

Abstract
In humans, genetic defects of the synthesis or regeneration of tetrahydrobiopterin (BH4), an essential cofactor in hydroxylation reactions, are associated with severe neurological disorders. The diagnosis of these conditions relies on the determination of BH4, dihydrobiopterin (BH2), and dihydroneopterin (NH2) in cerebrospinal fluid (CSF). As MS/MS is less sensitive than fluorescence detection (FD) for this purpose, the most widely used method since 1980 involves two HPLC runs including two differential off-line chemical oxidation procedures aiming to transform the reduced pterins into their fully oxidized fluorescent counterparts, biopterin (B) and neopterin (N). However, this tedious and time-consuming two-step indirect method underestimates BH4, BH2, and NH2 concentrations. Direct quantification of BH4 is essential for studying its metabolism and for monitoring the efficacy of BH4 supplementation in patients with genetic defects. Here we describe a single step method to simultaneously measure BH4, BH2, B, NH2, and N in CSF by HPLC coupled to FD after postcolumn coulometric oxidation. All target pterins were quantified in CSF with a small volume (100 μL), and a single filtration step for sample preparation and analysis. As compared to the most widely used method in more than 100 CSF samples, this new assay is the easiest route for accurately determining in a single run BH4, BH2, and NH2 in CSF in deficit situations as well as for monitoring the efficacy of the treatment.
Keywords: Biopterin, neopterin, hydoxylases, cerebrospinal fluid, metabolism, neurologic disorders
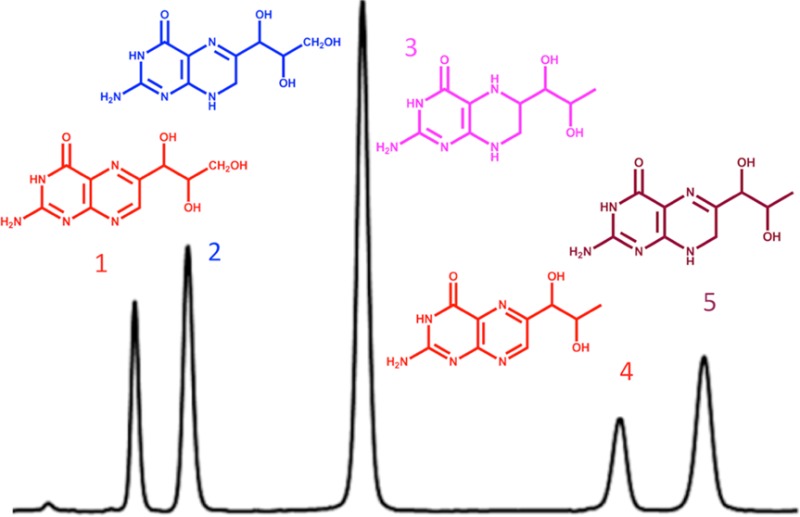
In mammals, tetrahydrobiopterin (BH4) (Figure 1) is an essential cofactor for aromatic amino acid hydroxylases, glyceryl-ether mono-oxygenase, and three nitric oxide synthases.1 Thus, BH4 plays an important role in monoamine neurotransmitters metabolism. In humans, defects in the metabolism or recycling of BH4 (Figure 1) leading to BH4 deficiency are associated with neurological deterioration including progressive mental and physical retardation, central hypotonia and peripheral spasticity, seizures, and microencephaly.2 Lack of BH4 synthesis or regeneration can be due to one of five known inborn errors of metabolism leading to the deficiency of five enzymes (Figure 1). The diagnosis of these conditions relies on the accurate determination of neurotransmitters3 and pterins, notably BH4, dihydrobiopterin (BH2), and dihydroneopterin (NH2) in the cerebrospinal fluid (CSF).4
Figure 1.

Metabolism pathway of tetrahydrobiopterin (BH4). N (neopterin); B (biopterin); BH2 (dihydrobiopterin); NH2 (dihydroneopterin). GTP (guanosine triphosphate); GTPCH (guanosine triphosphate cyclohydrolase EC 3.5.4.16); PTPS (6-pyruvoyl-tetrahydropterin synthase EC 4.2.3.12); SR (sepiapterin reductase EC 1.1.1.153); PCD (pterin-4α-carbinolamine dehydratase EC 3.5.4.16); DHPR (dihydropteridin reductase EC 1.5.1.34). (Dashed arrows: nonenzymatic.)
As oxidized pterins are fluorescent while their reduced counterparts are not, the most widely used method for pterin determination in CSF requires carrying out two HPLC (high performance liquid chromatography) runs coupled to fluorescence detection (FD) including two off-line iodine oxidation procedures for complete analysis.5−8 The first run involves oxidation under acidic conditions of both BH4 and BH2 into biopterin (B), and NH2 into neopterin (N) prior to injection into the chromatograph. The second run involves the oxidation under alkaline conditions of BH2 into B and BH4 into pterin (P). Although time-consuming, this indirect chemical oxidation (ICO) method has been widely used since 1980, notably because it has appropriate sensitivity.5−8 Total B can be also determined after MnO2 oxidation but this method does not allow distinguishing BH4 from BH2.8
Direct quantification of BH4, BH2, and NH2 is essential for studying BH4 and neurotransmitter metabolism (Figure 1). This area of research is very active as reflected in a recent description of an unknown genetic disorder of neurotransmitter translocation into synaptic vesicles.9 Besides, as BH4 is used as a therapeutic agent in patients with BH4 deficiency,7,10 a direct and accurate method of BH4 quantification in biological fluids and tissues is needed for pharmacokinetic studies and for determining the efficacy of the treatment. Notably, two recent papers raised doubts about the ability of BH4 at usual doses (20 or 40 mg/kg/day) to cross the blood brain barrier in patients with genetic BH4 recycling disorder.11,12 In such situations, only treatments with neurotransmitter precursors can induce a significant change of the CSF neurotransmitters profile.11,12
Some methods of direct quantification through FD after chemical or photochemical postcolumn oxidation have been implemented.13−17 However, the sensitivities of these methods were not satisfactory as compared to those of ICO methods.5−8
More recently, LC-MS/MS (liquid chromatography coupled to tandem mass spectrometry) was used to determine B concentrations in plasma by applying an oxidation conversion ratio.18 An enzymatic approach was also developed to distinguish BH4 from B with a two-step procedure.19 These indirect methods requiring two run procedures are still tedious and time-consuming. LC-MS/MS was also used for the simultaneous detection of BH4, BH2, and B in urine samples, cell extracts, and rat brains.20−22 However, according to the authors, MS/MS detection is less sensitive than FD for pterin quantification.22
In the mid-1980s, sequential electrochemical detection (ECD) and FD were proposed for the simultaneous quantification of BH4 and BH2 in CSF.23,24 BH4 was detected electrochemically, while BH2 was detected by FD after postcolumn coulometric oxidation (PCCO-FD) into B. Due to the large number of electroactive compounds in CSF, detecting specifically BH4 by ECD is a challenge. Actually, ECD is less robust than FD for routine analyses;8 besides this method does not allow the detection of N and NH2.
Here we focus on the effects of pH on the separation of the target pterins as a function of the stationary phase and on their electro-oxidation to develop a single method capable of simultaneous determination of all forms of target pterins present in CSF samples by HPLC coupled to PCCO-FD with a single filtration step for sample preparation and analysis.
For comparison, the proposed method was applied in parallel with the most widely used ICO method5−8 to analyze CSF samples collected from infants, children, and young patients aged 2 months to 20 years (median 5.8 years, n = 94) and adults aged from 20 to 41 years (median 30.8 years, n = 5) with a normal metabolic profile. We also included samples from patients aged 2 months to 38 years (median 15.6, n = 9) with defects in pterin metabolism, including a known sepiapterin reductase deficiency (SR),11 an amino acid decarboxylase deficiency (AADC), a tyrosine hydroxylase deficiency (TH), a secondary biopterin deficiency, and five cases of neopterin increase of different origins.
Results and Discussion
Characterization of Standard Pterins
As most standards are reduced species, which are prone to oxidation, we systematically checked their identities by high resolution mass spectrometry (HRMS). We also checked the structure of BH4, the least stable standard, by NMR.
MS experiments confirmed the m/z ratios of all standards. The most abundant protonated ions in the spectra of BH4, BH2, and B were at m/z 242.1244, 240.1094, and 238.0942, respectively. As for NH2 and N, the most abundant protonated ions were at m/z 256.1037 and 254.0885, respectively.
1H NMR, and 1H–13C heteronuclear correlation NMR confirmed the structure of BH4 [1H NMR (400 MHz, D2O) δ (ppm): 1.06 (d, J = 4.3 Hz, 3H, CH3), 3.32 (dd, J = 13.7 Hz, J′ = 9.7 Hz, 1H, CH7′), 3.51 (dd, J = 13.8 Hz, J′ = 4.0 Hz, 1H, CH7′), 3.57 (m, 2H, CH1′, CH2′), 3.58 (m, 1H, CH6); and 13C NMR (400 MHz, D2O, 13C resonances measured on a 13C–1H 2D HSQC spectrum) δ (ppm): 19.5 (1C, CH3), 36.8 (1C, C7H2), 53.5 (1C, C6H), 68.0 (1C, C1′H)]. Typical correlation patterns were observed on 1H–13C edited HSQC and 1H–1H COSY 2D spectra, indicating the presence of a fragment H3C–C2′H–C1′H–C6H–C7H2 that can be unambiguously assigned to the structure of BH4 (Figure 1).
Method Development
Figure 2 shows UV–visible chromatographic profiles of a standard solution of the target pterins obtained on different stationary phases. To evaluate the efficiency of each column, we determined the N value (number of theoretical plates) for the last eluting compound in each chromatogram. N is then used to calculate h [reduced plate height (h = H/dp), where H is the theoretical plate height (column length/N) and dp is the particle diameter both expressed in the same length unit)], which allows comparing efficiencies of different columns with different geometries filled with different particle sizes.
Figure 2.

Pterin separation as a function of stationary phase and pH. (a) Atlantis dC18 (NBH2 = 19 230); (b) Eclipse XDB C18 (NBH2 = 11 510); (c) XTerra (NBH2 = 2080); (d) Zic-HILIC (NNH2 = 16 250). NBH2 and NNH2 = theoretical plate number for BH2 or NH2, respectively. Mobile phase for panels (a)–(c): pH 7.4, 0.05 M sodium citrate/methanol (97/3, v/v). Mobile phase for panel (d): pH 7.4, 0.2 M ammonium formiate/acetonitrile (20/80, v/v). Flow rate 0.6 mL/min at 30 °C and UV detection at 260 nm for all columns (AU = absorbance units).
As the target pterins are basic compounds with low hydrophobicity, we used a polar embedded C18 column (Atlantis) to separate them without using an ion pairing reagent. At pH 7.4, the Atlantis column showed better efficiency (hBH2 = 3.6) (Figure 2a) than the Eclipse XDB column designed for reverse-phase separation of basic compounds (hBH2 = 4.5) (Figure 2b).
To investigate the effects of pH on pterin separation and detection between pH 2 and 10, we used the XTerra column filled with hybrid particles having a widened useable pH range.
As expected in HILIC (hydrophilic interaction liquid chromatography) separations, the ZIC-HILIC column showed different selectivity as compared to RP-columns (Figure 2d). While this column showed high efficiency for the standard mixture (hN = 2.65), injecting aqueous CSF samples resulted in a significant loss of efficiency (hN = 5.71) due to solvent-mobile phase incompatibility (data not shown). Hence, further studies were carried out on the Atlantis column (Figure 2b).
Figure 3 shows the hydrodynamic voltammograms and the oxidation yields as a function of pH of the target pterins as determined by HPLC-PCCO-FD. While high oxidation yields (≥50%) of BH2 and NH2 can be obtained irrespective of the pH range between pH 3 and pH 8, a sufficient yield (about 50%) only occurs at pH higher than 7.4 for BH4 (Figure 3b). At pH less than 6 the BH4 oxidation yield is about 20% and then it decreases to become negligible at pH equal to or less than 4. The limited oxidation yield of BH4 at pH less than 6.0 could explain why previous methods did not detect BH4 by PCCO-FD.23
Figure 3.

Fluorescence detection after electrochemical oxidation of NH2, BH2 and BH4. (a) Hydrovoltammograms at pH 7.4. (b) Oxidation yields as a function of pH at +400 mV. (a and b) HPLC conditions: Atlantis dC18 (4.6 × 150 mm, 3 μm); mobile phase: pH 7.4, 0.05 M sodium citrate/methanol (97/3, v/v); flow rate: 0.6 mL/min at 30 °C; detection: λex 350 nm; λem 450 nm after coulometric oxidation. (c) Retention factors as a function of pH on the same column and with the same mobile phase as for (a) and (b). Flow rate 0.6 mL/min at 30 °C and UV detection at 260 nm. X2 is an interfering compound coeluting with B (See Figure 4).
Figure 3c also shows that the best separation of the six pterins on the Atlantis column is obtained at pH 7.4 corresponding to the best oxidation yield of BH4 (Figure 3b) under the useable pH range for this column. Assuming that the oxidation yield of BH4 at pH 7.4 is sufficient to obtain an appropriate sensitivity for CSF analyses, we fixed the pH of the mobile phase for the proposed method at pH 7.4. It is possible to improve the oxidation yield of BH4 by increasing the pH of the mobile phase but this will reduce the stability of the stationary phase.
To optimize the selected chromatographic conditions, it was mandatory to investigate the applicability of the developed method to authentic CSF samples.
Applicability to CSF Samples
Figure 4 shows the chromatographic profiles of a pooled CSF sample (n = 5) before and after ICO25 or PCCO-FD under the selected chromatographic conditions. In agreement with previous observations,23 these profiles show that the CSF samples contain only reduced forms of petrins, mainly NH2, BH4, and BH2. Before oxidation, N and B concentrations in all analyzed samples never reached the LOD (Limit of Detection) of the method (Figure 4b1, dashed line).
Figure 4.

Chromatographic profiles of a standard mixture and a pooled CSF sample (n = 5) under the proposed chromatographic conditions: column = Atlantis dC18 (4.6 × 150 mm, 3 μm); mobile phase: pH 7.4, 0.05 M sodium citrate/methanol (97/3, v/v); flow rate: 0.6 mL/min at 30 °C, detection: λex 350 nm, λem 450 nm (FU: fluorescence units (arbitrary units)). (a) Standard mixture after postcolumn oxidation at +600 mV. Pooled CSF sample (b1, dashed line) without oxidation; (b2, solid line) after off-line chemical oxidation according to ref (25); (c) after postcolumn coulometric oxidation at +400 mV; and (d) after postcolumn oxidation at +600 mV allowing the disappearance of the interfering peaks X1 and X2.
While only N and B peaks were detected after ICO (Figure 4b2, solid line) several peaks including NH2, BH2 and BH4 peaks and some unidentified peaks (X1, X2) appeared after PCCO (Figure 4c). The X2 unidentified peak eluting at 13.5 min corresponding to the retention time of B cannot be attributed to this oxidized form because it was not detected without oxidation (Figure 4b1, dashed line). As this interfering peak was detected in all analyzed samples after PCCO-FD, we investigated its electrochemical and fluorescence behavior.
Figure 3a shows that while the PCCO product of X2 was fluorescent at an electrode potential lower than 500 mV, further oxidation at higher potentials results in decreased fluorescence intensity. At an electrode potential of +600 mV, this interference was no longer detected under our experimental conditions (Figure 4d). Thus, to avoid this interference, we decided to oxidize the effluent at 600 mV for pterin analyses. At this potential, the chromatographic profile of the same pooled CSF sample clearly shows that the three circulating forms of pterins are well separated and detected without interfering peaks (Figure 4d).
Proposed Method
Considered together, these results led us to adopt the following chromatographic conditions: separations were performed on an Atlantis dC18 (4.6 × 150 mm, 3 μm) column with a mobile phase consisting of pH 7.4, 0.05 M sodium citrate/methanol (97/3, v/v) delivered at a flow rate of 0.6 mL/min at 30 °C. The fluorimetric detection was performed at λex 350 nm and λem 450 nm after coulometric oxidation at +600 mV. The final run time was 16 min.
Method Evaluation
The identification of pterins in authentic CSF samples was based on their retention factors, increases in peak areas after spiking with known amounts of each pterin, and on electrochemical and fluorescence properties.
The stability of the samples at 10 °C in the dark in the injector was checked after several hours. We did not observe significant differences after 6 h. After 8 h, NH2 and BH4 concentrations decreased between 4% and 31% of their initial values (n = 10, data not shown). Thus, using this method, samples should be analyzed not more than 6 h.
We checked the stability of pterins in CSF samples (n = 40) frozen at −80 °C immediately after collection. As compared with the initial values determined at day 7 after collection, we did not observe significant differences after 24 months of storage. Thus, we avoided adding a preservative agent to the samples facilitating the collection procedure.
The comparison of within-run data between artificial and authentic CSF samples, spiked under the same conditions with each pterin, showed no significant differences between the two matrices (Table S-1, Supporting Information). The recoveries were equal to or higher than 88% irrespective of pterin concentration (Table S-1). Thus, we used an artificial CSF rather than authentic pooled CSF samples for method calibration.
The method was linear for all five pterins, up to 100 nmol L–1, which allowed direct BH4 determination in pharmacokinetic studies. The LOQs (limits of quantification) enabled the determination of all pterins even in deficit situations, and the CV (coefficient of variation) of the method never exceeded 9.9% (Table S-1).
As the sample preparation consisted of a single dilution in the mobile phase before protein filtration, and as precision was equal to or better than 9.9% in all instances (Table S-1), we did not use an internal standard for method validation. However, in clinical practice, there is a need for internal standards, thus we included lumazine as an internal standard to demonstrate the robustness of the method (Figure 5).
Figure 5.

Chromatographic profiles of a standard mixture and a pooled CSF sample (n = 5) under the proposed chromatographic conditions: column = Atlantis dC18 (4.6 × 150 mm, 3 μm); mobile phase: pH 7.4, 0.05 M sodium citrate/methanol (97/3, v/v); flow rate: 0.6 mL/min at 30 °C; postcolumn oxidation at +600 mV detection: λex 350 nm, λem 450 nm (FU: fluorescence units (arbitrary units)). (a) Standard mixture; (b) pooled CSF without internal standard; (c) pooled CSF with lumazine as internal standard (IS).
Experimental Determination of Pterins in Human CSF
Table 1 summarizes pterin concentrations, categorized by age and sex, of 99 CSF samples analyzed by the proposed method. The patients did not suffer from any known inborn error of metabolism.
Table 1. Pterin Concentrations (nmol/L) in Human CSF As Determined by the Proposed Method, Categorized by Age and Sex (M: male; F: female).
| age (years) | N | M | F | mean | SD | med | max | min |
|---|---|---|---|---|---|---|---|---|
| BH4 | ||||||||
| 0–0.33 | 2 | 2 | 0 | 61.3 | 19.2 | 61.3 | 74.9 | 47.7 |
| 0.34–0.66 | 4 | 3 | 1 | 39.7 | 18.2 | 37.5 | 64.0 | 19.8 |
| 0.67–1.00 | 6 | 5 | 1 | 46.7 | 13.6 | 46.8 | 63.9 | 28.6 |
| 1.1–5.00 | 29 | 13 | 16 | 33.6 | 13.2 | 33.4 | 76.3 | 12.8 |
| 5.001–20 | 53 | 27 | 26 | 31.7 | 11.9 | 30.8 | 61.1 | 7.6 |
| 20–42 | 5 | 4 | 1 | 16.1 | 3.5 | 14.7 | 20.8 | 12.3 |
| all | 99 | 54 | 45 | 33.3 | 13.9 | 32.3 | 76.3 | 7.6 |
| BH2 | ||||||||
| 0–0.33 | 2 | 2 | 0 | 11.5 | 2.5 | 11.5 | 13.2 | 9.7 |
| 0.34–0.66 | 4 | 3 | 1 | 9.6 | 1.4 | 10.0 | 10.8 | 7.6 |
| 0.67–1.00 | 6 | 5 | 1 | 11.4 | 1.7 | 11.6 | 13.4 | 8.9 |
| 1.1–5.00 | 29 | 13 | 16 | 9.7 | 3.0 | 9.1 | 17.6 | 4.2 |
| 5.001–20 | 53 | 27 | 26 | 9.3 | 2.7 | 9.3 | 16.4 | 3.1 |
| 20–42 | 5 | 4 | 1 | 8.4 | 3.1 | 9.8 | 11.3 | 4.6 |
| all | 99 | 54 | 45 | 9.6 | 2.8 | 9.5 | 17.6 | 3.1 |
| NH2 | ||||||||
| 0–0.33 | 2 | 2 | 0 | 14.1 | 3.3 | 14.1 | 16.4 | 11.7 |
| 0.34–0.66 | 4 | 3 | 1 | 13.0 | 1.3 | 12.7 | 14.8 | 11.9 |
| 0.67–1.00 | 6 | 5 | 1 | 14.2 | 7.0 | 11.3 | 23.5 | 7.7 |
| 1.1–5.00 | 29 | 13 | 16 | 18.4 | 10.0 | 15.6 | 47.3 | 7.6 |
| 5.001–20 | 53 | 27 | 26 | 14.4 | 5.2 | 13.4 | 29.9 | 3.9 |
| 20–42 | 5 | 4 | 1 | 18.2 | 5.6 | 17.8 | 25.3 | 11.8 |
| all | 99 | 54 | 45 | 15.7 | 7.1 | 13.5 | 47.3 | 3.9 |
Although the number of samples is not sufficient to establish definitive normative values according to age and sex (Figure 6a), the average concentrations were close to those obtained by Hyland et al.26 However, our results were systematically higher than those obtained by HPLC after either MnO2 or I2 ICO.25 When we compared the mean values, they were 38% and 67% higher than those reported by Komori et al.6 and Ormazabal et al.,27 respectively. When we compared the median values, they were 98% higher than those observed by Verbeek et al.25 However, ICO methods are known to underestimate amounts of reduced forms of N and B.15,23 To evaluate the proposed method more fully, we compared the results of total biopterin (BH4 + BH2) obtained by the proposed method with those obtained with the most widely used method involving ICO.25
Figure 6.
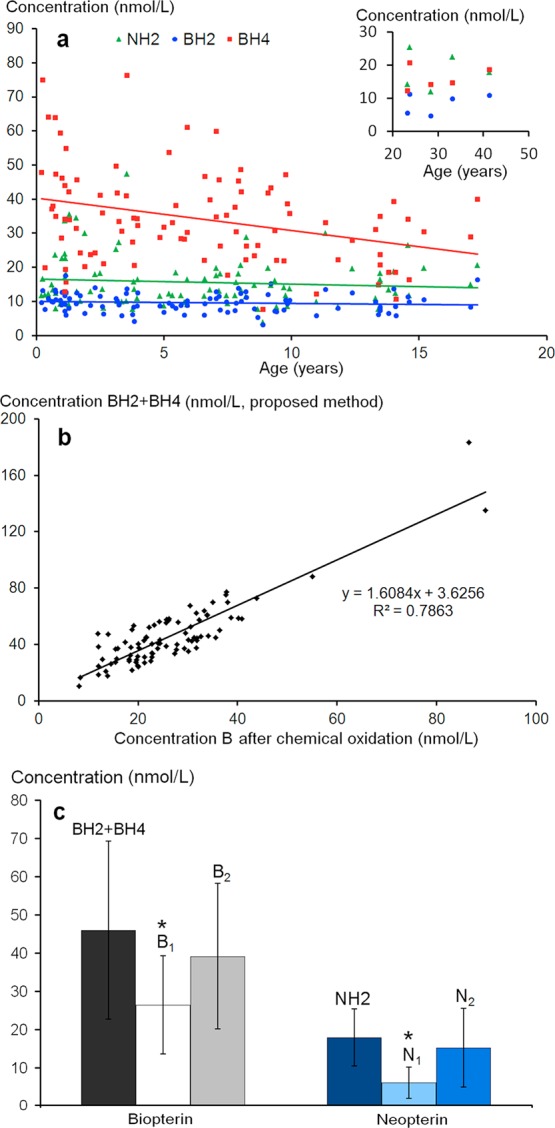
(a) Relationship between age and BH4 (y = −0.95x + 40.24; R2 = 0.106), BH2 (y = −0.057x + 10.00; R2 = 0.009), and NH2 (y = −0.15x + 16.53; R2 = 0.009) concentrations. (Inset: patients over 20 years.) (b) Comparison between B and N levels in 99 CSF samples determined by the proposed method (BH4 + BH2 = total biopterin) or by off-line chemical oxidation according to ref (25) . (c) Comparison between total biopterin (BH4+BH2) and NH2 as determined with the proposed method (see Figure 4); B1, B2 and N1, N2 refer to biopterin and neopterin levels as determined by offline chemical oxidation after calibration of the method with B and N calibrators or with BH4 and NH2 calibrators, respectively. Asterisk (*) indicates significant difference (p < 0.01) as compared to both methods used to determine (BH4 + BH2, and B1, and NH2, and N1, respectively).
Figure 6b shows that while there is a strong correlation between total B measured by both methods, the amounts of B and N determined by ICO are significantly lower than those determined by the proposed method (Figure 6c). This agrees with the assumption that total B measured by ICO underestimates the real amount of B and N.16,23 To confirm this assumption, we studied the ICO yields of the six pterins at three concentration levels. We observed that the slopes of the curves corresponding to equimolar solutions of BH4, BH2, and NH2 were weaker than those of B and N (Figure S-1). The calculated oxidation yields were approximately 63% for BH4 and BH2 and only about 41% for NH2, suggesting that the oxidation mechanism of reduced forms of pterins leads to a more complex mixture than a simple oxidation into B and N. As no other peaks were detected by fluorescence at the usual emission and excitation wavelengths, we assume that BH2, BH4, and NH2 might also give rise to nonfluorescent byproducts after chemical oxidation. These results agree with those previously observed.15,22
Figure 6c shows that using reduced forms of pterins to calibrate B and N determinations can attenuate the differences with the proposed PCCO-FD method. Nevertheless, as already reported23 within-run CVs for ICO are high irrespective of pterin concentration, they ranged from 6.4% to 35% (n = 6) under our experimental conditions.
Figure 7 shows the chromatographic profiles of CSF samples from patients with defects of pterin metabolism. The first panel (Figure 7a) shows the chromatogram of a CSF sample from a patient with a TH deficiency exhibiting a normal pterin profile. Figure 7b and c shows chromatograms associated with an AADC deficiency. It is noteworthy that this profile with normal pterin levels exhibited two additional intense peaks corresponding to 3 OMD (3-ortho-methyl-dihydroxyphenylalanine) and 5 OHT (5-hydroxytryptophan) in the presence of severe HVA (Homovanillic acid) and 5 HIAA (5-hydroxy-indole-acetic acid) deficiencies, which makes it characteristic of AADC deficiency. Panel (d) shows the chromatographic profile of a known SR deficiency.11,28 This chromatogram clearly shows the absence of BH4 accompanied by an elevated level of BH2. The chromatographic profile of panel (e) shows secondary BH4 and BH2 deficiencies in an adult suffering from extrapyramidal symptoms of unknown causes. In this patient, the NH2 CSF level was normal while the dopaminergic and serotoninergic deficiencies were non-DOPA responsive. Figure 7f shows the chromatographic profile of an example from five patients with very high levels of NH2, probably due to an intracerebral immune system activation.29
Figure 7.

Chromatographic profile obtained with the proposed method for CSF samples from patients with various metabolic disorders. (a) CSF sample from a patient with tyrosine hydroxylase deficiency. (b and c) CSF sample from a patient with amino acid decarboxylase deficiency showing two additional peaks corresponding to 3 OMD (3-ortho-methyl dopa) and 5 OHT (5-hydroxytryptophan). (d) CSF sample from a patient with sepiapterin reductase deficiency. (e) CSF sample from a patient with a secondary defect in BH4 synthesis. (f) CSF sample from a patient suffering from leukodystrophy of unknown origin. HPLC conditions: Atlantis dC18 (4.6 × 150 mm, 3 μm). Mobile phase: pH 7.4, 0.05 M sodium citrate/methanol (97/3, v/v). Flow rate: 0.6 mL/min at 30 °C. Oxidation: + 600 mV. Detection: λex 350 nm; λem 450 nm.
Taken together, these examples highlight the applicability of the proposed method for the diagnosis of pterin metabolic disorders.
Finally, Figure 8 shows the feasibility of the translation of the proposed method to ultrahigh performance liquid chromatography (UHPLC). This technique has been recently proposed for the quantification of neurotransmitters in microdialysis fractions.30 Due to extra-column effects, the separation is less efficient (hBH2 = 12) than that obtained with the conventional HPLC system. Nevertheless, if we assume that a resolution of about 1.3 between the unidentified peak X1 and BH4 is acceptable, the run time is shortened to about 3.5 min, which allows analyzing more than 100 samples during the stability period (6 h). Additional optimization and validation of this UHPLC method are in progress.
Figure 8.

Chromatographic profiles of a CSF sample (1, solid line) before and (2, dashed line) after spiking with with B (5 nM), N (5 nM), BH2 (20 nM), NH2 (20 nM), and BH4 (50 nM), showing the feasibility of the proposed method under UHPLC conditions. Chromatographic conditions: Acquity UPLC HSS T3 column (2.1 × 100 mm, 1.8 μm); flow rate: 0.5 mL/min at 30 °C. The mobile phase and the detection conditions are the same as for the proposed method (see Figure 4). X1 and X2 are unknown peaks.
Conclusions
An HPLC method for the simultaneous determination of circulating forms of biopterin and neopterin in cerebrospinal fluid is presented. The use of an embedded-polar-group bonded phase (Atlantis) allows separation of the target pterins without using an ion pairing reagent. Setting the pH of the mobile phase at 7.4 allows the postcolumn coulometric oxidation of the reduced pterin forms enabling their simultaneous determination by fluorescence detection.
All target pterins were quantified in a small volume (100 μL) of CSF using a single filtration step for sample preparation and analysis.
As the samples are stable for a maximum of 6 h in an autosampler at 10 °C and as the run time is 16 min (20 min for AADC deficiency), the number of samples including calibrators and quality control that can be analyzed per run is 20. Vitamin C or dithiotreithol (DTT) are usually added in large excess to stabilize BH4 in biological media.25 While vitamin C had no effect on BH4 stability under our chromatographic conditions, DTT entailed a significant increase of BH4 stability up to 10 h. However, this lengthened the run time by 14 min because 30 min was necessary to completely elute DTT. The translation of the proposed method to UHPLC is one avenue to improve overall throughput of this method.
Methods
Chemicals and Reagents
All reagents were purchased from Sigma (Saint-Quentin Fallavier, France) and were used without further purification.
Patient Samples
CSF samples were collected by lumbar puncture from patients suffering several neurological disorders with initially unknown etiology including movement disorders with or without encephalopathy, epileptic or neurodegenerative encephalopathy, and meningo-encephalitis.
Lumbar punctures were performed at the Department of Pediatric Neurology of the Hôpital Trousseau (Paris, France) as part of normal clinical management with the written informed consent of parents or legal representatives of each patient. This study using only CSF sample residues was performed according to French Public health regulations (Code de la santé publique - Article L1121-3, modified by Law n°2011-2012, December 29 2011 - Article 5).
Samples were collected in five fractions of 0.5 mL each. The first three fractions were processed for quantitative bacterial culture and routine biochemical tests, including amino acids, lactate, and pyruvate determinations. The two remaining fractions devoted to neurotransmitters, folate, and pterin metabolism were immediately frozen in liquid nitrogen and stored at −80 °C until analysis. To avoid a possible effect of the ventriculo-spinal gradient on CSF parameters, all pterin investigations were performed on the fourth fraction.
Exclusion criteria were traumatic punctures, inadequate collection and preservation of the samples, and L-dopa and BH4 treatment. Determinations of 5-methyl tetrahydrofolate by HPLC-FD25 and of neurotransmitter metabolites by HPLC-ECD27 were performed for all analyzed CSF samples before pterin determinations.
HPLC, MS, and NMR
Basic separations with diode array detection were performed on a Dionex Summit HPLC system (Les Ulis, France). For method development and pterin determinations, the HPLC system was coupled to PCCO-FD. A Jasco FP 920 detector equipped with a 16 μL standard cell was used to measure the fluorescence (λex 350 nm; λem 450 nm) of the oxidized products. PCCO was performed with a model 5011 cell controlled by a Coulochem 5100A module (ESA, France). For UHPLC experiments, we used a Waters ACQUITY UPLC system connected to an ESA Coulochem III electrochemical detector equipped with a coulometric cell model 6011 followed by an Acquity UPLC FLR detector.
Four columns were tested for chromatographic separations: a Zorbax Eclipse XDB-C18 (4.6 × 250 mm, 5 μm) from Agilent, an Atlantis dC18 (4.6 × 150 mm, 3 μm) protected by a precolumn filled with the same stationary phase, an XTerra C18 column (4.6 × 150 mm, 5 μm) both from Waters, and an ZIC-HILIC column (4.6 × 150 mm, 5 μm) from Merck. For UHPLC experiments, we used an Acquity UPLC HSS T3 (2.1 × 100 mm, 1.8 μm) column.
For RP-mode separations, the mobile phase was a mixture of 0.05 M citrate buffer and methanol (97/3 v/v). NaOH was used to adjust the pH to the required level. For HILIC-mode separations, the mobile phase was a mixture of 0.2 M ammonium formate buffer and acetonitrile (20/80 v/v).
HRMS studies were performed on an Exactive Orbitrap instrument equipped with an ESI system. NMR studies were performed on a Bruker Avance 400 MHz spectrometer.
Calibrators, Quality Control, and Sample Preparation
Stock standard solutions were prepared by dissolving about 400 μM B, N, BH2, and NH2 in 0.1 M HCl. For BH4, we added 30 mM ascorbic acid in 0.1 M HCl. Aliquots of stock solutions were then immediately stored at -80 °C until use.
For calibration, we used an artificial CSF matrix (A-CSF) consisting of 0.9% NaCl, 5 mM glucose, and 4 g L–1 albumin in a 10 mM NaH2PO4 buffer adjusted to pH 7.4 (with NaOH).
To plot the calibration curves, we spiked aliquots of A-CSF with N, NH2, B, BH2, and BH4 to give final concentrations of 5, 20, 50, and 100 nM of each pterin.
For recovery and precision studies, we used aliquots of a pooled CSF (p-CSF) sample (n = 10) spiked with 5, 20, 50, and 100 nM of each pterin. The basal pterin content of the p-CSF sample was determined through the standard-addition method.31 Aliquots of p-CSF frozen at −80 °C until analysis were used as quality control (QC).
For sample preparation, 100 μL of CSF sample or calibrator or QC sample were diluted (1:2 dilution ratio) in the mobile phase before filtration in a 5000 MWCO PES Vivaspin 500 filter (Sartorius, Aubagne, France) by centrifugation (10 min at 12 000g and 7 °C). Then 100 μL of the resulting filtrate were injected into the chromatograph.
Acidic iodine oxidations were performed as previously described.25
Statistics
The Shapiro–Wilk test was used to check the normality of data distribution. Comparisons were performed with a two-tailed Student’s t test, after checking the homogeneity of variances using Fisher’s test. P-values < 0.01 were considered statistically significant.
Glossary
Abbreviations
- BH4
tetrahydrobiopterin
- BH2
dihydrobiopterin
- NH2
dihydroneopterin
- CSF
cerebrospinal fluid
- HPLC
high performance liquid chromatography
- B
biopterin
- N
neopterin
- P
pterin
- ICO
indirect chemical oxidation
- FD
fluorescence detection
- ECD
electrochemical detection
- PCCO-FD
postcolumn coulometric oxidation-fluorescence detection
- LC-MS/MS
liquid chromatography coupled to tandem mass spectrometry
- SR
sepiapterin reductase
- AADC
amino acid decarboxylase
- TH
tyrosine hydroxylase
- HRMS
high resolution mass spectrometry
- NMR
nuclear magnetic resonance
- HILIC
hydrophilic interaction liquid chromatography
- LOD
limit of detection
- LOQ
limit of quantification
- CV
coefficient of variation
- 3 OMD
3 ortho-methyl dopa
- 5 OHT
5 hydroxy tryptophan
- HVA
homovanillic acid
- 5 HIAA
5 hydroxy indole acetic acid
- UHPLC
ultra high performance chromatography
- QC
quality control
- DTT
dithiothreitol
Supporting Information Available
Additional experimental data on method validation. This material is available free of charge via Internet at http://pubs.acs.org.
Author Contributions
Pierre Guibal, Nathalie Lévêque, and Nicolas Giraud performed research and analyzed data. Diane Doummar, Emmanuel Roze, Diana Rodriguez, and Thierry Billette De Villemeur managed children and collected CSF samples. Remy Couderc provided material support. Fathi Moussa designed research and analyzed data. Pierre Guibal and Fathi Moussa wrote the paper. All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Koshimura K.; Murakami Y.; Tanaka J.; Kato Y. (2000) The role of 6R-tetrahydrobiopterin in the nervous system. Prog. Neurobiol. 61, 415–438. [DOI] [PubMed] [Google Scholar]
- Hoekstra R.; Fekkes D. (2002) Pteridines and affective disorders. Acta Neuropsychiatr. 14, 120–126. [DOI] [PubMed] [Google Scholar]
- Perry M.; Li Q.; Kennedy R. T. (2009) Review of recent advances in analytical techniques for the determination of neurotransmitters. Anal. Chim. Acta 653, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau N., and Thöny B. (2008) Pterins and related enzymes. In Laboratory guide to the methods in biochemical genetics (Blau N., Duran M., and Gibson K. M., Eds.), pp 665–701, Springer, Berlin, Heidelberg. [Google Scholar]
- Fukushima T.; Nixon J. C. (1980) Analysis of reduced forms of biopterin in biological tissues and fluids. Anal. Biochem. 102, 176–188. [DOI] [PubMed] [Google Scholar]
- Komori H.; Matsuishi T.; Yamada S.; Ueda N.; Yamashita Y.; Kato H. (1999) Effect of age on cerebrospinal fluid levels of metabolites of biopterin and biogenic amines. Acta Paediatr. 88, 1344–1347. [DOI] [PubMed] [Google Scholar]
- Fiege B.; Ballhausen D.; Kierat L.; Leimbacher W.; Goriounov D.; Schircks B.; Thöny B.; Blau N. (2004) Plasma tetrahydrobiopterin and its pharmacokinetic following oral administration. Mol. Genet. Metab. 81, 45–51. [DOI] [PubMed] [Google Scholar]
- Werner E. R.; Werner-Felmayer G.; Wachter H. (1996) High-performance liquid chromatographic methods for the quantification of tetrahydrobiopterin biosynthetic enzymes. J. Chromatogr. B: Biomed. Sci. Appl. 684, 51–58. [DOI] [PubMed] [Google Scholar]
- Rilstone J. J.; Alkhater R. A.; Minassian B. A. (2013) Brain dopamine-serotonin vesicular transport disease and its treatment. N. Engl. J. Med. 368, 543–550. [DOI] [PubMed] [Google Scholar]
- Cerone R.; Schiaffino M. C.; Fantasia A. R.; Perfumo M.; Birk Moller L.; Blau N. (2004) Long-term follow-up of a patient with mild tetrahydrobiopterin-responsive phenylketonuria. Mol. Genet. Metab. 81, 137–139. [DOI] [PubMed] [Google Scholar]
- Leu-Semenescu S.; Arnulf I.; Decaix C.; Moussa F.; Clot F.; Boniol C.; Touitou Y.; Levy R.; Vidailhet M.; Roze E. (2010) Sleep and Rhythm Consequences of a Genetically Induced Loss of Serotonin. Sleep 33, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin C. R. 2nd; Hyland K.; Randall R.; Ficicioglu C. (2012) Dihydropteridine reductase deficiency and treatment with tetrahydrobiopterin: a case report. J. Inherited Metab. Dis. Rep. 202, 53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K.; Owada M. (1991) A simple and sensitive method for the determination of pterins in cerebrospinal fluid. Clinical usefulness for management of tetrahydrobiopterin deficiency. J. Inherited Metab. Dis. 14, 825–830. [DOI] [PubMed] [Google Scholar]
- Tani Y.; Ohno T. (1993) Analysis of 6R- and 6S-tetrahydrobiopterin and other pterins by reversed-phase ion-pair liquid-chromatography with fluorimetric detection by post-column sodium nitrite oxidation. J. Chromatogr. 617, 249–255. [DOI] [PubMed] [Google Scholar]
- Tani Y.; Kanai T. (1994) Decrease in 6R-5,6,7,8-tetrahydrobiopterin content in cerebrospinal fluid of autistic patients. Neurosci. Lett. 181, 169–172. [DOI] [PubMed] [Google Scholar]
- Espinosa-Mansilla A.; Muñoz de la Peña A.; Cañada-Cañada F.; Mancha de Llanos A. (2008) LC determination of biopterin reduced forms by UV-photogeneration of biopterin and fluorimetric detection. Talanta 77, 844–851. [Google Scholar]
- Cañada-Cañada F.; Espinosa-Mansilla A.; Muñoz de la Peña A.; Mancha de Llanos A. (2009) Determination of marker pteridins and biopterin reduced forms, tetrahydrobiopterin and dihydrobiopterin, in human urine, using a post-column photoinduced fluorescence liquid chromatographic derivatization method. Anal. Chim. Acta 648, 113–122. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Cao J.; Chen Y. S.; Zhu Y.; Patrick C.; Chien B.; Cheng A.; Foehr E. D. (2009) Detection of tetrahydrobiopterin by LC–MS/MS in plasma from multiple species. Bioanalysis 1, 895–903. [DOI] [PubMed] [Google Scholar]
- Kim H. L.; Kim D. H.; Lee Y. K.; Park S. O.; Lee Y. W.; Kwon O. S.; Park Y. S. (2010) An enzymatic method to distinguish tetrahydrobiopterin from oxidized biopterins using UDP-glucose:tetrahydrobiopterin glucosyltransferase. Anal. Biochem. 397, 79–83. [DOI] [PubMed] [Google Scholar]
- Kim H. R.; Kim T. H.; Hong S. H.; Kim H. G. (2012) Direct detection of tetrahydrobiopterin (BH4) and dopamine in rat brain using liquid chromatography coupled electrospray tandem mass spectrometry. Biochem. Biophys. Res. Commun. 419, 632–637. [DOI] [PubMed] [Google Scholar]
- Fismen L.; Eide T.; Djurhuus R.; Svardal A. M. (2012) Simultaneous quantification of tetrahydrobiopterin, dihydrobiopterin and biopterin by liquid chromatography coupled electrospray tandem mass spectrometry. Anal. Biochem. 430, 163–170. [DOI] [PubMed] [Google Scholar]
- Jiménez Girón A.; Martín-Tornero E.; Hurtado Sánchez M. C.; Durán Merás I.; Espinosa-Mansilla A. (2012) A simple HPLC-ESI-MS method for the direct determination of ten pteridinic biomarkers in human urine. Talanta 101, 465–472. [DOI] [PubMed] [Google Scholar]
- Hyland K. (1985) Estimation of tetrahydro, dihydro and fully oxidised pterins by high-performance liquid chromatography using sequential electrochemical and fluorometric detection. J. Chromatogr., Biomed. Appl. 343, 35–41. [DOI] [PubMed] [Google Scholar]
- Howells D. W.; Smith I.; Hyland K. (1986) Estimation of tetrahydrobiopterin and other pterins in cerebrospinal fluid using reversed-phase high-performance liquid chromatography with electrochemical and fluorescence detection. J. Chromatogr., Biomed. Appl. 381, 285–294. [DOI] [PubMed] [Google Scholar]
- Verbeek M. M.; Blom A. M.; Wevers R. A.; Lagerwerf A. J.; Van de Geer J.; Willemsen M. A. A. P. (2008) Technical and biochemical factors affecting cerebrospinal fluid 5-MTHF, biopterin and neopterin concentrations. Mol. Genet. Metab. 95, 127–132. [DOI] [PubMed] [Google Scholar]
- Hyland K.; Surtees R. A. H.; Heales S. J. R.; Bowron A.; Howells D. W.; Smith I. (1993) Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr. Res. 34, 10–14. [DOI] [PubMed] [Google Scholar]
- Ormazabal A.; Garcia-Cazorla A.; Fernandez Y.; Fernandez-Alvarez E.; Campistol J.; Artuch R. (2005) HPLC with electrochemical and fluorescence detection procedures for the diagnosis of inborn errors of biogenic amines and pterins. J. Neurosci. Methods 142, 153–158. [DOI] [PubMed] [Google Scholar]
- Neville B. G.; Parascandalo R.; Farrugia R.; Felic A. (2005) Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain 128, 2291–2296. [DOI] [PubMed] [Google Scholar]
- Murr C.; Widner B.; Wirleitner B.; Fuchs D. (2002) Neopterin as a marker for immune system activation. Curr. Drug Metab. 3, 175–187. [DOI] [PubMed] [Google Scholar]
- Reinhoud N. J.; Brouwer H.-J.; Van Heerwaarden L. M.; Korte-Bouws G. A. H. (2013) Analysis of glutamate, GABA, noradrenaline, dopamine, serotonin and metabolites using microbore UHPLC with electrochemical detection. ACS Chem. Neurosci. 4, 888–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog D. A., West D. M., and Holler F. J. (1996) In Fundamentals of Analytical Chemistry, 7th ed., pp 572–575, International ed., Saunders College Publishing, Philadelphia. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.