catecholics. Molecules 12, 878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe K. H. (2000) Synaptic structure and function in transgenic APP mice. Ann. N.Y. Acad. Sci. 924, 39–41. [DOI] [PubMed] [Google Scholar]
- a Liu Y.; Dargusch R.; Maher P.; Schubert D. (2008) A broadly neuroprotective derivative of curcumin. J. Neurochem. 105, 1336–1345. [DOI] [PubMed] [Google Scholar]; b Ishige K.; Schubert D.; Sagara Y. (2001) Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radicals Biol. Med. 30, 433–446. [DOI] [PubMed] [Google Scholar]; c Maher P.; Salgado K. F.; Zivin J. A.; Lapchak P. A. (2007) A novel approach to screening for new neuroprotective compounds for the treatment of stroke. Brain Res. 1173, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargusch R.; Schubert D. (2002) Specificity of resistance to oxidative stress. J. Neurochem. 81, 1394–1400. [DOI] [PubMed] [Google Scholar]
- Valera E.; Dargusch R.; Maher P. A.; Schubert D. R. (2013) Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J. Neurosci. 33, 10512–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T. H.; Miyamoto M.; Sastre A.; Schnaar R. L.; Coyle J. T. (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558. [DOI] [PubMed] [Google Scholar]
- Tan S.; Schubert D.; Maher P. (2001) Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506. [DOI] [PubMed] [Google Scholar]
- Davis J. B.; Maher P. (1994) Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 652, 169–173. [DOI] [PubMed] [Google Scholar]
- Lewerenz J.; Baxter P.; Kassubek R.; Albrecht P.; Liefferinge J. V.; Westhoff M. A.; Halatsch M. E.; Karpel-Massler G.; Meakin P. J.; Hayes J. D.; Aronica E.; Smolders I.; Ludolph A. C.; Methner A.; Conrad M.; Massie A.; Hardingham G. E.; Maher P. (2014) Phosphoinositide 3-Kinases Upregulate System xc– via Eukaryotic Initiation Factor 2α and Activating Transcription Factor 4 – A Pathway Active in Glioblastomas and Epilepsy. Antioxid. Redox Signaling 20, 2907–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi D. W. (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. [DOI] [PubMed] [Google Scholar]
- a Currais A.; Maher P. (2013) Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox Signaling 19, 813–822. [DOI] [PubMed] [Google Scholar]; b Maher P. (2009) Modulation of multiple pathways involved in the maintenance of neuronal function during aging by fisetin. Genes Nutr. 4, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray W. J.; Yao M.; Nowotny P.; Mumm J.; Zhang W.; Wu J. Y.; Kopan R.; Goate A. M. (1999) Evidence for a physical interaction between presenilin and Notch. Proc. Natl. Acad. Sci. U.S.A. 96, 3263–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S. J.; Watson J. J.; Shoemark D. K.; Barua N. U.; Patel N. K. (2013) GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 138, 155–175. [DOI] [PubMed] [Google Scholar]
- Olanow C. W. (2014) Parkinson disease: Gene therapy for Parkinson disease-a hope, or a dream?. Nat. Rev. Neurol. 10, 186–187. [DOI] [PubMed] [Google Scholar]
- Tuszynski M. H.; Thal L.; Pay M.; Salmon D. P.; U H. S.; Bakay R.; Patel P.; Blesch A.; Vahlsing H. L.; Ho G.; Tong G.; Potkin S. G.; Fallon J.; Hansen L.; Mufson E. J.; Kordower J. H.; Gall C.; Conner J. (2005) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11, 551–555. [DOI] [PubMed] [Google Scholar]
- Nelson N. (2008) Monkeys in the Middle: How One Drug Company Kept a Parkinson’s Disease Breakthrough Out of Reach, p 272, BookSurge Publishing, North Charleston, SC. [Google Scholar]
- Abe K.; Takayanagi M.; Saito H. (1990) Effects of recombinant human basic FGF and its modified protein CS23 on survival of primary cultured neurons from various regions of fetal rat brain. Jpn. J. Pharmacol. 53, 221–227. [DOI] [PubMed] [Google Scholar]
- a Chen J.; Zacharek A.; Zhang C.; Jiang H.; Li Y.; Roberts C.; Lu M.; Kapke A.; Chopp M. (2005) Endothelial nitric oxide synthase regulates brain-derived neurotrophic factor expression and neurogenesis after stroke in mice. J. Neurosci. 25, 2366–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Olin D.; MacMurray J.; Comings D. E. (2005) Risk of late-onset Alzheimer’s disease associated with BDNF C270T polymorphism. Neurosci. Lett. 381, 275–278. [DOI] [PubMed] [Google Scholar]; c Tapia-Arancibia L.; Aliaga E.; Silhol M.; Arancibia S. (2008) New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res. Rev. 59, 201–220. [DOI] [PubMed] [Google Scholar]
- Pezet S.; Malcangio M. (2004) Brain-derived neurotrophic factor as a drug target for CNS disorders. Expert Opin. Ther. Targets 8, 391–399. [DOI] [PubMed] [Google Scholar]
- Massa S. M.; Yang T.; Xie Y.; Shi J.; Bilgen M.; Joyce J. N.; Nehama D.; Rajadas J.; Longo F. M. (2010) Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest. 120, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J.; Higgins M.; Halliday G.; Garner B. (2012) Amyloid beta selectively modulates neuronal TrkB alternative transcript expression with implications for Alzheimer’s disease. Neuroscience 210, 363–374. [DOI] [PubMed] [Google Scholar]
- Rossler O. G.; Giehl K. M.; Thiel G. (2004) Neuroprotection of immortalized hippocampal neurones by brain-derived neurotrophic factor and Raf-1 protein kinase: role of extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase. J. Neurochem. 88, 1240–1252. [DOI] [PubMed] [Google Scholar]
- Warren R. E.; Frier B. M. (2005) Hypoglycaemia and cognitive function. Diabetes, Obes. Metab. 7, 493–503. [DOI] [PubMed] [Google Scholar]
- Winkler B. S.; Sauer M. W.; Starnes C. A. (2003) Modulation of the Pasteur effect in retinal cells: implications for understanding compensatory metabolic mechanisms. Exp. Eye Res. 76, 715–723. [DOI] [PubMed] [Google Scholar]
- a Rego A. C.; Areias F. M.; Santos M. S.; Oliveira C. R. (1999) Distinct glycolysis inhibitors determine retinal cell sensitivity to glutamate-mediated injury. Neurochem. Res. 24, 351–358. [DOI] [PubMed] [Google Scholar]; b Reiner P. B.; Laycock A. G.; Doll C. J. (1990) A pharmacological model of ischemia in the hippocampal slice. Neurosci. Lett. 119, 175–178. [DOI] [PubMed] [Google Scholar]; c Reshef A.; Sperling O.; Zoref-Shani E. (1997) Activation and inhibition of protein kinase C protect rat neuronal cultures against ischemia-reperfusion insult. Neurosci. Lett. 238, 37–40. [DOI] [PubMed] [Google Scholar]; d Sigalov E.; Fridkin M.; Brenneman D. E.; Gozes I. (2000) VIP-Related protection against iodoacetate toxicity in pheochromocytoma (PC12) cells: a model for ischemic/hypoxic injury. J. Mol. Neurosci. 15, 147–154. [DOI] [PubMed] [Google Scholar]; e Sperling O.; Bromberg Y.; Oelsner H.; Zoref-Shani E. (2003) Reactive oxygen species play an important role in iodoacetate-induced neurotoxicity in primary rat neuronal cultures and in differentiated PC12 cells. Neurosci. Lett. 351, 137–140. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M.; Green K. N.; Oddo S. (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509. [DOI] [PubMed] [Google Scholar]
- Chui D. H.; Tanahashi J.; Ozawa K.; Ikeda S.; Checler F. (1999) Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat. Med. 5, 560–564. [DOI] [PubMed] [Google Scholar]
- a Billings L. M.; Oddo S.; Green K. N.; McGaugh J. L.; LaFerla F. M. (2005) Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. [DOI] [PubMed] [Google Scholar]; b Oakley H.; Cole S. L.; Logan S.; Maus E.; Shao P.; Craft J.; Guillozet-Bongaarts A.; Ohno M.; Disterhoft J.; Van Eldik L.; Berry R.; Vassar R. (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tampellini D.; Capetillo-Zarate E.; Dumont M.; Huang Z.; Yu F.; Lin M. T.; Gouras G. K. (2010) Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer’s disease transgenic mice. J. Neurosci. 30, 14299–14304. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wirths O.; Multhaup G.; Czech C.; Blanchard V.; Moussaoui S.; Tremp G.; Pradier L.; Beyreuther K.; Bayer T. A. (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 306, 116–120. [DOI] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Tran L.; Lambert M. P.; Glabe C. G.; Klein W. L.; LaFerla F. M. (2006) Temporal profile of amyloid-beta (Aβ) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J. Biol. Chem. 281, 1599–1604. [DOI] [PubMed] [Google Scholar]
- Himeno E.; Ohyagi Y.; Ma L.; Nakamura N.; Miyoshi K.; Sakae N.; Motomura K.; Soejima N.; Yamasaki R.; Hashimoto T.; Tabira T.; LaFerla F. M.; Kira J. (2011) Apomorphine treatment in Alzheimer mice promoting amyloid-beta degradation. Ann. Neurol. 69, 248–256. [DOI] [PubMed] [Google Scholar]
- Sopher B. L.; Fukuchi K.; Smith A. C.; Leppig K. A.; Furlong C. E.; Martin G. M. (1994) Cytotoxicity mediated by conditional expression of a carboxyl-terminal derivative of the beta-amyloid precursor protein. Mol. Brain Res. 26, 207–217. [DOI] [PubMed] [Google Scholar]
- Maezawa I.; Hong H. S.; Wu H. C.; Battina S. K.; Rana S.; Iwamoto T.; Radke G. A.; Pettersson E.; Martin G. M.; Hua D. H.; Jin L. W. (2006) A novel tricyclic pyrone compound ameliorates cell death associated with intracellular amyloid-beta oligomeric complexes. J. Neurochem. 98, 57–67. [DOI] [PubMed] [Google Scholar]
- Hong H. S.; Maezawa I.; Yao N.; Xu B.; Diaz-Avalos R.; Rana S.; Hua D. H.; Cheng R. H.; Lam K. S.; Jin L. W. (2007) Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Aβ oligomer-induced cytotoxicity. Brain Res. 1130, 223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner B. A.; Duffy L. K.; Kirschner D. A. (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250, 279–282. [DOI] [PubMed] [Google Scholar]
- Klein W. L. (2013) Synaptotoxic amyloid-beta oligomers: a molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease?. J. Alzheimer’s Dis. 33(Suppl 1), S49–65. [DOI] [PubMed] [Google Scholar]
- a Behl C.; Davis J.; Cole G. M.; Schubert D. (1992) Vitamin E protects nerve cells from amyloid beta protein toxicity. Biochem. Biophys. Res. Commun. 186, 944–950. [DOI] [PubMed] [Google Scholar]; b Behl C.; Davis J. B.; Lesley R.; Schubert D. (1994) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77, 817–827. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Peterson D. A.; Kimura H.; Schubert D. (1997) Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide MTT reduction. J. Neurochem. 69, 581–593. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Schubert D. (1997) Cytotoxic amyloid peptides inhibit cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. J. Neurochem. 69, 2285–2293. [DOI] [PubMed] [Google Scholar]
- Cole G. M.; Teter B.; Frautschy S. A. (2007) Neuroprotective effects of curcumin. Adv. Exp. Med. Biol. 595, 197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiruta C.; Zhao Y.; Tang F.; Wang T.; Schubert D. (2013) Metabolism of a potent neuroprotective hydrazone. Bioorg. Med. Chem. 21, 2733–2741. [DOI] [PubMed] [Google Scholar]
- Pajouhesh H.; Lenz G. R. (2005) Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P.; Akaishi T.; Schubert D.; Abe K. (2010) A pyrazole derivative of curcumin enhances memory. Neurobiol. Aging 31, 706–709. [DOI] [PubMed] [Google Scholar]
- Lapchak P. A.; Schubert D. R.; Maher P. A. (2011) Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke. J. Neurochem. 116, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu A.; Ying Z.; Schubert D.; Gomez-Pinilla F. (2011) Brain and spinal cord interaction: a dietary curcumin derivative counteracts locomotor and cognitive deficits after brain trauma. Neurorehabil. Neural Repair 25, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sharma S.; Ying Z.; Gomez-Pinilla F. (2010) A pyrazole curcumin derivative restores membrane homeostasis disrupted after brain trauma. Exp. Neurol. 226, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sharma S., Wu A., Ying L., Schubert D., and Gomez-Pinilla F. (2009) Dietary curcumin derivative promotes membrane repair after brain trauma. Neuroscience 2009 (Society for Neuroscience), Abstact No. 2009-S-10843-SfN. [Google Scholar]
- Narumoto O.; Matsuo Y.; Sakaguchi M.; Shoji S.; Yamashita N.; Schubert D.; Abe K.; Horiguchi K.; Nagase T.; Yamashita N. (2012) Suppressive effects of a pyrazole derivative of curcumin on airway inflammation and remodeling. Exp. Mol. Pathol. 93, 18–25. [DOI] [PubMed] [Google Scholar]
- Maher P.; Akaishi T.; Abe K. (2006) Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc. Natl. Acad. Sci. U.S.A. 103, 16568–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M.; Leypoldt F.; Lewerenz J.; Birkenmayer G.; Orozco D.; Ludewig P.; Thundyil J.; Arumugam T. V.; Gerloff C.; Tolosa E.; Maher P.; Magnus T. (2012) The flavonoid fisetin attenuates post-ischemic immune cell infiltration, activation and infarct size after transient cerebral middle artery occlusion in mice. J. Cereb. Blood Flow Metab. 32, 835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P.; Dargusch R.; Bodai R.; Gerard P.; Purcell J.; Marsh J. L. (2011) ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 20, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P.; Dargusch R.; Ehren J. L.; Okada S.; Sharma K.; Schubert D. (2011) Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One 6, e21226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A.; Prior M.; Dargusch R.; Armando A.; Ehren J.; Schubert D.; Quehenberger O.; Maher P. (2014) Modulation of p25 and Inflammatory Pathways by Fisetin Maintains Cognitive Function in Alzheimer’s Disease Transgenic Mice. Aging Cell 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currais A.; Chiruta C.; Goujon-Svrzic M.; Costa G.; Santos T.; Batista M. T.; Paiva J.; Madureira M. d. C.; Maher P. (2014) Screening and identification of neuroprotective compounds relevant to Alzheimer’s disease from medicinal plants of S. Tomé e Principe. J. Ethnopharmacol., (in press). [DOI] [PubMed] [Google Scholar]