

Abstract
Reversing abnormal gene silencing in cancer cells due to DNA hypermethylation of promoter CpG islands may offer new cancer prevention or therapeutic approaches. Moreover, such approaches may be broadly applicable to enhance the efficacy of radiotherapy, chemotherapy or immunotherapy. Here we demonstrate the powerful utility of a novel gene reporter system to permit studies of the dynamics, mechanisms, and translational relevance of candidate therapies of this type in human colon cancer cells. The reporter system is based on in situ modification of the endogenous locus of the tumor suppressor gene SFRP1, a pivotal regulator of the Wnt pathway that is silenced by DNA hypermethylation in many colon cancers. The modified SFRP1-GFP reporter allele employed remained basally silent, like the unaltered allele, and it was activated only by drug treatments that de-repress gene silencing by reversing DNA hypermethylation. We employed the established DNA methyltransferase inhihibitor (DNMTi) 5-aza-deoxycitidine (DAC) to show how this system can be used to address key questions in the clinical development of epigenetic cancer therapies. First, we defined conditions for which clinically relevant dosing could induce sustained induction of RNA and protein. Second, we found that, in-vivo, a more prolonged drug exposure than anticipated was essential to de-repress gene silencing in significant cell numbers and this has implications for generating effective anticancer responses in patients with hematopoietic or solid tumors. Finally, we discovered how histone deacetylase inhibitors (HDACi) alone, when administered to cells actively replicating DNA, can robustly re-express the silenced gene with no change in promoter methylation status. Taken together, our findings offer a new tool and insights for devising optimal clinical experiments to evaluate DNMTi and HDACi, alone or in combination, and with other cancer treatments, as agents for the epigenetic management and prevention of cancer.
INTRODUCTION
The role of epigenetic abnormalities as drivers of tumorigenesis has been increasingly recognized in recent years (1–3). Hundreds of genes in individual cancers are known to be differentially silenced in association with DNA hypermethylation of CpG islands in promoter regions (1–3). A key group of these genes are known to function as tumor suppressors (1–3). Targeted reversal of tumor suppressor gene silencing is an attractive strategy for cancer prevention and therapy (4–6). A substantial effort is now underway to use existing drugs, and develop new drugs, for this purpose (7). These efforts will require quantitative, high throughput screening systems for drug identification and refinement, and for understanding how such drugs might be optimally employed.
Previous approaches to this problem have utilized exogenous reporter gene systems in which the introduced construct is DNA methylated prior to cell introduction (8). This exogenous reporter approach does not necessarily mimic promoters in their endogenous setting in cancer cells with respect to sequential events for acquisition of the aberrant DNA methylation and/or numerous regional chromatin modifications which contribute to the evolution and maintenance of this gene silencing. Herein, we describe a human colon cancer cell system in which a fluorescence-based reporter has been homologously recombined into an exon region of a tumor suppressor gene downstream from its endogenous, DNA hypermethylated, promoter region. The reporter then remains silenced until drug induced re-expression, which can be monitored at the single cell level, by multiple assay approaches. We provide first examples of how we have used this system for deriving new, highly translationally relevant, insights into the actions of DNA methylatransferase inhibitors (DNMTi’s) and histone deactylase inhibiting (HDACi) drugs. The findings should prove important for better utilizing such drugs in the clinic, alone and together.
MATERIALS AND METHODS
Construction of the SFRP1-GFP recombinant reporter system
The recombinant construct utilizes an AAV shuttle vector in which both 700 bp 5′ and 1,000 bp 3′ homologous arms flanking the SFRP1 gene and the CpG island in the proximal promoter region were inserted from wild type Hct116 genomic DNA (Suppl Fig. 1). PCR reactions were utilized to assemble, between the arms, Lox sites flanking a TK-NeoR cassette, and IRES sequences preceding the GFP gene (Suppl. Figs. 1 and 2). The total insert size for the above elements is 4.7 kb. This recombinant AAV vector was then co-transfected, using Lipo-fectamine 2000, into AAV-293 cells with recombinant pAAV-RC & pHelper sequences to produce AAV virus particles which were then utilized to infect wild type Hct116 cells at 37C for 2–3 hrs. Cells were then neomycin selected to be screened for clones positive for proper recombinants as validated by the PCR strategy shown in Suppl Fig. 2. Two clones with the proper insertion of the construct into exon 2, #90 and #1.4, of endogenous SRFP 1 were selected for all further studies. To activate the Lox sites and delete the NeoR sequences between these, the recombinant clones were infected with virus containing Cre recombinase.
FACS analysis
Using a Becton Dickinson, FACS-Calibur flow cytometer, expression of the SFRP1-GFP allele, was measured as maximum fluorescent intensities before and after drug treatments, and responses were recorded as the percentage numbers of GFP positive cells. We also used this technology, in selected experiments, to sort for GFP positive and negative cells. In selected experiments, we compared the GFP signals to signals from cells treated with 10 μM EDU (5-ethynyl-2′-deoxyuridine) (from Invitrogen) to assess the status of cells for DNA synthesis and compare this to levels of SFRP1-GFP expression. Since the buffer for detection of EDU contents CuSO (4) which induces fluorescence quenching, cells were always sorted first for GFP prior to analyses for detecting EDU. In all of these studies, we also assessed cells stained with 7-aminoactinomycin D (7AAD) for detecting and quantitating cell nuclei and cell cycle position by FACS analysis. For every experiment, we used a GFP negative cell-line control versus drug treated GFP positive cells to gate and set the boundary for negative versus positive GFP cells after drug treatments.
Time Lapse video microscopy
For selected studies, we monitored drug induction of SFRP-GFP expression over a time course by performing timed video microscopy, using a Nikon Eclipse TE2000-E camera combined with a NIS-Elements AR 3.10 program, in the Imaging Core facility in the Sidney Kimmel Comprehensive Cancer Center at the Johns Hopkins Medical Institutions. Frames were captured at 15 min intervals with magnification = 20X. For analysis of percent positive cells at each time point, TIFF format pictures were counted for all cells in the field.
Cell culture and drug treatment
All culturing of HCT116 cells was performed in McCoy medium with 10% FBS. DAC treatment conditions varied with experimental questions but doses ranged from 50 to 500 nM, generally administered daily with fresh drug and medium each day. The HDACi, trichostatin (TSA), was given at doses of 300 nM for 24 hrs.
DNA extraction and DNA methylation analysis
DNA extraction and bisulfite conversion were performed with the EZ DN Methylation Kit from Zymo Research. Methylation Specific PCR (MSP) analysis was performed as previously described using primers located in the endogenous promoter CpG island of SFRP1 with a product size of 126 bp (9, 10).
Real-time–PCR
RNA was isolated using TRIzol (Invitrogen) and converted to cDNA using Superscript III (invitrogen). qRT-PCR assays were performed on a Bio-Rad Real-Time PCR System using SYBR Green Mix (Bio-Rad) for detection. All mRNA levels were normalized for endogenous SFRP1, or for SFRP1-GFP sequences, to levels for GAPDH using qRT values for 40 cycles without addition of Superscript III as a zero point. The sFRP1 product is 156 bp and PCR primers are located in exon1 (catgcagttcttcggcttct) and exon2 (gatttcaactcgttgtcacagg). For SFRP-GFP, the product is 192 bp and primers are located inside the recombinant GFP sequences, (catccccgactacttcaagc/cccatggtcttcttctgcat).
Chromatin Immunoprecipitation (CHIP)
106 cells per assay were crosslinked using 1% formaldehyde (Sigma). Nuclear extracts were obtained using CEBN buffer (11, 12)and sonication was performed for using 30 cycles (10 s on 30 s off). Chip was performed overnight at 4°C using 50–80 ug chromatin DNA and 4 different antibodies (from Millipore) consisting of an IgG control, histone H3, H3K4me3, and H3K27me3 respectively. This was followed by incubation with Dynal Magnetic beads (Invitrogen) for 3 hours and then by 2 washes each with low salt buffer, high salt buffer and LiCl buffer. Chromatin associated DNA was then eluted from the beads during a 65°C overnight incubation with 50 mM NaHCO3 buffer with 1% SDS to reverse the crosslinks. IP specific products were then amplified using previously reported (11) RT-PCR forward and reverse primers (gcaccgcagctagagaaccga/ctgcttcctaatttcaaccaacagccc) located in the sFRP1 proximal promoter region (−770 to −700 bp relative to TSS site).
In-vivo Studies of Response to DAC and Immunocytochemistry (ICC) Studies
Hct116-sFRP1-GFP (#1.4) cells, 106 per site, were injected subcutaneously into 2 flank sites/mouse using 6 weeks old, female, NOD-SCID mice. Ten mice were used, each, for mock and DAC treatment experiments using 0.5 mg/kg DAC for 5 days per week for 4 weeks. Palpable tumors were allowed to form before treatment was begun. Five mice were sacrificed for tumor analysis for mock and DAC treatment groups at 2 and 4 weeks. All tumors were removed and fixed with 4% paraformaldehyde overnight at 4°C, followed by immersion in 30% sucrose for 10–12 hours. Frozen sections (20 μm) were cut using cryostats and mounted on slides. Portions of sections were prepared on coverslips and stained with antifade DAPI reagent (Life Technology) and also used for GFP fluorescence detection. Companion slides were used for ICC with incubation with anti-GFP antibody (1:1000; Life Technologies A11122) at 4°C overnight. For controls, samples were incubated with Alexa488 conjugated IgG at room temperature for 3 h.
RESULTS
Construction of the model system
Our reporter system was derived using gene targeting for an allele of SFRP1 in the colorectal cancer cell line, HCT 116. This tumor suppressor gene, which can inhibit Wnt signaling (13), is transcriptionally silenced in association with promoter, CpG island, DNA hypermethylation in this cell line, as it is very frequently in colorectal cancer and other cancers types (10, 14). To introduce a reporter tag directly into this locus, we employed a recombinant adeno-associated virus (rAAV)-based approach (15) to disrupt the native second exon with a construct containing a neomycin transferase minigene driven by a highly active Thymidine Kinase (TK) promoter plus a GFP reporter downstream of this drug selection cassette (Fig. 1A). This neo cassette is flanked by LoxP sites to facilitate its excision following transient expression of Cre recombinase. An Internal Ribosomal Entry Sequence (IRES) was positioned 5′ to the GFP open reading frame, facilitating its expression as a bicistronic element. This approach allowed selection of neo-expressing cells in which the construct had homologously recombined at the SFRP1 locus (Fig. 1A). Following the identification and expansion of the desired recombinant clones, the neo cassette was excised to create transcriptional competence for the GFP allele after infection of target cells with Cre-recombinase expressing adenovirus. The resulting cells, however, should exhibit little or no detectable basal expression of GFP prior to any drug treatment due to maintenance of the silenced state of the recombinant SFRP1 allele in association with its aberrant, CpG island, DNA hypermethylation (Fig. 1A). This is, in fact, the case as only after treatment of the HCT 116 cells with the DNA demethylating agent, DAC, can we detect reactivated expression of the SFRP1-GFP allele (Fig. 1B) and this occurs concomitantly with partial DNA demethylation of SFRP1 alleles (Fig. 1C). The fact that both wild type and recombinant alleles maintain a basal status of dense DNA methylation is indicated by a lack of any signal for non-DNA methylated alleles by the very sensitive, MSP assay being utilized for the present studies (Fig. 1C). Previous comparisons of MSP to extensive bisulfite sequencing, for the DNA methylation of the promoter sequences being studied indicates that methylated signal in the absence of any un-methylated signal is seen for the MSP assay of SFRP1 alleles only when dense DNA methylation is present (10, 14).
Fig. 1. Schema for construction and performance of the SFRP1-GFP recombinant allele in Hct116 cells.

A. Homologous recombination technology is used to knock in a GFP reporter, inclusive of flanking LOXP sites, a Neo resistant cassette, and IRES sequences ahead of GFP, into exon 2 of transcriptionally silenced, and promoter DNA hypermethylated, SFRP1. The potential performance of the resulting recombinant allele, after inducing Cre recombination is shown in the bottom panel – ie no predicted GFP expression until drug induced reactivation of the endogenously silenced gene. B. Validation the SFRP1-GFP expression of the #90 and #1.4 recombinant clones. FACS fluorescence analysis of SFRP1-GFP expression is performed comparing cells before and after inducing Cre recombination and with and without drug treatment. The positive region is within the enclosed rectangle with the Y axis = GFP negative channel and the X-axis = fluorescence for GFP. GFP is also visualized, in the inset panel, by fluorescence microscopy with phase contrast view of all cells in the background Mag = 20x). C. MSP analysis of the DNA methylation status of the endogenous SFRP1 promoter (M = methylated alleles; U = unmethylated alleles) performed for HCT116 wild type and cells and the two clones with the recombinant SFRP1-GFP allele after Cre recombination. HCT 116 DKO cells are used as controls for complete demethylation of the SFRP1 promoter and have a genetic knockout of DNMT1 and DNMT3B. (− lanes) = no DAC treatment; (+ lanes) = DAC treatment; dH2O = control with MSP primers, but no DNA templates. MSP primers for unmethylated and methylated sequences are those previously reported for analysis of the SFRP1 promoter, CpG island region 6 with a product size of 126 bp (10).
Quantification and timing of DAC induced SFRP1-GFP expression
The SFRP1-GFP expression in the above studies can be precisely quantified, as monitored by FACS flow analysis, for protein levels and timing of gene reactivation by varying doses of the drug (Fig. 2A, B). Using a typical protocol for in-vitro, DAC induced, reactivation of hypermethylated genes, in which .01 to 1.0 uM drug is administered daily in fresh media to offset its short half-life in aqueous solution, for 3 days, the number of GFP-positive cells (Fig. 2A) and the overall fluorescence intensity (Fig. 2B), both peaked at 3 days after the start of DAC treatment and then fell progressively thereafter. This is a time course typically observed for RNA expression of DNA hypermethylated genes in this type of short term protocol using delivery of fresh drug and fresh media every 24 hours (10).
Fig. 2. Quantitation by FACS analysis of cell SFRP1-GFP fluorescence following DAC treatment.
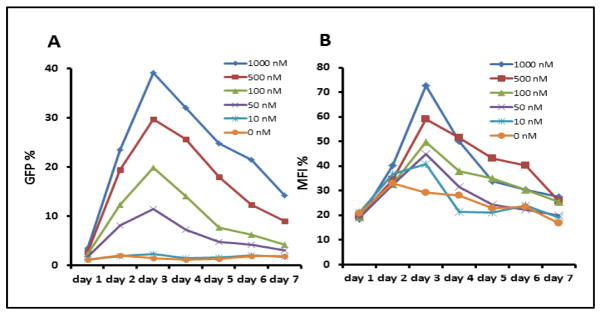
A. Dose response and time curve - cells were treated for 3 days with the doses shown and with fresh drug and media added daily. The percent GFP positive cells is shown on the Y-axis and days on the X-axis. B. The maximum fluorescence intensity (MFI) for the same experiment as in A is shown on the Y-axis.
Unlike in the standard protocol above with media change daily, when DAC is used in patients as cancer therapy, nutrient supply to tumor is initially constant as is the cycling time of tumor cell subpopulations. Furthermore, half-life of the drug in the cancer cells, and especially once incorporated into DNA is likely much longer. We could test for different such conditions using the property of our system of allowing time-lapse fluorescence microscopy. With such an approach some surprisingly different and probably clinically important differences in timing and kinetics for activation of the recombinant SFRP1-GFP allele are detected in response to a relatively low dose of 500 nM. Thus, after a single day dose, followed by a media change at 24 hours, cells positive for GFP expression reached 14% by 5 days (Fig. 3A). However, if media is not changed after this dose, GFP expression is more progressive and levels of ~36% cells are achieved by 5 days (Fig. 3B). Without media change after three days of drug administration, 53% of cells were positive at 5 days (Fig. 3C). Thus, the colon cancer cells without fresh media addition, and with only very short drug administration, are capable of progressively re-expressing SFRP1-GFP far beyond the 3 day peak seen in the usual protocols.
Fig. 3. Time course for induction of SFRP1-GFP expression induced by different treatment conditions of cells with 500 nM DAC.

A. Frames from time lapse video microscopy of cells with the SFRP1-GFP allele treated with DAC for 24 hours, followed by a media change, and then onset of the microscopy. Time points listed are for the video frames taken after the media change and no further addition of drug and all the cells in the field were manually counted for fluorescence for each frame shown with the % positive cells indicated at 120 hours. ( mag = 20x) B. Video microscopy is performed as in A after cells are again treated for 24 hours but without any further media change following drug administration and before video microscopy. Time frames start just after drug treatment with all conditions for counting of GFP+ cells as in A. C. Studies as in A and B with 500 nm DAC treatment for 3 days, with media change each day for the first two doses and video microscopy without media change after the final drug administration.
Use of the model to explore the effects of histone deacetylase inhibitor on SFRP1-GFP expression
We next used our reporter system to study features of a laboratory paradigm, which is being tested in clinical trials (5, 10, 14, 16–19), in which DNA demethylating agents and histone deactylase inhibitors (HDACi’s) are combined for re-expression of DNA hypermethylated genes (14, 16). Cancer genes with densely DNA hypermethylated, CpG island promoters, generally will not reactivate well with administration of HDACi’s alone but these drugs can provide additive or synergistic effects if given after a DNA demethylating agent (14, 16). When examined at the protein level as quantitated for our reporter system by FACs analysis, and when compared to RNA results by RT-PCR, TSA augments SFRP1-GFP reactivation at all DAC doses ranging from .01 to 1 μM (Fig. 4A).
Fig. 4. Studies of the response of SFRP1-GFP expression induced by the HDACi, TSA, with and without adding DAC treatment and and under different cell growth conditions.

A. TSA administered at 300 nM for 24 hours either alone, or following various doses of DAC given for 3 days, added as fresh drug and media each day. Y-axis = % + GFP cells as analyzed by FACS and fully described in previous experiments. Experiment is performed with initial high cell seeding; X-axis in the left panel = doses of each drug given below the bars with the top value for DAC and bottom values for combination doses with 300 nM constant for TSA. Y-axis = % + SFRP1 GFP + cells as in Fig. 3. Bars = response to the DAC dose alone (blue) and the drug combination (red) - note lack of, or very minimal, expression, initially, for no treatment with either drug (1st blue bar in each of 3 panels). Middle panel = the identical experiment but with simultaneously extracted RNA assayed with qRT-PCR using primers to monitor endogenous SFRP1 sequences and values for fold change normalized to GAPDH values on the Y-axis. Far right panel = same RNA as in the middle panel with qRT- PCR primers for GFP sequences in the SFRP1-GFP transcripts and fold changes on the y-axis. The error bar represent +/− SEM. B. Time-lapse video microscopy frames, at 12 hour intervals over 5 days, following a single, 300 nM dose of TSA administered to cells seeded at high cell confluence. Percentage of GFP+ cells was determined identically as in Fig. 3, and the values are indicated above each time point panel. Inverted phase images of the cells are shown beneath the fluorescence images in each case. Mag = 20x. C. Identical experiment to that in B except cells were seeded at low confluence before TSA administration. D. Quantitation of the GFP positive cells (Y-axis) versus time (X-axis) for studies in B and C with the left panel and right panels depicting the initial high confluence and low confluence seeding conditions, respectively. E. Induction of SFRP1-GFP in cells treated with 300 nM TSA for 24 hours after initial seeding at different densities ranging from 5–50%. Y-axis = % GFP positive cells as determined by FACS analysis; X-axis = levels of initial seeding density. First lane labeled 1.4− represents cells at 5% confluence initially and not treated with drug.
A recent study, using an exogenous reporter system, challenges the paradigm that HDACi’s cannot, alone, re-express DNA hypermethylated, cancer genes and finds that the DNA methylation rather limits sustained expression by facilitating re-recruitment of repressive chromatin (8). We now use our system to find that this situation is even more complex and that re-expression with HDACi alone fully depends on a window of vulnerability determined by cells having active DNA replication. In this regard, we do observe conditions under which TSA treatment, alone, can and cannot reactivate our silenced gene with promoter, DNA methylation. In time lapse fluorescence microscopy where media is not changed after a single 24 hr administration of 300 nM TSA, cells remain viable over a 5 day period (Fig. 4B, C). When seeded at high density, where little proliferation occurs before and after 24 hours of TSA administration, only a very small number of cells express SFRP1-GFP and this number then decreases rapidly (Fig. 4B). In striking contrast, when seeded at low density, SFRP1-GFP expression increases steadily from 24 hours on, reaching a much higher, and stable, peak over 2 to 4 days before declining slightly at 5 days (Fig. 4C). This dependency of the cells on seeding density is further highlighted when this study is quantitatively analyzed (Fig. 4D) and when various seeding densities are analyzed versus percentage of cells induced by TSA to express SFRP-GFP (Fig. 4E).
We hypothesized from the immediately above results, that the low density conditions might have revealed a previously not recognized requirement for DNA replication for sustained gene re-expression after TSA treatment. We studied this for cells seeded at low density by combining TSA treatment for 24 hours with short exposure to EdU for two hours, to monitor DNA replication, and subsequent sorting of cells for positive (GFP+ cells) versus negative (GFP− cells) levels (Fig. 5A–D). Indeed, GFP+ cells after TSA were more than 9-fold enriched for EdU incorporation than GFP− cells reflective of their being in the S-phase of the cell cycle (Fig. 5C, D). Importantly, this S-phase dependent activity of the drug occurs in the absence of any change in DNA methylation status in the CpG island surrounding the transcription start site of SFRP1 for either the wild type or recombinant SFRP1 alleles since we detect no appearance of any methylation signal by the very sensitive MSP assay (Fig. 5E).
Fig. 5. Relationships between response of SFRP1-GFP expression to TSA and the growth kinetics, and DNA replication status, of cells.

A. FACS sorting for GFP intensity after cells, seeded at low densities as in Fig. 4C, have been treated with 300 nM TSA for 24 hours. The cells collected for subsequent studies which have the lowest 20% expression are labeled P5 (pink color dots in far left box) and those which have the highest 25 % expression are labeled P4 (orange colored dots in far right box). Y- axis = GFP negative channel; X-axis = GFP fluorescence intensity as in previous figures. B. FACS analysis of non-TSA treated cells incubated with EDU for 2 hours (Y-axis = intensity of Pacific Blue staining for EDU; X-axis = intensity of staining with 7-AAD). High intensity EDU cells ( black dots) are located in S-phase. C. EDU labeling 120 min following 300 nM TSA administration (Y-axis) and cell cycle position by 7-AAD staining (X-axis) for the GFP low cells from A. Note low EDU uptake (square) in the S phase region of the cycle. D. Identical analysis as in C. but for the GFP high group from A. Note high EDU labeling (square) in the S phase position of the cycle. E. MSP analyses of the DNA methylation status of the endogenous promoter CpG island of SFRP1 and SFRP1-GFP alleles after cells have been treated with either 10 or 300 nM TSA and before and after Cre-recombination. Both the #90, and #1.4 recombinant clones have been analyzed in the top panel and the # 1.4 clone in the bottom panel. Primer locations are as detailed in Fig. 1C legend. M = methylated alleles; U = unmethylated alleles and controls are as per Fig. 1C. The upper panel is data for seeding of cells at high confluence with low GFP expression. The lower panel is low cell confluence with high GFP expression. Note the presence of the M band only throughout indicating that unmethylated alleles are not detected for any of the conditions analyzed. F. ChIP analysis of H3K4me3 and H3K27me3 ( middle and lower panels, respectively), and IgG control ( top panel) in the proximal sFRP1 promoter region ( primer set spanning (−770 to −700 bp ) in clone #1.4 cells. Untreated total cells = #1.4− and total cells treated with 300 nM TSA for 24 h = #1.4+. GFP− and GFP+ = GFP low and high cells, respectively, post TSA treatment and fractionated in the same experiment by FACS sorting as in panels A–D. The error bars represent +/− SEM.
For the above studies of TSA, we also investigated some key aspects of relevant chromatin changes at the key promoter sequences of the densely, DNA hypermethylated SFRP1. These studies are important in assessing whether the basal, chromatin mediated repression of the recombinant allele is similar to that for the wild type allele before TSA addition. In this regard, we have previously determined for the wild type alleles that even DAC, alone, when inducing re-exprexprssion of the silenced gene, increases histone acetylation at such sites with and without addition of TSA (20). Also, the best indication of basal transcriptional chromatin status for densely, DNA hypermethylated, genes is very low levels of the active chromatin mark, H3K4me2 or H3K4me3 methylation. Also, this mark increases dramatically when such genes are induced to re-express with DAC or when a gene which is only very partially DNA methylated is induced to increase expression with TSA (21). Indeed, as previously published for promoter sequence sites for SFRP1, when DNA hypermethylated in HCT116 cells (21, 22), we find by ChIP analyses of total cells that the H3K4me3 in the above studies has a very low level of H3K4me3 which increases 3-fold after TSA is administered to proliferating cells seeded at low density (Fig. 5, panel F). Furthermore, we find, as previously (21, 22), the repressive mark, H3K27me3 to be readily detectable at these same sequences and to drop by over 2-fold with TSA treatment (Fig. 5, panel F). The same trends were seen in the sorted high and low GFP expressing cells after TSA (Fig. 5, panel F) but inability in the experiments to fully purify these cell populations probably diminishes the detectable differences by the very sensitive ChIP assays of the chromatin marks.
In these above ChIP studies, we cannot, for certain, separate the status of the WT alleles from that of endogenous, promoter sequences in the recombinant SFRP1-GFP alleles. This is because the distinguishing exogenous sequences for the GFP cassette in the latter are too far downstream to link to the, above, upstream proximal promoter sequences in the Chip assay. However, the match of the very low basal levels of H3K4me3 and detectable H3K27me3 levels to our previous analyses of the densely, DNA hypermethylated SFRP1 in WT HCT116 cells (21, 22) firmly suggest to us that the endogenous promoter sequences in both the WT and recombinant SFRP1 alleles have a repressed chromatin status in the setting of retaining dense, basal, DNA hypermethylation.
Use of the model to study the kinetics of DAC induced SFRP1-GFP expression in-vivo
Our ability to visualize, at the cellular level, the re-expression of a cancer specific, DNA hypermethylated gene offered us the opportunity to examine DAC induced re-expression of SFRP1-GFP in vivo. The studies offer an important and somewhat surprising glimpse of a time course for this event during chronic DAC treatment that may be highly relevant for understanding use of this drug in cancer therapy.
We treated immune-incompetent mice, bearing established tumors after HCT 116-SFRP1-GFP, cell explanation, with 0.5 mg/kg DAC for 5 days per week for 4 weeks. Tumors were removed at 2 and 4 weeks during treatment and examined by monitoring of both GFP fluorescence and immunohistochemistry for GFP (Fig. 6). Surprisingly, only a slight increase in tumor cells with detectable GFP was seen in DAC versus mock treated mice after 2 weeks. However, a striking increase in such cells, as detected by both fluorescence and immunohistochemistry, was observed after 4 weeks of treatment (Fig. 6). These kinetics, compared to the prolonged time course for increases in SFRP1-GFP expression in the earlier, in-vitro studies, as discussed further below, may be very important to clinical use of drugs like DAC in cancer management.
Fig. 6.

Image of GFP positive cells in tumors. In 2 wks DAC treated animals, a certain number of GFP positive cells were observed. In 4 week treated animals, GFP positive cells dramatically increased as compared to the 2 week treated group group. ICC staining (GFP-Ab) showed that more GFP immune-labeled cells were seen than GFP-fluorescence cells (from the sFRP1-GFP transgenic cell line itself) in the tissue without ICC. In both the 2 week and 4 week time-points for mock animals, very few GFP positive cells were detected. Scale bar = 50 μm.
Discussion
We have built an assay system which we suggest has the utility for rapid, quantitative, and visual readout for effects of “epigenetic therapy” agents on abnormally silenced, DNA hypermethylated, cancer genes. It has already allowed us to suggest several new aspects concerning reactivation of such genes in terms of timing, duration, and DNA synthesis dependency of gene expression responses to DNA demethylating and HDACi’s. Our observations then suggest that our new system may have much potential for studying how low doses of the drugs studied, and other drugs, can reprogram cancer cells to provide anti-tumor effects. Moreover, it should be extremely useful for providing a rapid and quantitative readout for high throughput screens for new agents, or modifications of existing agents, which may benefit efforts to develop epigenetically based strategies for cancer prevention and treatment.
Our results on timing of DAC effects in our model already raise important implications for translational use of DNA demethylating agents and histone deactylase inhibitors in the clinic. For DNA demethylating agents like DAC, our findings for a 24 hour cell exposure followed by no change in culture media versus a typical 3 day exposure changing drug and media each day, are revealing. The former exposure indicates a much more prolonged response to the short does than the short half-life of the drug in aqueous solution would suggest. These in-vitro results are robustly amplified in the in-vivo results depicted in Fig. 6 wherein robust increase in numbers of cells with DAC induced expression of SFRP1-GFP occurred only between 2 and 4 weeks of chronic drug administration at a relatively low, clinically relevant, dose. All of these results may provide one explanation for the course of cellular re-programming results with low doses of DAC in our recent pre-clinical studies (7), and in the clinical responses to DAC in the setting of myelodysplasia/AML of the elderly (MDS/AML) where DNA demethylating agents have been approved by the FDA for efficacy (6, 23) - and for promising responses in the setting of advanced lung cancer (19). In the clinic, as well characterized in MDS/AML, patients typically take multiple cycles of therapy, over 2 to 3 months, before responses are seen (6, 23). We have speculated from pre-clinical work, that effect on exhaustion of stem-like cells, kinetics of drug uptake, ect might help explain such results. The present studies, both the in-viitro and in-vivo studies with our model system, suggest another component may be the long buildup of drug effects on accumulation of cells with re-expression of a silenced, DNA hypermethylated gene. While the half-life of currently used DNA demethylating agents in therapy is very short in the circulation (6, 23), our studies could suggest that this may not be the case for targeting DNMT’s once the drugs are incorporated into DNA in dividing cells. This step is pre-requisite for inhibiting the catalytic activity of these proteins and probably triggering their degradation (24). Our prolonged cellular effects, and possibly the prolonged course for therapeutic efficacy might then reflect kinetics wherein cumulative effects on DNA de-methylation, and gene re-expression, occur as cells come into cycle during chronic treatment.
Our findings are also important for considering the application of HDACi to tumor cells and have implications for use of such agents, alone, and in combination with DNA demethylating agents in the clinic. We now find an apparent heavy dependency for potency of TSA activation of a cancer-specific, densely DNA hypermethylated gene on ongoing DNA replication. Also, the gene reactivation without the loss of DNA methylation is similar to results achieved with removal of methyl cytosine binding proteins complexes, which are known to help mediate transcriptional repression through their interaction with, and targeting of, HDAC’s to DNA hypermethylated genes in cancer cells (25). The mechanisms explaining precisely how DNA replication and chromatin assembly are altered by HDAC inhibition in the current paradigm merits much further investigation. The kinetics demonstrated here indicates that, for clinical application, the timing for joint administration of DNA de-methylating drugs and HDACi may have to be heavily considered. Both drugs may depend on active cell replication for optimal onset and durability of their effects on re-expression of DNA hypermethylated genes. Both also, with different timing, decrease cell replication at some point after their interaction with cancer cells (7, 26, 27). Thus, care may have to be taken to avoid each drug from interfering with the other in clinical settings in terms of any efficacy based on their additive or synergistic effects for gene re-expression.
Supplementary Material
Acknowledgments
This paper was supported with SBB grant support from National Institute of Environmental Health Sciences (NIEHS) ES011858, National Cancer Institute (NCI) CA043318 and BioNumerik Pharmaceuticals, Inc. Video studies were produced in the Cell Imaging Core Facility at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins and to Kathy Bender for manuscript preparation.
Footnotes
Possible conflict of interest:
S.B.B. consults for MDxHealth. MSP is licensed to MDxHealth in agreement with Johns Hopkins University (JHU) and SBB and JHU are entitled to royalty shares received from sales.
References
- 1.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 3.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 5.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours-lessons from the past. Nat Rev Clin Oncol. 2013;10:256–66. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–46. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell. 2012;21:430–46. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Si J, Boumber YA, Shu J, Qin T, Ahmed S, He R, et al. Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res. 2010;70:6968–77. doi: 10.1158/0008-5472.CAN-09-4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–22. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 11.Cai Y, Geutjes EJ, de Lint K, Roepman P, Bruurs L, Yu LR, et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene. 2013 doi: 10.1038/onc.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, et al. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rattner A, Hsieh JC, Smallwood PM, Gilbert DJ, Copeland NG, Jenkins NA, et al. A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc Natl Acad Sci U S A. 1997;94:2859–63. doi: 10.1073/pnas.94.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–9. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 15.Rago C, Vogelstein B, Bunz F. Genetic knockouts and knockins in human somatic cells. Nat Protoc. 2007;2:2734–46. doi: 10.1038/nprot.2007.408. [DOI] [PubMed] [Google Scholar]
- 16.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 17.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–9. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 18.Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol. 2008;45:23–30. doi: 10.1053/j.seminhematol.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fahrner JA, Eguchi S, Herman JG, Baylin SB. Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 2002;62:7213–8. [PubMed] [Google Scholar]
- 21.McGarvey k, Van Neste L, Cope L, Ohm J, Herman J, Van Criekinge W, Schuebel K, Baylin S. Defining a Chromatin Pattern That Characterizes DNA Hypermethylated Genes in Colon Cancer Cells. Cancer Research. 2008;68:5753–9. doi: 10.1158/0008-5472.CAN-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–19. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Issa JP. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: dose, duration, and patient selection. Nat Clin Pract Oncol. 2005;2 (Suppl 1):S24–9. doi: 10.1038/ncponc0355. [DOI] [PubMed] [Google Scholar]
- 24.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–41. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Ballestar E, Esteller M. Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem Cell Biol. 2005;83:374–84. doi: 10.1139/o05-035. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Manero G, Issa JP. Histone deacetylase inhibitors: a review of their clinical status as antineoplastic agents. Cancer Invest. 2005;23:635–42. doi: 10.1080/07357900500283119. [DOI] [PubMed] [Google Scholar]
- 27.Robert C, Rassool FV. HDAC Inhibitors: Roles of DNA Damage and Repair. Adv Cancer Res. 2012;116:87–129. doi: 10.1016/B978-0-12-394387-3.00003-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.