

Abstract

Recent progress in the discovery of mGlu1 allosteric modulators has suggested the modulation of mGlu1 could offer possible treatment for a number of central nervous system disorders; however, the available chemotypes are inadequate to fully investigate the therapeutic potential of mGlu1 modulation. To address this issue, we used a fluorescence-based high-throughput screening assay to screen an allosteric modulator-biased library of compounds to generate structurally diverse mGlu1 negative allosteric modulator hits for chemical optimization. Herein, we describe the discovery and characterization of a novel mGlu1 chemotype. This series of succinimide negative allosteric modulators, exemplified by VU0410425, exhibited potent inhibitory activity at rat mGlu1 but was, surprisingly, inactive at human mGlu1. VU0410425 and a set of chemically diverse mGlu1 negative allosteric modulators previously reported in the literature were utilized to examine this species disconnect between rat and human mGlu1 activity. Mutation of the key transmembrane domain residue 757 and functional screening of VU0410425 and the literature compounds suggests that amino acid 757 plays a role in the activity of these compounds, but the contribution of the residue is scaffold specific, ranging from critical to minor. The operational model of allosterism was used to estimate the binding affinities of each compound to compare to functional data. This novel series of mGlu1 negative allosteric modulators provides valuable insight into the pharmacology underlying the disconnect between rat and human mGlu1 activity, an issue that must be understood to progress the therapeutic potential of allosteric modulators of mGlu1.
Keywords: Allosteric modulator, glutamate, mGlu1, metabotropic
Glutamate is the major excitatory neurotransmitter in the central nervous system, exerting its effects through the activation of two classes of glutamate receptors, the ionotropic and the metabotropic glutamate (mGlu) receptors. The mGlu receptors belong to the family C G protein coupled receptors (GPCRs) and distinguish themselves from other GPCRs by the presence of a large extracellular N-terminal agonist binding domain. There are eight known subtypes of the mGlu receptor family, divided into three groups based on sequence homology, pharmacology, and coupling to downstream signaling pathways.1 Group I mGlu receptors include mGlu1 and mGlu5, are primarily localized postsynaptically, and couple to Gαq and subsequent increases in intracellular calcium. Group II (mGlu2 and mGlu3) and group III (mGlu4, mGlu6, mGlu7, and mGlu8) mGlu receptors are largely presynaptic and couple to Gαi/o and associated effectors such as inhibition of adenylyl cyclase.
Previous studies suggest that modulation of mGlu1 could offer possible treatment for a number of central nervous system (CNS) disorders including addiction,2−4 anxiety,5−7 epilepsy,8,9 pain,5,10−12 and psychotic disorders.13−16 BAY36-7620 (1) (Figure 1) was one of the first mGlu1 negative allosteric modulators (NAMs) shown to be centrally active;17−19 however, its potency is too low to be of use therapeutically. JNJ1625968520 (2) is also systemically active and was shown to be efficacious in a rat model of anxiety7 and various models of addiction.3 Antipsychotic activity has also been observed for mGlu1 negative allosteric modulators, including 5-(1-(2,4-difluorophenyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-isopropylisoindolin-1-one (DFMTI) (3), which was efficacious in disrupting prepulse inhibition when dosed orally in rats.15,16 In addition, recent studies have highlighted the possibility that mGlu1 plays a role in the development of melanoma21−23 and certain types of breast cancer,24 and it has been proposed that mGlu1 NAMs could offer a novel therapeutic avenue for treatment of these cancers.
Figure 1.

mGlu1 NAM compounds in the literature.
Recent progress in the discovery of mGlu1-selective NAMs has been exciting; however, the number and diversity of chemotypes available to investigate the role of mGlu1 for potential therapeutics greatly lags behind other mGlu receptors such as mGlu5.25,26 Recent advancements targeting discovery of novel chemotypes include the novel piperazine, VU0469650 (4), which demonstrated excellent potency and selectivity as well as good CNS exposure following intraperitoneal dosing in rats.27 Chemical optimization also produced a related novel scaffold exemplified by VU0470300 (5), a promising lead for future development.28 To further address this issue, we used a fluorescence-based high-throughput screening assay to screen an allosteric modulator-biased library of compounds to generate novel, structurally diverse mGlu1 NAM hits for chemical optimization. Herein, we describe the discovery of a novel mGlu1 chemotype, exemplified by compound 6 (VU0410425) (Figure 2). This succinimide-based series of NAMs exhibited potent inhibitory activity at rat mGlu1. Interestingly, the series did not display significant antagonist activity when tested at human mGlu1. This dichotomy prompted us to further characterize the activity of VU0410425 and other diverse mGlu1 NAMs at mutant rat and human mGlu1 receptors. We aimed to provide a better understanding of the molecular components of mGlu1 species selectivity, a critical aspect of the development of therapeutics based on mGlu1 receptor modulation.
Figure 2.

mGlu1 NAM succinimide hit.
Results and Discussion
Novel mGlu1 Scaffold Discovered through Screening of Allosteric Modulator-Biased Library
In recent years, mGlu1 NAMs have been developed for potential treatment of CNS disorders as well as for cancers including melanoma and certain types of breast cancer. Figure 1 highlights some of the mGlu1 NAM chemotypes investigated to date. In pursuit of structurally diverse mGlu1 NAMs, we utilized a functional cell-based assay that measures rat mGlu1 receptor-induced mobilization of intracellular calcium to screen an in-house library of allosteric modulator-biased compounds. Succinimide hit 6 (VU0410425) (Figure 2) was identified, exhibiting an initial mGlu1 potency of 229 nM. This compound was originally described as a positive allosteric modulator (PAM) of mGlu4 during the development of VU0400195, an mGlu4 PAM with oral efficacy in an antiparkinsonian animal model.29 In the mGlu4 setting, compounds within this scaffold demonstrated excellent pharmacokinetic profiles and brain penetration in rodents, indicating promise as a lead for mGlu1. Resynthesis of the compound confirmed activity, leading to an optimization program based on succinimide 6.
Chemical Optimization of Succinimide Hit 6 (VU0410425)
The mGlu1 NAM optimization effort of this succinimide scaffold was initiated with a functional cell-based assay using cells expressing rat mGlu1,27,28 and structure–activity relationship (SAR) trends are presented using results from that assay herein, as well as functional data that was subsequently collected in an analogous assay run with cells expressing human mGlu1. Recognizing that the picolinamide was a preferred moiety for engendering mGlu4 PAM activity, we began development of SAR around mGlu1 NAM activity in that region (R3) of the chemotype, seeking potent modifications (Table 1). Concomitant to that work, a limited evaluation of the impact of substitution on the phenyl core adjacent to the succinimide group (R1 and R2) was made. We quickly discovered that the des-chloro analogue (7) of hit compound 6 was only marginally less potent. While acetamide 8 was only moderately potent, the isobutyramide in analogue 9 proved to be a competent replacement for the picolinamide moiety. The mGlu1 potency of analogue 9 was further enhanced through addition of chloro groups adjacent to the succinimide moiety (10 and 11). Several analogues with substituted benzamides were prepared in the context of the unsubstituted phenyl core (R1 = R2 = H), and the 3-substituted derivatives (12–14) demonstrated excellent potency. Interestingly, 4-fluorobenzamide 15 was a clear partial antagonist with a CRC that plateaued well above baseline. Surprisingly, while several analogues in this series demonstrated excellent potency at rat mGlu1, most analogues were inactive up to the top concentration tested (30 μM) at human mGlu1. The only exceptions were compounds 9 and 10, which were weak antagonists, and analogue 11, which was a partial antagonist with modest potency. Even these three compounds were substantially less potent (>20-fold) at human mGlu1 than rat mGlu1.
Table 1. Initial Amide and Phenyl Core SAR.
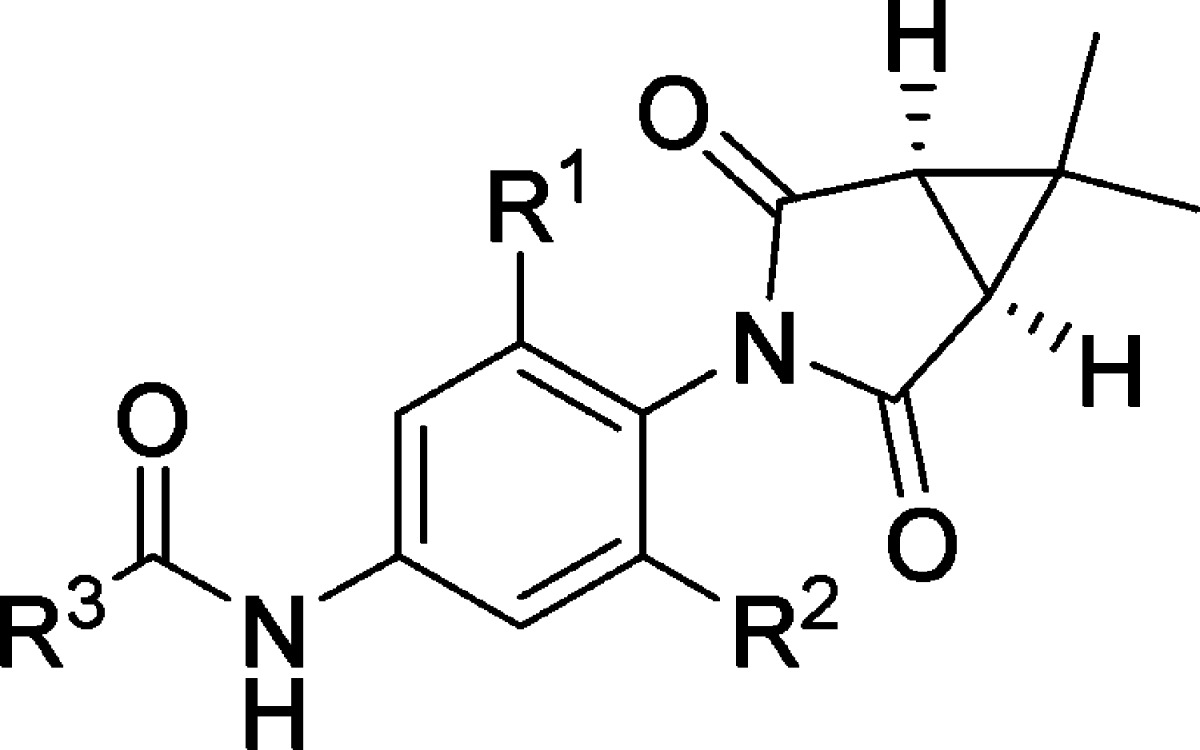
| entry | R1 | R2 | R3 | rat mGlu1 pIC50 (±SEM)a | rat mGlu1 IC50 (nM) | rat % Glu Max (±SEM)ab | human mGlu1 pIC50 (±SEM)a | human mGlu1 IC50 (nM) | human % Glu Max (±SEM)ab |
|---|---|---|---|---|---|---|---|---|---|
| 6 | Cl | H | 2-pyridyl | 6.85 ± 0.02 | 140 | 2.4 ± 0.1 | <4.5 | >30 000 | |
| 7 | H | H | 2-pyridyl | 6.66 ± 0.02 | 220 | 2.0 ± 0.3 | <4.5 | >30 000 | |
| 8 | H | H | methyl | 5.46 ± 0.17 | 3480 | 2.9 ± 1.3 | <4.5 | >30 000 | |
| 9 | H | H | isopropyl | 6.60 ± 0.03 | 249 | 1.4 ± 0.2 | <5.0c | >10 000 | 55.1 ± 7.1 |
| 10 | Cl | H | isopropyl | 6.69 ± 0.02 | 204 | 2.9 ± 0.5 | <5.0c | >10 000 | 44.5 ± 8.1 |
| 11 | Cl | Cl | isopropyl | 6.87 ± 0.05 | 136 | 3.0 ± 0.6 | 5.54 ± 0.02 | 2910 | 62.9 ± 2.2 |
| 12 | H | H | 3-fluorophenyl | 7.03 ± 0.08 | 94 | 3.0 ± 0.7 | <4.5 | >30 000 | |
| 13 | H | H | 3-chlorophenyl | 6.92 ± 0.05 | 122 | 8.1 ± 5.2 | <4.5 | >30 000 | |
| 14 | H | H | 3-methylphenyl | 7.13 ± 0.02 | 75 | 3.3 ± 0.7 | <4.5 | >30 000 | |
| 15 | H | H | 4-fluorophenyl | 6.72 ± 0.08 | 189 | 45.0 ± 10.4 | <4.5 | >30 000 | |
| 16 | H | H | 4-chlorophenyl | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 17 | H | H | 4-methylphenyl | <4.5 | >30 000 | <4.5 | >30 000 |
Values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
CRC does not plateau.
Having established isobutyramide 9 as a potent mGlu1 NAM at the rat receptor, that moiety was held constant to evaluate alternative succinimide groups (Table 2). Unfortunately, none of these modifications were tolerated (18–22), illustrating a high sensitivity to modification in that portion of the chemotype. These new succinimide analogues were also inactive up to the top concentration tested at human mGlu1. Finally, returning to the amide region of the chemotype, the effects of reversing the amide bond were examined (Table 3). Such a modification was thought advantageous as it would lead to a less electron-rich phenyl core that lacked the potential to form reactive quinone metabolites. The simple N-phenyl derivative 23 offered an encouraging early result with moderate potency; however, the N-methyl analogue 24 was inactive up to the top concentration tested. Substitution of the N-phenyl ring of 23 was subsequently evaluated (25–31) with some positive results. Both 2-fluorophenyl analogue 25 and 3-chlorophenyl analogue 29 demonstrated enhanced potency relative to unsubstituted comparator 23. Turning our attention to aliphatic amine analogues (32–38) was less effective at improving upon 23. Unfortunately, the only aliphatic amine analogue with submicromolar mGlu1 NAM activity was N-3,3-dimethylbutyl analogue 38. Not surprisingly, none of these reverse amides demonstrated appreciable potency at human mGlu1. In fact, only analogues 30 and 38 demonstrated any measurable human mGlu1 antagonist activity; however, these compounds were very weak. We examined the selectivity profiles of all active analogues relative to mGlu4 and mGlu5, the receptor subtypes most likely to show overlap with mGlu1 activity, by testing a range of concentration of compound in a calcium mobilization assay. Compound 7, des-chloro analogue of 6, exhibited weak mGlu4 PAM activity (IC50 > 10 μM) but all other analogues of 6 were inactive at mGlu4 and mGlu5 up to 30 μM, the highest concentration tested. Analog 9 was selected as a representative of the series for testing for selectivity against the other members of the mGlu family. Gratifyingly, 9 was devoid of any activity as evidenced by the inability of a 10 μM concentration of the compound to induce a shift in the agonist concentration–response curve (CRC) in cells expressing mGlus2–8.
Table 2. Succinimide SAR.
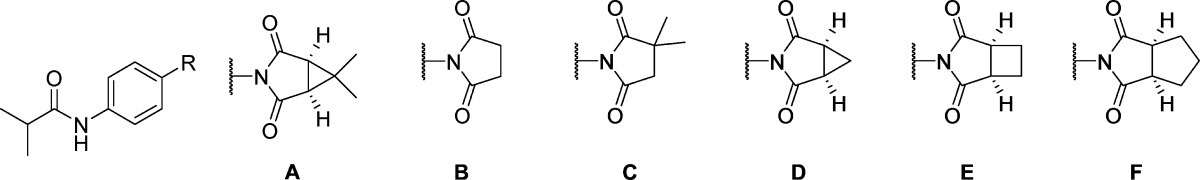
| entry | R | rat mGlu1 pIC50 (±SEM)a | rat mGlu1 IC50 (nM) | rat % Glu Max (±SEM)ab | human mGlu1 pIC50 (±SEM)a | human mGlu1 IC50 (nM) | human % Glu Max (±SEM)ab |
|---|---|---|---|---|---|---|---|
| 9 | A | 6.60 ± 0.03 | 249 | 1.4 ± 0.2 | <5.0c | >10 000 | 55.1 ± 7.1 |
| 18 | B | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 19 | C | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 20 | D | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 21 | E | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 22 | F | <4.5 | >30 000 | <4.5 | >30 000 |
Values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
CRC does not plateau.
Table 3. Reverse Amide SAR.
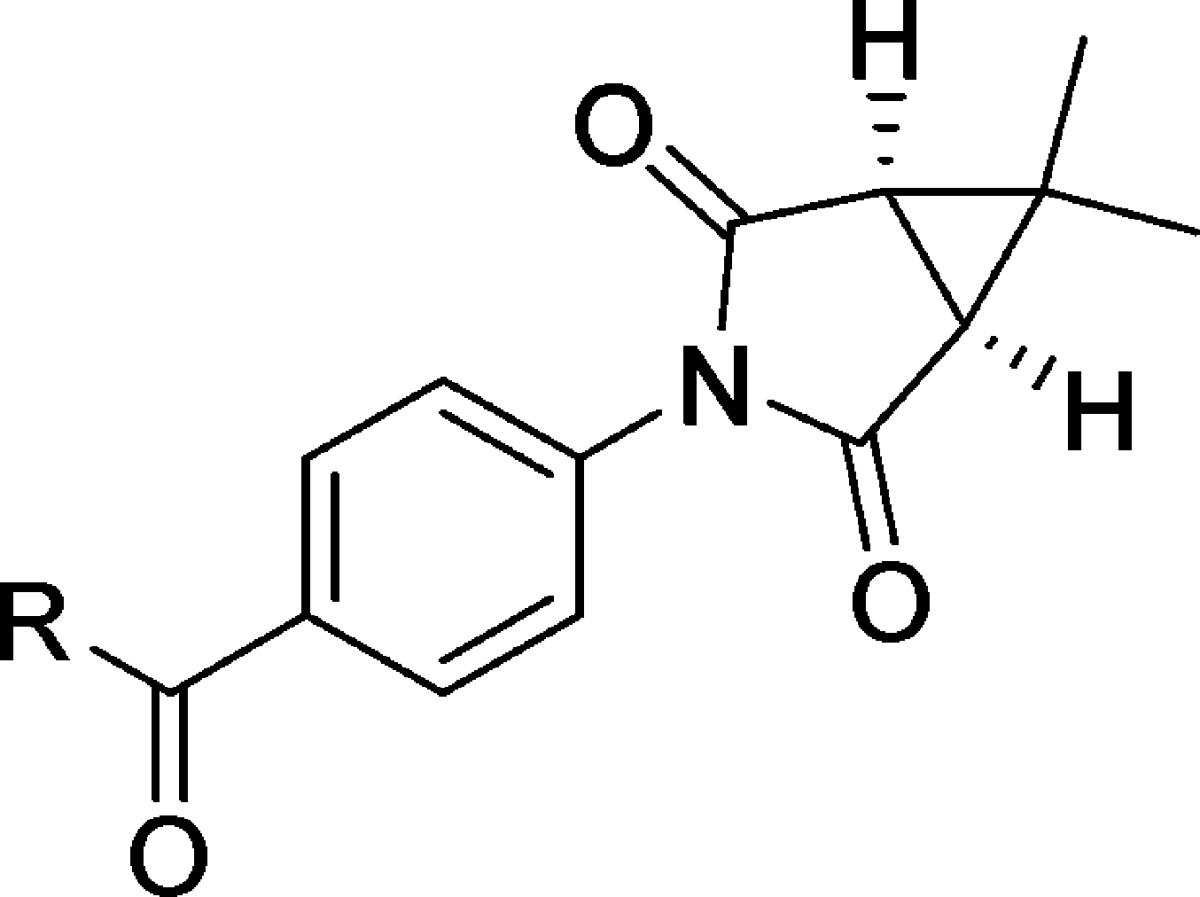
| entry | R | rat mGlu1 pIC50 (±SEM)a | rat mGlu1 IC50 (nM) | rat % Glu Max (±SEM)ab | human mGlu1 pIC50 (±SEM)a | human mGlu1 IC50 (nM) | human % Glu Max (±SEM)ab |
|---|---|---|---|---|---|---|---|
| 23 | N-phenyl | 5.92 ± 0.10 | 1190 | 4.8 ± 1.7 | <4.5 | >30 000 | |
| 24 | N-methyl-N-phenyl | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 25 | N-2-fluorophenyl | 6.64 ± 0.10 | 230 | 2.5 ± 0.7 | <4.5 | >30 000 | |
| 26 | N-2-chlorophenyl | 5.93 ± 0.18 | 1180 | 2.2 ± 0.6 | <4.5 | >30 000 | |
| 27 | N-2-methylphenyl | <5.0c | >10 000 | 21.1 ± 7.7 | <4.5 | >30 000 | |
| 28 | N-3-fluorophenyl | 6.11 ± 0.17 | 768 | 9.5 ± 3.3 | <4.5 | >30 000 | |
| 29 | N-3-chlorophenyl | 6.35 ± 0.03 | 443 | 58.9 ± 12.3 | <4.5 | >30 000 | |
| 30 | N-3-methylphenyl | 5.74 ± 0.25 | 1830 | 1.1 ± 0.8 | <5.0c | >10 000 | 69.9 ± 2.7 |
| 31 | N-4-fluorophenyl | 5.96 ± 0.01 | 1100 | 31.8 ± 3.1 | <4.5 | >30 000 | |
| 32 | N-isopropyl | <5.0c | >10 000 | 19.3 ± 8.2 | <4.5 | >30 000 | |
| 33 | N-cyclohexyl | 5.80 ± 0.02 | 1570 | 5.4 ± 0.4 | <4.5 | >30 000 | |
| 34 | N-(trans)-4-methylcyclohexyl | <4.5 | >30 000 | <4.5 | >30 000 | ||
| 35 | N-cycloheptyl | <5.0c | >10 000 | 20.7 ± 5.6 | <4.5 | >30 000 | |
| 36 | (R)-N-1-cyclohexylethyl | <5.0c | >10 000 | 62.0 ± 5.2 | <4.5 | >30 000 | |
| 37 | (S)-N-1-cyclohexylethyl | <5.0c | >10 000 | 16.9 ± 7.8 | <4.5 | >30 000 | |
| 38 | N-3,3-dimethylbutyl | 6.13 ± 0.05 | 735 | 3.1 ± 0.4 | <5.0c | >10 000 | 70.0 ± 3.1 |
Values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3.
CRC does not plateau.
Compounds That Potently Inhibit Glutamate-Induced Calcium Mobilization at Rat mGlu1 Exhibit Distinct Functional Effects at Human mGlu1
While SAR is often developed using rat or mouse receptors since many behavioral models are performed in rodents, it is important that compounds are also active in the human receptor as the ultimate goal is development of human therapeutics. As described above, the succinimide series exhibited quite distinct human activity compared to rat, with the majority of compounds having no appreciable activity at the human receptor. A small number of compounds did exhibit inhibitory activity and Figure 3 illustrates examples of three subtle, but distinct, types of human mGlu1 activity when compared to the corresponding rat mGlu1 activity.
Figure 3.

Succinimide analogues exhibit a large disconnect between rat and human mGlu1 activity in a calcium mobilization assay using cells expressing rat (closed triangles) and human (closed circles) mGlu1.
Original hit 6 (Figure 3a) exemplifies the majority of the compounds, which acted as potent, full antagonists at the rat receptor but showed no activity at human mGlu1 at concentrations up to 30 μM, the highest concentration tested. In Figure 3b, 10 highlights a second category of compounds, those that showed significantly weaker human mGlu1 potency compared to rat. The potency difference between rat and human mGlu1 was greater than 50-fold in this case. 11 (Figure 3c) acted as a full antagonist at the rat receptor, but displayed weak, partial antagonist activity when tested in cells expressing human mGlu1 receptor. While not previously noted for an mGlu1 NAM, the partial antagonist profile has been documented for a number of mGlu5 NAMs from a variety of different scaffolds and it has proven difficult to predict this pharmacological profile using SAR, even within a scaffold.30−35 The partial antagonist activity exhibited by this compound (and 15, Table 1) was distinct from the weak antagonist activity seen with 10 in that 11 displayed a clear plateau in the CRC, reaching a maximal inhibition of approximately 60% maximal glutamate response. The difference between a partial and full antagonist in vivo is unknown at this time; however, it is hypothesized that advantages such as an improved side effect profile could be possible by tailoring the desired level of inhibition of a partial antagonist.33 While potency differences between species are common, we were surprised by the dramatic lack of activity by the majority of compounds in the succinimide series. There are reports of mGlu1 NAMs and PAMs showing specificity for rat versus human mGlu1,36,37 but a comprehensive comparison of multiple chemical scaffolds across both species has not been discussed in the literature. This prompted an investigation into the species differences of both the novel succinimide series of mGlu1 NAMs as well as a set of chemically diverse, previously published mGlu1 NAMs. VU0410425 and analogue 11 were selected as succinimide NAMs to represent the series in a more detailed pharmacological characterization.
Previously Identified mGlu1 NAMs Exhibit Multiple Profiles Across Species
We set out to test VU0410425 and the other mGlu1 chemotype NAMs in the calcium mobilization assay at both rat and human mGlu1 receptors to determine if the disparity in the potency of compounds between rat and human was specific to the succinimide scaffold or more widespread. A set of chemically diverse mGlu1 NAMs were either purchased from commercial vendors or synthesized in-house according to literature procedures. These NAMs were reported to exhibit rat mGlu1 potencies ranging from 2.1 to 160 nM, specifically, 7,9-diamino-3-(p-tolyl)thieno[2,3-d:4,5-d′]dipyrimidin-4(3H)-one (DATTP)38 and BAY36-7620 (1), respectively. In the case of BAY36-7620, a significant decrease in potency has previously been reported at human mGlu1 when compared to rodent receptor activity.39 Results are summarized in Table 4. Figure 4 depicts four CRCs that exemplify the variety of profiles displayed by this set of mGlu1 NAMs. DATTP (Figure 4a) stood alone as the only compound that exhibited enhanced human potency (27 nM) compared to rat (56 nM), a small 2-fold increase. The rat and human potencies of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide (FITM)15,40 were equivalent (23 versus 28 nM) as seen in Figure 4b. LY456236,8,41 DFMTI, and JNJ16259685 all exhibited a moderate decrease in human potency of approximately 3-fold when compared to rat, exemplified by DFMTI in Figure 4c. The remaining compounds (R214127,42 5-(4-(hydroxymethyl)piperidin-1-yl)-N-((1r,4r)-4-methylcyclohexyl)pyrazine-2-carboxamide (HPMCP),43N-cycloheptylthieno[2,3-d]pyrimidin-4-amine (CHTPA),44 BAY36-7620, and VU0412425) all displayed a significant disconnect between rat and human potency, approaching or above a 10-fold difference, with BAY36-7620 and VU0410425 showing the most significant difference of >30-fold (Figure 4d). The range of profiles seen within this small set of diverse mGlu1 NAMs emphasizes the importance of testing compounds at multiple species of receptor. While the objective is activity in humans, activity at rodent receptors is often critical for preclinical evaluation. For our purposes, we altered our strategy to incorporate screening at human mGlu1 as our tier one assay followed by periodic testing at rat mGlu1 for compounds being considered for testing in rodent models. These findings prompted additional investigation of the molecular differences between the rat and human mGlu1 receptors.
Table 4. Literature mGlu1 NAM Activitya.
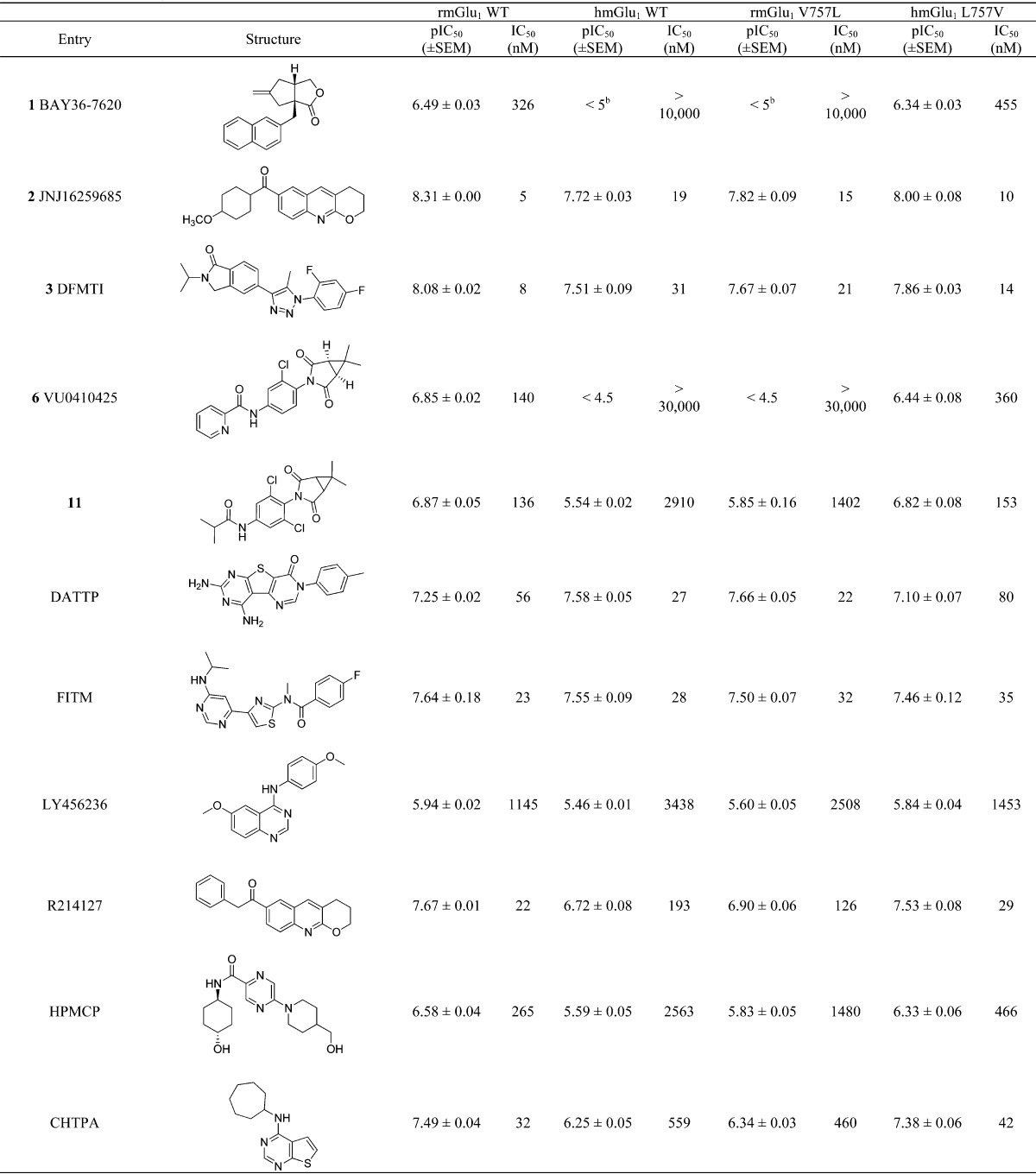
Values are average of n ≥ 3.
CRC does not plateau.
Figure 4.

Literature mGlu1 NAMs exhibit multiple profiles across species in a calcium mobilization assay using cells expressing rat (closed triangles) and human (closed circles) mGlu1. (a) DATTP exhibits enhanced human potency (27 nM) compared to rat (56 nM). (b) FITM has equivalent potency at human and rat mGlu1 (28 vs 23 nM). (c) DFMTI exhibits a moderate decrease in human potency (31 nM) compared to rat (8 nM). (d) BAY36-7620 displays a significant decrease in activity at human mGlu1 compared to rat (>30-fold).
mGlu1 NAM Activity Depends upon the Amino Acid Present at Position 757
As previously described, only one amino acid in the transmembrane domain (TMD) differs between rat and human mGlu1, position 757 (rmGlu1 V757 and hmGlu1 L757).36,37,45 In fact, this leucine is conserved across all other human mGlu receptors including the other group I receptor, mGlu5. We hypothesized that this residue may play a critical role in the activity of these mGlu1 NAMs and could shed light into the structural determinants that differentiate their species profile. We constructed stable cell lines containing a valine to leucine mutation in the case of rat mGlu1 (rmGlu1 V757L) or leucine to valine mutation in the case of human mGlu1 (hmGlu1 L757V). For compounds that maintained similar potencies between species, we expected no change in potency value when tested on the mutant receptors. For compounds with moderate to large differences between rat and human potencies, we anticipated loss of activity at rmGlu1 V757L to levels similar to humanlike activity, and at hmGlu1 L757V, gain of activity to levels comparable to ratlike activity. Table 4 presents a comparison of calcium assay results using HEK cells expressing either rat mGlu1 (WT or V757L) or human mGlu1 (WT or L757V). In rmGlu1 V757L, the potency of DATTP was enhanced 2–3-fold (56 vs 22 nM), a value consistent with the hmGlu1 WT potency (27 nM). In hmGlu1 L757V, the potency was decreased to a value of 80 nM, a number in line with the rat WT potency. Consistent with WT activity, the rat and human potencies of FITM were equivalent in the mutants, suggesting that residue 757 does not play a role in the activity of this compound. The mutations had a small, but opposite, effect on the potencies of mGlu1 NAMs LY456236, DFMTI, and JNJ16259685. For each compound, we saw a 2–3-fold decrease in potency when tested at rmGlu1 V757L, with IC50 values nearing those obtained when tested at hmGlu1 WT. When tested in cells expressing hmGlu1 L757V, an increase in potency was observed, in each case returning to a value consistent with that obtained at rmGlu1 WT. The mutations had a more robust effect on the potencies of R214127, HPMCP, CHTPA, BAY36-7620, VU0410425, and compound 11. When tested at rmGlu1 V757L, large decreases in potency were observed when compared to WT: R214127 (6-fold), HPMCP (6-fold), CHTPA (14-fold), and nearly abolished activity in the case of BAY36-7620 (>30-fold). A complete loss of activity was observed for VU0410425 when rmGlu1 WT was compared to V757L. The hmGlu1 L757V mutant was able to rescue activity in each case to levels comparable to those of rmGlu1 WT. In the case of compound 11, a large decrease in potency (10-fold) as well as efficacy (53% maximal glutamate) was observed when tested at rmGlu1 V757L, reflecting a retention of the partial antagonist activity observed in hmGlu1 WT. The hmGlu1 L757V mutant was able to return both potency and efficacy levels to values comparable to those of rmGlu1 WT. With the exception of FITM, the mutants had a clear effect on the activities of the mGlu1 NAMs. At the rmGlu1 V757L mutant, a dramatic reduction in potency was evident for CHTPA, BAY36-7620, and VU0410425, whereas at the hmGlu1 L757V an enhancement in potency was observed for these compounds. These results suggested that residue 757 is critical for the NAM activity of CHTPA, BAY36-7620, and VU0410425. Figure 5a and b depicts the CRCs for BAY36-7620 and VU0410425, exemplifying this mode of activity. For the majority of other compounds, the mutants had a subtle, but consistent, effect as described above, suggesting that residue 757 is likely to play a role in the NAM activity of these compounds; however, other residues also must contribute in some manner. DFMTI is representative of this category of activity (see Figure 5c).
Figure 5.

mGlu1 NAM activity of certain NAMs is dependent on residue 757 in a calcium mobilization assay using cells expressing rat (closed triangles) and human (closed circles) WT mGlu1 and rat V757L (open triangles) and human L757V (open circles) mGlu1. (a) A large decrease in potency is observed at rmGlu1 V757L compared to WT for BAY36-7620 which is rescued by hmGlu1 L757V. (b) A complete loss of activity is observed at rmGlu1 V757L compared to WT for VU0410425. The hmGlu1 L757V mutant restores activity to levels similar to those seen for rmGlu1 WT. (c) A small decrease in potency is observed for DFMTI at rmGlu1 V757L (21 nM) compared to WT (8 nM). The hmGlu1 L757V mutant restores activity to levels similar to those seen for rmGlu1 WT.
The Presence of Valine or Leucine in Position 757 Is Important for the Species Selectivity of Some mGlu1 NAMs
If an antagonist acts as a noncompetitive inhibitor of mGlu1, increasing concentrations of compound should shift the glutamate concentration response curve to the right and decrease the maximal signal of glutamate. We further evaluated the contribution of residue 757 by examining NAM effects in this manner. If NAM activity is dependent on residue 757, then mutation of this residue should disrupt or decrease the rightward shift of the glutamate CRC in the presence of NAM. Three compounds were selected for progressive fold shift analyses that represent different chemical scaffolds and modes of activity. BAY36-7620 and DFMTI were selected to exemplify literature compounds with high and low dependence on residue 757, respectively. VU0410425 was selected for characterization to represent a compound typical of our novel succinimide series. Recently, the use of the operational model of allosterism was validated as a method for estimation of modulator binding affinities.31,46 This model offers an effective way to estimate affinity values directly from functional assays. It is especially useful for derivation of predicted affinities of modulators that act via binding sites for which radioligands have not been developed. Our results described above suggest the possibility of multiple binding sites, therefore use of the model could provide valuable insight into interactions at mGlu1. Progressive fold shift assays were utilized to derive affinity values from shifts in the glutamate CRC in the presence of fixed concentrations of NAM using the operational model as previously described by Gregory et al.31
In Figure 6, the effect of multiple fixed concentrations of BAY36-7620 on the glutamate CRCs of WT and mutant mGlu1 receptors is shown. Expectedly, increasing concentrations of BAY36-7620 in rmGlu1 WT cells (Figure 6a) induced a rightward shift and reduced the maximal effect of glutamate, behavior consistent with a noncompetitive allosteric antagonist. Introduction of the V757L mutation into rmGlu1 nearly abolished the NAM activity of BAY36-7620 (Figure 6b). A small right shift of the CRC was observed at high concentrations, a result very similar to that seen in the case of hmGlu1 WT (Figure 6c). Introduction of L757V into hmGlu1 rescued the NAM activity of BAY36-7620 in dramatic fashion (Figure 6d), returning activity to levels similar to those seen for rmGlu1 WT. Affinity estimates were then derived by globally fitting the data set to the operational model of allosterism. Table 5 summarizes the results derived from the operational model and compares the estimated affinity values to the functional potencies determined in the calcium assays. The V757L mutation in rmGlu1 induced a large reduction of BAY36-7620 predicted affinity (242 nM versus >10 μM). The estimated affinity value of BAY36-7620 for hmGlu1 WT was also very weak (>10 μM), demonstrating the human receptor-like behavior of the rat V757L mutant. Introduction of L757V into hmGlu1 greatly enhanced the affinity of BAY36-7620 (667 nM), a value approaching the affinity of BAY36-7620 for rmGlu1 WT. In the case of each WT and mutant receptor, the IC50 determined experimentally for BAY36-7620 was comparable (<2-fold) to the affinity value derived from the model, suggesting binding affinity and functional activity are highly correlated. These results were in agreement with the antagonist CRC results that suggest the activity of BAY36-7620 was highly dependent on amino acid 757.
Figure 6.

rmGlu1 V757L mutation results in significant loss of activity for BAY36-7620 while hmGlu1 L757V mutation shows gain of activity similar to rat WT. The effects of multiple fixed concentrations of BAY36-7620 on the glutamate CRCs of (a) rmGlu1 WT, (b) rmGlu1 V757L, (c) hmGlu1 WT, and (d) hmGlu1 L757V in a calcium mobilization assay using cells expressing mGlu1 are shown.
Table 5. Comparison of mGlu1 NAM Activitya with Affinity Estimate from Operational Model of Allosterism.
| rmGlu1 WT |
hmGlu1 WT |
rmGlu1 V757L |
hmGlu1 L757V |
|||||
|---|---|---|---|---|---|---|---|---|
| entry | pIC50 (IC50) | est pKB (KB) | pIC50 (IC50) | est pKB (KB) | pIC50 (IC50) | est pKB (KB) | pIC50 (IC50) | est pKB (KB) |
| 1 BAY36-7620 | 6.49 ± 0.03 | 6.62 ± 0.08 | <5 | <5 | <5 | <5 | 6.34 ± 0.03 | 6.18 ± 0.09 |
| (326 nM) | (242 nM) | (>10 μM) | (>10 μM) | (>10 μM) | (>10 μM) | (455 nM) | (667 nM) | |
| 3 DFMTI | 8.08 ± 0.02 | 8.06 ± 0.06 | 7.51 ± 0.09 | 7.75 ± 0.07 | 7.67 ± 0.07 | 7.65 ± 0.09 | 7.86 ± 0.03 | 8.19 ± 0.05 |
| (8 nM) | (9 nM) | (31 nM) | (18 nM) | (21 nM) | (22 nM) | (14 nM) | (7 nM) | |
| 6 VU0410425 | 6.85 ± 0.02 | 6.43 ± 0.04 | <4.5 | 5.17 ± 0.06 | <4.5 | 5.31 ± 0.08 | 6.44 ± 0.08 | 6.45 ± 0.04 |
| (140 nM) | (371 nM) | (>30 μM) | (6.8 μM) | (>30 μM) | (4.8 μM) | (360 nM) | (358 nM) | |
Calcium mobilization assay; values are average of n ≥ 3.
In Figure 7, the effect of multiple concentrations of DFMTI on the glutamate CRCs of the four mGlu1 receptors is shown. Increasing concentrations of DFMTI in cells expressing rmGlu1 WT (Figure 7a) induced a rightward shift in the CRC and reduced the maximal effect of glutamate, as seen with BAY36-7620, although much lower concentrations of DFMTI effectively decreased the glutamate response. In contrast to BAY36-7620, DFMTI was still able to completely block the rmGlu1 L757V glutamate response, although significantly higher concentrations were required to induce blockade (Figure 7b). A similar pattern of activity was observed in both the hmGlu1 WT and L757V receptor cell lines (Figure 7c and d). The 757 mutational effects in progressive fold shift experiments were more subtle for DFMTI than BAY36-7620, as we observed in NAM CRC assays; however, the affinity estimates from the operational model were able to quantify the small changes introduced by the mutations. Again, the IC50 values determined experimentally for DFMTI were comparable (≤2-fold) to the affinity values derived from the model, suggesting binding affinity and functional activity are correlated as was observed for BAY36-7620 (see Table 5). In contrast to BAY36-7620 where a significant loss of affinity was noted, the V757L mutation in rmGlu1 induced a much smaller reduction of DFMTI affinity (9 nM vs 22 nM). The estimated affinity values of DFMTI for rmGlu1 V757L and hmGlu1 WT were very similar (22 nM vs 18 nM), again emphasizing the human receptorlike behavior of the rat V757L mutant. Introduction of L757V into hmGlu1 slightly enhanced the affinity of DFMTI (7 nM) to a value nearly identical to the affinity of DFMTI for rmGlu1 WT. As observed in the case of the antagonist CRC assay, the activity of DFMTI was not highly regulated by residue 757. A subtle correlation does exist, but the interaction does not appear to be critical for functional activity or binding.
Figure 7.

rmGlu1 V757L mutation results in slight decrease in DFMTI NAM activity while hmGlu1 L757V mutation gains activity similar to rat WT. The effects of multiple fixed concentrations of DFMTI on the glutamate CRCs of (a) rmGlu1 WT, (b) rmGlu1 V757L, (c) hmGlu1 WT, and (d) hmGlu1 L757V in a calcium mobilization assay using cells expressing mGlu1 are shown.
In Figure 8, the effect of multiple concentrations of succinimide VU0410425 on the glutamate CRCs of the various mGlu1 receptors is shown. In a manner similar to the case of BAY36-7620, increasing concentrations of VU0410425 in rmGlu1 WT (Figure 8a) induced a rightward shift and reduced the maximal effect of glutamate nearly to baseline. Introduction of the V757L mutation into rmGlu1 blocked the inhibitory activity of VU0410425 (Figure 8b), mirroring the results seen for the hmGlu1 WT receptor (Figure 8c). Introduction of L757V into hmGlu1 significantly rescued the NAM activity of VU0410425 (Figure 8d), although maximal inhibitory activity plateaued at approximately 50%, a lower degree of inhibition than in the case of rmGlu1 WT (16%). Affinity estimates for each system were again derived by globally fitting the data set to the operational model of allosterism (see Table 5). The IC50 values determined experimentally for VU0410425 at rmGlu1 wt and hmGlu1 L757V were comparable to the affinity values derived from the model. The IC50 values determined experimentally for VU0410425 in rmGlu1 V757L and hmGlu1 WT were lower than the affinities derived from the model (>30 μM vs ∼5 μM), the first deviation we had observed. Comparing affinity values, the V757L mutation in rmGlu1 induced a large decrease in VU0410425 affinity (371 nM vs 4.8 μM). The estimated affinity values of VU0410425 for rmGlu1 V757L and hmGlu1 WT were both weak (4.8 and 6.8 μM, respectively) but were not devoid of activity as seen in the antagonist CRC assay. Introduction of L757V into hmGlu1 significantly increased the affinity of VU0410425 (358 nM) to a value nearly identical to the affinity of VU0410425 for rmGlu1 WT. The progressive fold shift results for VU0410425 resembled those seen for BAY36-7620, but a noteworthy difference exists for the two compounds. In the case of rmGlu1 WT and, more significantly, for hmGlu1 L757V, VU0410425 was unable to completely block the mGlu1 response to glutamate, suggesting a limited degree of negative cooperativity of the antagonist with glutamate. These results suggest that, while the activity of VU0410425 is highly dependent on residue 757 as in the case of BAY36-7620, a substantial difference exists mechanistically in how the two scaffolds mediate antagonism of the receptor.
Figure 8.

rmGlu1 V757L mutation results in significant loss of VU0410425 activity while hmGlu1 L757V mutation gains activity similar to that seen in rat WT. The effects of multiple fixed concentrations of VU0410425 on the glutamate CRCs of (a) rmGlu1 WT, (b) rmGlu1 V757L, (c) hmGlu1 WT, and (d) hmGlu1 L757V in a calcium mobilization assay using cells expressing mGlu1 are shown.
In summary, we utilized screening of an allosteric modulator-biased library to identify a novel succinimide mGlu1 NAM chemotype. Chemical optimization was performed to develop SAR trends within the series. The series exhibits potent inhibitory activity in cells expressing rat mGlu1 but is generally inactive in cells expressing human mGlu1. Prototypical succinimide VU0410425, along with a set of chemically diverse mGlu1 NAMs previously described in the literature, were selected to further characterize the disconnect between rat and human mGlu1 activity. Screening of these compounds in cells expressing WT or mutant mGlu1 receptors suggests that residue 757, the only residue in the TMD that differs between rat and human receptor, plays a role in their activity. The contribution of the residue, however, appears to be scaffold specific as we saw 757 mutations cause robust effects on activity of certain NAMs, such as VU0410425 and BAY36-7620, but have minimal effect on the activity of other compounds such as DFMTI. These data suggest the presence of multiple, possibly overlapping, allosteric binding sites for mGlu1 NAMs and emphasize the need to guide SAR and estimate affinity values from functional assays. Recent findings describing the crystal structure of human mGlu1 bound to FITM predicted the involvement of L757 in ligand–receptor interactions for a selection of analogs. Future docking studies utilizing the newly elucidated structure to understand the role of residue 757 would be extremely helpful in guiding future drug design.47 In conclusion, discovery of a novel mGlu1 NAM series based on the succinimide scaffold has provided valuable insight into the pharmacology underlying species differences in mGlu1 activity. An understanding of this issue will be critical to progress the therapeutic potential of allosteric modulation of mGlu1.
Methods
Materials
All reagents used in the cell culture medium were purchased from Invitrogen (Carlsbad, CA) except the tetracycline-tested fetal bovine serum from Atlantic Biologicals (Atlanta, GA). Tetracycline hydrochloride was purchased from Sigma. The synthesis of VU0410425 (6) is described in the literature.29 Synthetic procedures and characterization data for analogues 7–38 are described in the Supporting Information. BAY36-7620 and JNJ16259685 were purchased from Tocris Bioscience (Bristol, U.K.). 5-(1-(2,4-Difluorophenyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-isopropylisoindolin-1-one (DFMTI),16,48 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide (FITM),15 LY456236,49 7,9-diamino-3-(p-tolyl)thieno[2,3-d:4,5-d′]dipyrimidin-4(3H)-one (DATTP),38 R214127,42 5-(4-(hydroxymethyl)piperidin-1-yl)-N-((1r,4r)-4-methylcyclohexyl)pyrazine-2-carboxamide (HPMCP),43 and N-cycloheptylthieno[2,3-d]pyrimidin-4-amine (CHTPA)44 were all synthesized in-house using previously reported methods. Unless stated otherwise, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of analytical grade.
Cell Culture and Mutagenesis
To generate the tetracycline-inducible mGlu1 cell lines, TREx293 cells (Invitrogen) were transfected with human and rat mGlu1 expression plasmids in pcDNA5/TO using Fugene6 (Promega, Madison,WI) according to the manufacturer’s manual. Two days after transfection, the cells were exposed to 200 μg/mL hygromycin selection in the presence of 10 μg/mL blasticidin for maintaining the Tet repressor. After 14 days of selections, the resulting polyclones were used for the calcium mobilization assay described below. Reciprocal change in valine and leucine at 757 position of wild-type rat and human mGlu1, respectively, was made using site-directed mutagenesis (Quikchange II XL; Agilent Technologies, Santa Clara, CA), and this point-mutation was confirmed by sequencing. These mutant cell lines were generated in the same manner as those of WT. Wild-type and mutant cell lines were maintained at 37 °C in DMEM supplemented with 10% Tet-tested fetal bovine serum, 2 mM l-glutamine, 20 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, antibiotic/antimycotic solution, 100 μg/mL hygromycine, and 5 μg/mL blasticidin in the presence of 5% CO2.
Calcium Mobilization Assay
Rat and human mGlu1 (WT or mutant)-TREx293 cells were plated in black-walled, clear-bottomed, poly-d-lysine coated 384-well plates (BD Biosciences, San Jose, CA) at a density of 20 000 cells/well in 20 μL of assay medium (DMEM supplemented with 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate) containing tetracycline (TET) to induce the mGlu1 expression; 50 ng/mL TET was used except for rat mGlu1 WT which was induced with 10 ng/mL TET to achieve comparable levels of the receptor expression. The cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, cells were washed with assay buffer (Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid (Sigma-Aldrich, St. Louis, MO)) using an ELX405 microplate washer (BioTek) leaving 20 μL/well. Immediately, cells were incubated with 20 μL/well of Fluo-4 AM (Invitrogen) calcium indicator dye solution (1.15 μM final concentration) for 45 m at 37 °C. The Fluo-4 dye, prepared as a DMSO stock, was mixed in a 1:1 ratio with 10% pluronic acid F-127 and then diluted in assay buffer. The dye was then removed and washed with assay buffer using an ELX405, leaving 20 μL/well. Ca2+ flux was measured using the Functional Drug Screening System (FDSS7000, Hamamatsu, Japan).
NAM CRC Format
Compounds were serially diluted 1:3 in DMSO into 10 point concentration response curves and transferred to daughter plates using the Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) followed by further dilution into assay buffer to a 2× stock using a Thermo Fisher Combi (Thermo Fisher, Waltham, MA). In FDSS7000, compounds were added to cells at t = 3 s and the cells were incubated with these compounds for 2.3 min. Immediately, cells were stimulated with an EC20 concentration of glutamate, and 1.9 min later, an EC80/ECmax concentration of glutamate was added and readings taken for an additional 1.7 min.
Fold Shift Format
Compounds were diluted by half-log in DMSO and further diluted into assay buffer to a 2× stock which was applied to cells at t = 3 s. Cells were incubated with the test compounds for 2.3 min and then stimulated with varying concentrations of glutamate, and readings taken for an additional 1.7 min. Data were collected at 1 Hz. Concentration response curves were generated using a four point logistical equation with XLfit curve fitting software for Excel (IDBS, Guildford, U.K.) or GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
Selectivity
Rat mGlu5
HEK 293 cells stably expressing rat mGlu5 were plated in black-walled, clear-bottomed, poly-d-lysine coated 384-well plates in 20 μL of assay medium (DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate) at a density of 20K cells/well. The cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, medium was removed and the cells incubated with 20 μL of 2 μM Fluo-4 AM, prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127 and diluted in assay buffer (Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid, pH 7.4) for 45 min at 37 °C. Dye was removed, 20 μL of assay buffer was added, and the plate was incubated for 10 min at room temperature. Ca2+ flux was measured using the Functional Drug Screening System (FDSS7000, Hamamatsu, Japan) as described above for mGlu1 assays.
Human mGlu4
Human mGlu4/Gqi5/CHO cells (30 000 cells/20 μL/well) were plated in black-walled, clear-bottomed, TC-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in DMEM containing 10% dialyzed FBS, 20 mM HEPES, 100 U/mL penicillin/streptomycin, and 1 mM sodium pyruvate (plating medium). The cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, the medium was removed, and replaced using a Thermo Fisher Combi (Thermo Fisher Scientific, Waltham, MA), with 20 μL of 1 μM Fluo-4/acetoxymethyl ester (Invitrogen, Carlsbad, CA) prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) Pluronic acid F-127 and diluted in assay buffer (Hanks’ balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid; Sigma-Aldrich, St. Louis, MO) for 45 min at 37 °C. Dye was removed and 20 μL of assay buffer was added. Ca2+ flux was measured using the Functional Drug Screening System (FDSS7000, Hamamatsu, Japan) as described above for mGlu1 assays.
Rat mGlu2–4,7,8 and Human mGlu6
Compound 9 activity at the group II and group III mGlu receptors was assessed using thallium flux through G-protein-coupled inwardly rectifying potassium (GIRK) channels, a method that has been described in detail (Niswender et al.).50 These cell lines were grown in growth media containing 45% DMEM, 45% F-12, 10% FBS, 20 mM HEPES, 2 mM l-glutamine, antibiotic/antimycotic, nonessential amino acids, 700 μg/mL G418, and 0.6 μg/mL puromycin at 37 °C in the presence of 5% CO2. Briefly, HEK/GIRK cells expressing the mGlu subtypes 2, 3, 4, 6, 7, or 8 were plated into 384-well, black-walled, clear-bottom poly-d-lysine coated plates at a density of 15 000 cells/20 μL/well in assay medium and incubated overnight at 37 °C in the presence of 5% CO2. The following day, the medium from the cells and 20 μL/well of 1.7 μM concentration of the indicator dye BTC-AM (Invitrogen, Carlsbad, CA) in assay buffer was added. Cells were incubated for 1 h at room temperature, and the dye was replaced with 20 μL/well of assay buffer. After establishment of a fluorescence baseline for about 3 s, test compound was added to the cells at 2× final concentration, and the response in cells was measured. 2.3 min later the appropriate concentration of agonist (L-AP4 for mGlu7, glutamate for all other mGlu receptors) was added and readings taken for an additional 2.6 min. Agonists were diluted in thallium buffer (125 mM sodium bicarbonate, 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, 10 mM HEPES) at 5× the final concentration to be assayed. Data were analyzed as described in Niswender et al.
Data Analysis for Operational Model of Allosterism
Shifts of the glutamate concentration–response curves with allosteric modulator were globally fitted to an operational model of allosterism.31,46
where [A] is the molar concentration of orthosteric agonist glutamate, KA is the equilibrium dissociation constant of the orthosteric agonist glutamate, [B] is the molar concentration of the allosteric modulator, and KB is the allosteric modulator equilibrium dissociation constant. Affinity modulation is governed by the cooperativity factor α, and efficacy modulation is governed by cooperativity factor β. The parameters τA and τB are related to the ability of the orthosteric and allosteric ligands, respectively, to yield receptor activation. Em and n denote the maximal possible system response and the transducer function that links occupancy to response, respectively. For these simulations, the affinity of glutamate was held constant to a literature value (pKA = 6.47), modulator coupling efficiency (τB) was held constant to zero as defined for modulators devoid of agonist activity, efficacy modulation (β) was held constant to zero for systems where the NAM fully abolishes response to agonist, and all other constraints were derived from global fitting of glutamate concentration response curves in the absence and presence of allosteric modulators.
Acknowledgments
We thank Karen J. Gregory for assistance with affinity estimations using the operational model.
Glossary
Abbreviations
- GPCR
G-protein-coupled receptor
- mGlu
metabotropic glutamate receptor
- CNS
central nervous system
- NAM
negative allosteric modulator
- DFMTI
5-(1-(2,4-difluorophenyl)-5-methyl-1H-1,2,3-triazol-4-yl)-2-isopropylisoindolin-1-one
- PAM
positive allosteric modulator
- SAR
structure–activity relationship
- CRC
concentration–response curve
- DATTP
7,9-diamino-3-(p-tolyl)thieno[2,3-d:4,5-d′]dipyrimidin-4(3H)-one
- FITM
4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide
- HPMCP
5-(4-(hydroxymethyl)piperidin-1-yl)-N-((1r,4r)-4-methylcyclohexyl)pyrazine-2-carboxamide
- CHTPA
N-cycloheptylthieno[2,3-d]pyrimidin-4-amine
- TMD
transmembrane domain
- WT
wild-type
- HEK
human embryonic kidney
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethyl sulfoxide
- FDSS
functional drug screening system
- FBS
fetal bovine serum
- HBSS
Hanks’ balanced salt solution
- TET
tetracycline
Supporting Information Available
Synthetic procedures and characterization data for analogues 7–38. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
K.A.E. and C.W.L. oversaw and designed the chemistry. D.W.E. performed synthetic chemistry work. A.L.R., C.M.N, and P.J.C. oversaw, designed, and interpreted the molecular pharmacology experiments. H.P.C. and D.F.V. performed the molecular pharmacology experiments.
The authors thank the NIH (NS032373, MH062646, and MH097056) and Seaside Therapeutics (VUMC36176) for their support of our programs in the development of mGlu1 NAMs.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Niswender C. M.; Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlinska J. H.; Bochenski M.; Danysz W. (2011) The role of group I mGlu receptors in the expression of ethanol-induced conditioned place preference and ethanol withdrawal seizures in rats. Eur. J. Pharmacol. 670, 154–161. [DOI] [PubMed] [Google Scholar]
- Xie X.; Ramirez D. R.; Lasseter H. C.; Fuchs R. A. (2010) Effects of mGluR1 antagonism in the dorsal hippocampus on drug context-induced reinstatement of cocaine-seeking behavior in rats. Psychopharmacology 208, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravolina O. A.; Zakharova E. S.; Shekunova E. V.; Zvartau E. E.; Danysz W.; Bespalov A. Y. (2007) mGlu1 receptor blockade attenuates cue- and nicotine-induced reinstatement of extinguished nicotine self-administration behavior in rats. Neuropharmacology 52, 263–269. [DOI] [PubMed] [Google Scholar]
- Satow A.; Maehara S.; Ise S.; Hikichi H.; Fukushima M.; Suzuki G.; Kimura T.; Tanak T.; Ito S.; Kawamoto H.; Ohta H. (2008) Pharmacological effects of the metabotropic glutamate receptor 1 antagonist compared with those of the metabotropic glutamate receptor 5 antagonist and metabotropic glutamate receptor 2/3 agonist in rodents: detailed investigations with a selective allosteric metabotropic glutamate receptor 1 antagonist, FTIDC [4-[1-(2-fluoropyridine-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methy l-3,6-dihydropyridine-1(2H)-carboxamide]. J. Pharmacol. Exp. Ther. 326, 577–586. [DOI] [PubMed] [Google Scholar]
- Rorick-Kehn L. M.; Hart J. C.; McKinzie D. L. (2005) Pharmacological characterization of stress-induced hyperthermia in DBA/2 mice using metabotropic and ionotropic glutamate receptor ligands. Psychopharmacology 183, 226–240. [DOI] [PubMed] [Google Scholar]
- Steckler T.; Lavreysen H.; Oliveira A. M.; Aerts N.; Van Craenendonck H.; Prickaerts J.; Megens A.; Lesage A. S. (2005) Effects of mGlu1 receptor blockade on anxiety-related behaviour in the rat lick suppression test. Psychopharmacology 179, 198–206. [DOI] [PubMed] [Google Scholar]
- Shannon H. E.; Peters S. C.; Kingston A. E. (2005) Anticonvulsant effects of LY456236, a selective mGlu1 receptor antagonist. Neuropharmacology 49(Suppl 1), 188–195. [DOI] [PubMed] [Google Scholar]
- Barton M. E.; Peters S. C.; Shannon H. E. (2003) Comparison of the effect of glutamate receptor modulators in the 6 Hz and maximal electroshock seizure models. Epilepsy Res. 56, 17–26. [DOI] [PubMed] [Google Scholar]
- Bennett C. E.; Burnett D. A.; Greenlee W. J.; Knutson C. E.; Korakas P.; Li C.; Tulshian D.; Wu W. L.; Bertorelli R.; Fredduzzi S.; Grilli M.; Lozza G.; Reggiani A.; Veltri A. (2012) Fused tricyclic mGluR1 antagonists for the treatment of neuropathic pain. Bioorg. Med. Chem. Lett. 22, 1575–1578. [DOI] [PubMed] [Google Scholar]
- Mantell S. J.; Gibson K. R.; Osborne S. A.; Maw G. N.; Rees H.; Dodd P. G.; Greener B.; Harbottle G. W.; Million W. A.; Poinsard C.; England S.; Carnell P.; Betts A. M.; Monhemius R.; Prime R. L. (2009) In vitro and in vivo SAR of pyrido[3,4-d]pyramid-4-ylamine based mGluR1 antagonists. Bioorg. Med. Chem. Lett. 19, 2190–2194. [DOI] [PubMed] [Google Scholar]
- Zhu C. Z.; Baker S.; EI-Kouhen O.; Lehto S. G.; Hollingsworth P. R.; Gauvin D. M.; Hernandez G.; Zheng G.; Chang R.; Moreland R. B.; Stewart A. O.; Brioni J. D.; Honore P. (2008) Analgesic activity of metabotropic glutamate receptor 1 antagonists on spontaneous post-operative pain in rats. Eur. J. Pharmacol. 580, 314–321. [DOI] [PubMed] [Google Scholar]
- Satow A.; Suzuki G.; Maehara S.; Hikichi H.; Murai T.; Kawagoe-Takaki H.; Hata M.; Ito S.; Ozaki S.; Kawamoto H.; Ohta H. (2009) Unique antipsychotic activities of the selective metabotropic glutamate receptor 1 allosteric antagonist 2-cyclopropyl-5-[1-(2-fluoro-3-pyridinyl)-5-methyl-1H-1,2,3-triazol-4-yl]-2,3-dihydro-1H-isoindol-1-one. J. Pharmacol. Exp. Ther. 330, 179–190. [DOI] [PubMed] [Google Scholar]
- Hikichi H.; Nishino M.; Fukushima M.; Satow A.; Maehara S.; Kawamoto H.; Ohta H. (2010) Pharmacological effects of metabotropic glutamate receptor ligands on prepulse inhibition in DBA/2J mice. Eur. J. Pharmacol. 639, 99–105. [DOI] [PubMed] [Google Scholar]
- Satoh A.; Nagatomi Y.; Hirata Y.; Ito S.; Suzuki G.; Kimura T.; Maehara S.; Hikichi H.; Satow A.; Hata M.; Ohta H.; Kawamoto H. (2009) Discovery and in vitro and in vivo profiles of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorg. Med. Chem. Lett. 19, 5464–5468. [DOI] [PubMed] [Google Scholar]
- Ito S.; Hirata Y.; Nagatomi Y.; Satoh A.; Suzuki G.; Kimura T.; Satow A.; Maehara S.; Hikichi H.; Hata M.; Ohta H.; Kawamoto H. (2009) Discovery and biological profile of isoindolinone derivatives as novel metabotropic glutamate receptor 1 antagonists: a potential treatment for psychotic disorders. Bioorg. Med. Chem. Lett. 19, 5310–5313. [DOI] [PubMed] [Google Scholar]
- Lavreysen H.; Pereira S. N.; Leysen J. E.; Langlois X.; Lesage A. S. (2004) Metabotropic glutamate 1 receptor distribution and occupancy in the rat brain: a quantitative autoradiographic study using [3H]R214127. Neuropharmacology 46, 609–619. [DOI] [PubMed] [Google Scholar]
- Lavreysen H.; Janssen C.; Bischoff F.; Langlois X.; Leysen J. E.; Lesage A. S. (2003) [3H]R214127: a novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol. Pharmacol. 63, 1082–1093. [DOI] [PubMed] [Google Scholar]
- Carroll F. Y.; Stolle A.; Beart P. M.; Voerste A.; Brabet I.; Mauler F.; Joly C.; Antonicek H.; Bockaert J.; Muller T.; Pin J. P.; Prezeau L. (2001) BAY36–7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol. Pharmacol. 59, 965–973. [PMC free article] [PubMed] [Google Scholar]
- Lavreysen H.; Wouters R.; Bischoff F.; Nobrega Pereira S.; Langlois X.; Blokland S.; Somers M.; Dillen L.; Lesage A. S. (2004) JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology 47, 961–972. [DOI] [PubMed] [Google Scholar]
- Martino J. J.; Wall B. A.; Mastrantoni E.; Wilimczyk B. J.; La Cava S. N.; Degenhardt K.; White E.; Chen S. (2013) Metabotropic glutamate receptor 1 (Grm1) is an oncogene in epithelial cells. Oncogene 32, 4366–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani Y.; Harada T.; Funasaka Y.; Nakao K.; Takahara C.; Abdel-Daim M.; Sakai N.; Saito N.; Nishigori C.; Aiba A. (2008) Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene 27, 7162–7170. [DOI] [PubMed] [Google Scholar]
- Namkoong J.; Shin S. S.; Lee H. J.; Marin Y. E.; Wall B. A.; Goydos J. S.; Chen S. (2007) Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 67, 2298–2305. [DOI] [PubMed] [Google Scholar]
- Speyer C. L.; Smith J. S.; Banda M.; DeVries J. A.; Mekani T.; Gorski D. H. (2012) Metabotropic glutamate receptor-1: a potential therapeutic target for the treatment of breast cancer. Breast Cancer Res. Treat. 132, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A. (2009) Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr. Opin. Drug Discovery Dev. 12, 446–457. [PubMed] [Google Scholar]
- Rocher J. P.; Bonnet B.; Bolea C.; Lutjens R.; Le Poul E.; Poli S.; Epping-Jordan M.; Bessis A. S.; Ludwig B.; Mutel V. (2011) mGluR5 negative allosteric modulators overview: a medicinal chemistry approach towards a series of novel therapeutic agents. Curr. Top. Med. Chem. 11, 680–695. [DOI] [PubMed] [Google Scholar]
- Lovell K. M.; Felts A. S.; Rodriguez A. L.; Venable D. F.; Cho H. P.; Morrison R. D.; Byers F. W.; Daniels J. S.; Niswender C. M.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2013) N-Acyl-N′-arylpiperazines as negative allosteric modulators of mGlu1: identification of VU0469650, a potent and selective tool compound with CNS exposure in rats. Bioorg. Med. Chem. Lett. 23, 3713–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manka J. T.; Rodriguez A. L.; Morrison R. D.; Venable D. F.; Cho H. P.; Blobaum A. L.; Daniels J. S.; Niswender C. M.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2013) Octahydropyrrolo[3,4-c]pyrrole negative allosteric modulators of mGlu1. Bioorg. Med. Chem. Lett. 23, 5091–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. K.; Engers D. W.; Thompson A. D.; Field J. R.; Blobaum A. L.; Lindsley S. R.; Zhou Y.; Gogliotti R. D.; Jadhav S.; Zamorano R.; Bogenpohl J.; Smith Y.; Morrison R.; Daniels J. S.; Weaver C. D.; Conn P. J.; Lindsley C. W.; Niswender C. M.; Hopkins C. R. (2011) Discovery, synthesis, and structure-activity relationship development of a series of N-4-(2,5-dioxopyrrolidin-1-yl)phenylpicolinamides (VU0400195, ML182): characterization of a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu(4)) with oral efficacy in an antiparkinsonian animal model. J. Med. Chem. 54, 7639–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato R. J.; Felts A. S.; Rodriguez A. L.; Venable D. F.; Morrison R. D.; Byers F. W.; Daniels J. S.; Niswender C. M.; Conn P. J.; Lindsley C. W.; Jones C. K.; Emmitte K. A. (2013) Substituted 1-Phenyl-3-(pyridin-2-yl)urea negative allosteric modulators of mGlu5: discovery of a new tool compound VU0463841 with activity in rat models of cocaine addiction. ACS Chem. Neurosci. 4, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory K. J.; Noetzel M. J.; Rook J. M.; Vinson P. N.; Stauffer S. R.; Rodriguez A. L.; Emmitte K. A.; Zhou Y.; Chun A. C.; Felts A. S.; Chauder B. A.; Lindsley C. W.; Niswender C. M.; Conn P. J. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol. Pharmacol. 82, 860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts A. S.; Lindsley S. R.; Lamb J. P.; Rodriguez A. L.; Menon U. N.; Jadhav S.; Jones C. K.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2010) 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu(5): Identification of easily prepared tool compounds with CNS exposure in rats. Bioorg. Med. Chem. Lett. 20, 4390–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. L.; Nong Y.; Sekaran N. K.; Alagille D.; Tamagnan G. D.; Conn P. J. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol. 68, 1793–1802. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Rodriguez A. L.; Conn P. J.; Lindsley C. W. (2008) Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg. Med. Chem. Lett. 18, 4098–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. L.; Grier M. D.; Jones C. K.; Herman E. J.; Kane A. S.; Smith R. L.; Williams R.; Zhou Y.; Marlo J. E.; Days E. L.; Blatt T. N.; Jadhav S.; Menon U. N.; Vinson P. N.; Rook J. M.; Stauffer S. R.; Niswender C. M.; Lindsley C. W.; Weaver C. D.; Conn P. J. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol. 78, 1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemstapat K.; de Paulis T.; Chen Y.; Brady A. E.; Grover V. K.; Alagille D.; Tamagnan G. D.; Conn P. J. (2006) A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol. Pharmacol. 70, 616–626. [DOI] [PubMed] [Google Scholar]
- Knoflach F.; Mutel V.; Jolidon S.; Kew J. N.; Malherbe P.; Vieira E.; Wichmann J.; Kemp J. A. (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc. Natl. Acad. Sci. U.S.A. 98, 13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasikumar T. K.; Qiang L.; Burnett D. A.; Greenlee W. J.; Li C.; Grilli M.; Bertorelli R.; Lozza G.; Reggiani A. (2010) A-ring modifications on the triazafluorenone core structure and their mGluR1 antagonist properties. Bioorg. Med. Chem. Lett. 20, 2474–2477. [DOI] [PubMed] [Google Scholar]
- Suzuki G.; Kimura T.; Satow A.; Kaneko N.; Fukuda J.; Hikichi H.; Sakai N.; Maehara S.; Kawagoe-Takaki H.; Hata M.; Azuma T.; Ito S.; Kawamoto H.; Ohta H. (2007) Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide (FTIDC). J. Pharmacol. Exp. Ther. 321, 1144–1153. [DOI] [PubMed] [Google Scholar]
- Yamasaki T.; Fujinaga M.; Yoshida Y.; Kumata K.; Yui J.; Kawamura K.; Hatori A.; Fukumura T.; Zhang M. R. (2011) Radiosynthesis and preliminary evaluation of 4-[18F]fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide as a new positron emission tomography ligand for metabotropic glutamate receptor subtype 1. Bioorg. Med. Chem. Lett. 21, 2998–3001. [DOI] [PubMed] [Google Scholar]
- Varty G. B.; Grilli M.; Forlani A.; Fredduzzi S.; Grzelak M. E.; Guthrie D. H.; Hodgson R. A.; Lu S. X.; Nicolussi E.; Pond A. J.; Parker E. M.; Hunter J. C.; Higgins G. A.; Reggiani A.; Bertorelli R. (2005) The antinociceptive and anxiolytic-like effects of the metabotropic glutamate receptor 5 (mGluR5) antagonists, MPEP and MTEP, and the mGluR1 antagonist, LY456236, in rodents: a comparison of efficacy and side-effect profiles. Psychopharmacology 179, 207–217. [DOI] [PubMed] [Google Scholar]
- Mabire D.; Coupa S.; Adelinet C.; Poncelet A.; Simonnet Y.; Venet M.; Wouters R.; Lesage A. S.; Van Beijsterveldt L.; Bischoff F. (2005) Synthesis, structure-activity relationship, and receptor pharmacology of a new series of quinoline derivatives acting as selective, noncompetitive mGlu1 antagonists. J. Med. Chem. 48, 2134–2153. [DOI] [PubMed] [Google Scholar]
- Owen D. R.; Dodd P. G.; Gayton S.; Greener B. S.; Harbottle G. W.; Mantell S. J.; Maw G. N.; Osborne S. A.; Rees H.; Ringer T. J.; Rodriguez-Lens M.; Smith G. F. (2007) Structure-activity relationships of novel non-competitive mGluR1 antagonists: a potential treatment for chronic pain. Bioorg. Med. Chem. Lett. 17, 486–490. [DOI] [PubMed] [Google Scholar]
- Itahana H., Kamikubo T., Nozawa E., Kaku H., Okada M., Toya T., Nakamura A., and Nagai S. (2002) Thienopyrimidine Derivative. Patent WO02062803 (A1), Japan.
- Malherbe P.; Kratochwil N.; Knoflach F.; Zenner M. T.; Kew J. N.; Kratzeisen C.; Maerki H. P.; Adam G.; Mutel V. (2003) Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J. Biol. Chem. 278, 8340–8347. [DOI] [PubMed] [Google Scholar]
- Leach K.; Sexton P. M.; Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 28, 382–389. [DOI] [PubMed] [Google Scholar]
- Wu H.; Wang C.; Gregory K. J.; Han G. W.; Cho H. P.; Xia Y.; Niswender C. M.; Katritch V.; Meiler J.; Cherezov V.; Conn P. J.; Stevens R. C. (2014) Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 344, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuritani T.; Mizuno H.; Nonoyama N.; Kii S.; Akao A.; Sato K.; Yasuda N.; Mase T. (2009) Efficient synthesis of 1,4-diaryl-5-methyl-1,2,3-triazole, a potential mGluR1 antagonist, and the risk assessment study of arylazides. Org. Process Res. Dev. 13, 1407–1412. [Google Scholar]
- Ambler S. J., Baker S. R., Clark B. P., Coleman D. S., Foglesong R. J., Goldsworthy J., Jagdmann G. E. Jr., Johnson K. W., Kingston A. E., Owton W. M., Schoepp D. D., Hong J. E., Schkeryantz J. M., Vannieuwenhze M. S., and Zia-Ebrahimi M. S. (2001) Pharmaceutical Compounds. Patent WO0132632 (A2), USA.
- Niswender C. M.; Johnson K. A.; Weaver C. D.; Jones C. K.; Xiang Z.; Luo Q.; Rodriguez A. L.; Marlo J. E.; de Paulis T.; Thompson A. D.; Days E. L.; Nalywajko T.; Austin C. A.; Williams M. B.; Ayala J. E.; Williams R.; Lindsley C. W.; Conn P. J. (2008) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 74, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.