

Abstract
Acute graft-versus-host disease (GVHD) is a major complication of allogeneic hematopoietic cell transplantation (HCT) and the main cause of nonrelapse mortality during the first 100 days post-transplant. Although GVHD can be prevented by extensive removal of mature donor T cells from the donor hematopoietic stem cell population, doing so eliminates any potential allogeneic graft-versus-tumor (GVT) effect also mediated by donor T cells and results in unacceptable rates of cancer relapse. One potential solution to this problem of separating GVHD development from a GVT response is to prevent T cell–mediated GVHD in the intestinal tract (IT) while preserving systemic antihost alloreactivity of donor T cells that target residual tumor cells expressing host alloantigens. We examined the ability of the anti-inflammatory rho kinase inhibitor, fasudil, given orally and intraperitoneally, to prevent GVHD in a C3H → B6C3F1 mouse model of MHC-haploidentical bone marrow transplantation. Fasudil-treated recipients of anti-thy-1 mAb + C′ treated bone marrow (ATBM) cells plus T cells had a 73% 90-day survival compared with 25% among untreated ATBM + T cell recipients (P < .0001). Severe initial weight loss was similar in the 2 groups, but less diarrhea was observed among treated animals, and fasudil-treated survivors recovered more weight than untreated survivors. Skin inflammation occurred and resolved between weeks 2 and 8 with similar severity and kinetics in both treated and untreated surviving animals, indicating persistent alloreactivity. Day 10 posttransplantation splenocytes from fasudil-treated mice, containing mature donor T cells, and day 98 splenocytes, containing mature donor and de novo thymus-derived T cells, exhibited alloreactivity against host parental antigens, as assessed by in vitro IFN-γ production and rounds of allostimulated proliferation, respectively. These data support the idea that targeted treatment of the IT with rho kinase inhibitors can ameliorate lethal GVHD while preserving systemic alloreactivity. The results also suggest that similar mechanisms of IT-specific tolerance or resistance to GVHD operate in fasudil-treated and untreated long-term survivors of allogeneic ATBM + T cells.
Keywords: GVHD, Fasudil, Intestinal tract, Rho kinase, Haploidentical, Allogeneic
INTRODUCTION
Although multiple organs are typically involved, the main cause of death in acute graft-versus-host disease (GVHD) appears to be damage to the intestinal tract (IT), especially the small and large bowel [1,2]. Whereas skin involvement is more frequent, IT GVHD is more refractory to treatment and more predictive of nonrelapse mortality (NRM) [3–5]. Prevention of GVHD by purging donor hematopoietic cell transplants (HCTs) of mature lymphocytes before transplantation leads to untenable rates of tumor relapse because of the loss of a graft-versus- tumor (GVT) effect, also mediated by mature donor lymphocytes (primarily T cells). Indeed, a roughly inverse correlation between severity of GVHD and incidence of relapse has been documented [6–8]. This conundrum has spurred efforts to mitigate GVHD while preserving a GVT effect.
One strategy is to identify tumor-specific antigens and the T cell clones recognizing them so they can be selectively expanded while all other allogeneic clones are removed [9,10]. The limited number of cancers with well-defined tumor-specific antigens is an obstacle to this approach, but so, too, is the removal of alloreactivity, which comprises a much broader, stronger, and less readily evaded tumor response repertoire than that generated against a single tumor-specific antigen. Another approach is to augment regulatory T cells (Tregs) in the graft with additional donor Tregs; however, like immunosuppression in general or removal of mature donor T cells from the donor graft, this may carry the risk of increased relapse [11].
A very different conceptual strategy is to ameliorate GVHD in the most vulnerable organs while preserving alloreactivity in general and an alloreactive GVT effect in particular. The theoretical possibility of achieving this situation has been demonstrated in mouse models by use of donor T cells genetically deficient in receptors critical for gut homing [2,12] or in cytotoxic T lymphocyte (CTL) synapse formation [13]. More directly relevant to treatment, murine GVHD has been suppressed by immunodepletion of T cells expressing gut homing receptors from the transplant [12], injection of mAb targeting neovascularizing donor endothelial cells [14,15], or administration of inhibitors of T cell receptorecoupled protein kinase C (PKC)α and PKCθ proteins [13]. The latter approach had the disadvantage of diminishing (although, surprisingly, not eliminating) antitumor CTL By contrast, the homing receptor–based approaches appear to reduce the influx of inflammatory cells into the bowel mucosa while leaving the remaining systemic alloimmunity largely intact. This raises the possibility that alloreactive T cells might be redirected to the skin and non-IT organs, exacerbating morbidity and mortality from nondigestive tract GVHD. Although this was not observed in the studies just cited, there was some evidence of redirection of GVHD away from gut and into skin in mice fed a vitamin A–deficient diet leading to decreased CCR9 and α4β7 expression on lymphocytes [16].
Importantly, an approach that, despite its potential for systemic immunosuppression, seems to target the IT has recently been translated from animals to clinical studies. The CCR5 blocking agent, maraviroc, given orally to patients from −2 to +30 days post-HCT, resulted in dramatically reduced GVHD and NRM within the first 100 days as well as 1 year post-transplantation compared with historical control subjects at the same institution [5]. In particular, there was a NRM rate of 0% within the first 100 days and no grade III or IV IT or liver GVHD. CCR5 was first shown to be important as a marker of host-reactive T cells in mouse models of GVHD targeting skin, liver, and gut tissues [17,18]. Thus, despite the expression of CCR5 on alloreactive lymphocytes throughout the body, gut-sparing effects appeared to dominate the clinical results of this trial, accounting for most of the beneficial effects.
In light of the above studies, we took an approach with anticipated systemic effects but also with the potential to preferentially target the IT. We used a rho-associated coiled coil kinase inhibitor, fasudil, demonstrated to have suppressive effects on inflammatory cell activation, motility, and homing in the context of preclinical models of autoimmune disease [19] and tumor metastasis [20–28]. Although potentially systemic in impact, we reasoned that p.o. and i.p. administration might result in predominant protection of the IT because of concentration gradients and initial local gut mucosal and serosal uptake. Fasudil is of particular interest, because it has an excellent 20-year safety record of use in Japan for prevention of poststroke cerebral artery spasm [29,30].
METHODS
Mice
Eight-week-old male C3H/HeJ (C3H; H2k), (C57BL/6 × C3H)F1 (B6C3F1; H2b/k), DBA/2J (H2d), and BALB/cJ (H2d) mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and used at 10 to 20 weeks as cell recipients or donors. Mice were kept in a pathogen-free environment in autoclaved microisolator cages and were provided with autoclaved water and food ad libitum. All protocols used in this study were approved by the Hackensack University Medical Center’s Institutional Animal Care and Use Committee.
Transplantation Experiments
All preparative manipulations of donor cells were conducted in PBS supplemented with .1% bovine BSA. T cell–depleted (anti-thy-1 mAb-treated) bone marrow (ATBM) cells were prepared by flushing bone marrow cells from the femurs of donor mice, followed by incubation with J1j (anti-thy-1.2) mAb (1:100 dilution) and guinea pig complement (C′) (1:5) for 45 minutes at 37°C. T cell–enriched donor cell populations were prepared from pooled RBC-lysed spleen cell suspensions. B cells were removed by incubation with J11D2 mAb (1:500) and C′ for 45 minutes at 37°C. B6C3F1 host animals were exposed to 1100 cGy irradiation (split dose of 550 cGy × 2, 4 hours apart) using a 137Ce source (Gammacell 40 Exactor; MDS Nordion). Within 3 to 4 hours after irradiation was completed, the mice were transplanted via tail vein injection with 2 × 106 C3H ATBM cells. For GVHD induction, mice simultaneously received 5 × 106 C3H splenic enriched T cells. Mice were weighed twice per week and evaluated daily for clinical signs, including mobility, activity, hunching, grooming, diarrhea (during observation and handling), loss of fur, and skin lesions. Weight loss up to 30% of initial weight was permitted if mice remained active, because these animals often survived and recovered some or all of the lost weight over the observation period.
Fasudil Treatment
Fasudil-treated mice started the drug 24 hours before irradiation and transplant, receiving both i.p. (fasudil-hydrochloride, 200 μg twice daily) and p.o. (fasudil-dihydrochloride, 1 mg/mL drinking water, or ~3 mg/day). This dual mode of administration was continued for 10 days post-transplantation, after which time i.p. injections were discontinued but p.o. drug was maintained for the period of observation (up to 90 days).
In Vitro Assays
IFN-γ ELISpot
Spleens from mice 10 days post-transplantation were homogenized to single-cell suspensions, pooled, and cultured at 1 × 107 responder cells with 1 × 107 irradiated (3000 cGy) stimulator cells in 2 mL for 4 days. Cells were washed and plated at 2 × 105 overnight in triplicate on nitrocellulose EIA plates coated with rat anti-mouse IFN-γ mAb (50 μg/mL), developed with biotinylated rat anti-mouse IFN-γ (4 μg/mL), and ELISpots scored by an automated CTL Immunospot Series 3A reader.
CFSE proliferation and IFN-γ production in one-way mixed lymphocyte reactions
Twelve × 106 splenocytes from B6C3F1 hosts were harvested at day 98 post-transplantation, labeled with CFSE (5 μM), and mixed at a 3:1 responder-to-stimulator ratio with unlabeled 3000 cGy irradiated B6C3F1, C3H, or BALB/c splenocytes. On days 2, 3, and 5 of culture, supernatant samples (100 μL) were taken for batch EIA assay of IFN-γ (BD Biosciences), with fresh media replaced on days 2 and 3. Cells were recovered and assayed for CFSE intensity peaks using a flow cytometer (model FC500; Beckman Coulter).
Histopathology
Long-term survivor mice (12 to 13 weeks post-transplant) were killed and immediately dissected to remove liver, spleen, lung, tongue, and small intestines. Tongue was used as a substitute for skin, because it is much easier to prepare for histology and typically reflects the events simultaneously occurring in the dermis. Tissue was sliced into 2- to 5-mm sections, washed with PBS, and cryopreserved in OCT freezing media in molds floating in liquid nitrogen. After 10 minutes, frozen samples were transferred to −80°C and stored for subsequent thin sectioning and staining with H & E. Slides were scored using a published grading system [31] for focal or diffuse infiltration of inflammatory leuokocytes into lamina propria, crypts, with or without destruction of crypt and villous architecture: 0, rare inflammatory cells in the lamina propria; 1, increased numbers of granulocytes in the lamina propria; 2, confluence of inflammatory cells extending into the submucosa; 3, transmural extension of the inflammatory infiltrate. Crypt damage was scored as follows: 0, intact crypts; 1, loss of the basal one-third; 2, loss of the basal two-thirds; 3, entire crypt loss; 4, change of epithelial surface with erosion; 5, confluent erosion. Ulceration was scored as follows: 0, absence of ulcer; 1, 1 or 2 foci of ulcerations; 2, 3 or 4 foci of ulcerations; 3, confluent or extensive ulceration. Values were added to give a maximal histological score of 11.
Statistics
Survival curves were estimated using Kaplan-Meier’s product limit method. Comparison of survival curves was performed using a 2-sided log-rank test. Comparisons of treatment group weights over time were made using the nonparametric Kolmogorov-Smirnov test, with no a priori assumption of normal distribution. Proportions were compared by a 2-sided z test for significant differences. Survival data analysis was performed using SAS 9.2 (SAS Institute Inc., Cary, NC).
RESULTS
Survival
As shown in Figure 1, fasudil-treated B6C3F1 mice had significantly greater 90-day survival after injection of C3H ATBM + T cells compared with untreated recipients (73% versus 25%, P < .0001). In the GVHD control group, most mice (70%) succumbed to disease between days 8 and 28 posttransplantation. In the fasudil-treated group, most fatalities (5 of 26 mice) also occurred early post-transplantation (days 8 to 18), and only 2 additional deaths occurred later, between days 47 and 56. As expected, mice transplanted with only C3H ATBM cells all survived, and mice exposed to lethal irradiation alone without receiving any donor ATBM cells succumbed within a narrow window, 12 to 16 days postirradiation.
Figure 1.
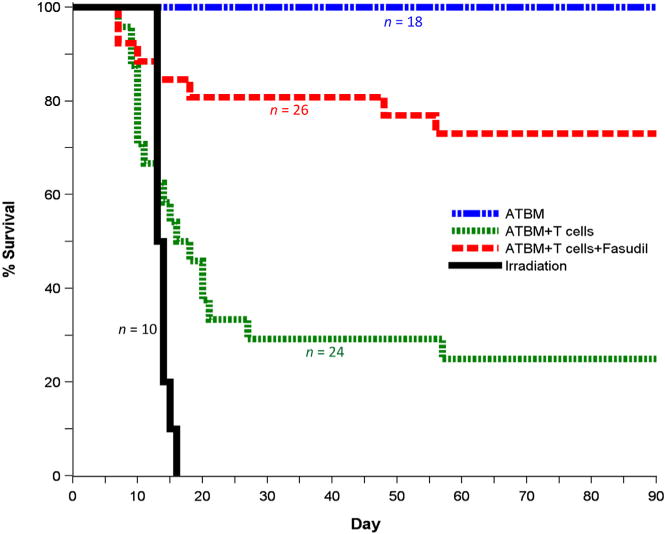
Fasudil decreases mortality from GVHD. Fasudil administered i.p. for 10 days and in drinking water for the duration of the experiment starting 1 day before lethal irradiation and transfer of ATBM + donor T cells significantly increased 90-day survival from 25% to 73% (P < .0001). Irradiated mice not receiving donor cells showed different early survival rates, with all deaths occurring within a relatively narrow window of 12 to 16 days. Data were pooled from 3 separate replicate experiments involving total sample sizes of 10 to 26 for the different groups, as indicated.
Weight Loss
With or without fasudil treatment, ATBM + T mice experienced significant weight loss within the first 2 weeks (25% to 30%) compared with ATBM-only recipients (10%). On average, fasudil-treated and untreated surviving mice lost the same percentage of weight in the first 1 to 2 weeks, with comparable kinetics of weight stabilization and gradual increase among 1-month survivors. There was continued gradual, but incomplete, weight recovery over the remaining period of observation (Figure 2). Among these progressively more self-selected animals in both groups surviving beyond day 28, weight recovery of fasudil-treated mice was significantly greater than among untreated mice receiving ATBM + T cells (P = .001).
Figure 2.
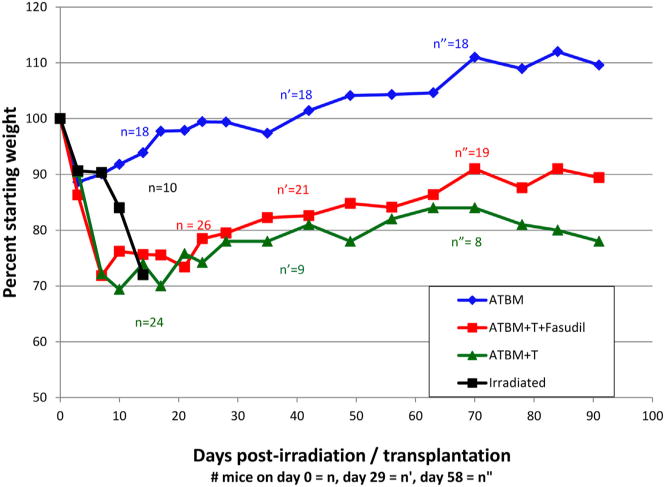
Fasudil does not fully prevent weight loss during acute GVHD. Despite impressive reduction in mortality, mice receiving fasudil along with ATBM + donor T cells were not spared significant weight loss, comparable with untreated ATBM + T recipients, during the first month post-transplantation. In the first week, these groups lost 2 to 3 times as much weight as did ATBM only recipients or as irradiated mice receiving no donor cells. Fasudil-treated mice surviving the first month of ATBM + T cell transplantation, as well as far fewer untreated surviving mice, gradually gained back more of their lost weight than the fewer surviving untreated mice (P = .001), but never caught up with the ATBM-only group (P < .0001).
Irradiated mice receiving no stem cell replacement closely matched the weight curve of ATBM transplants for the first 7 days, losing only 10% of starting weight rather than the 25% to 30% lost by mice receiving ATBM + mature donor T cells. However, although the latter groups stabilized or gradually increased their weights during the second and subsequent weeks post-transplant, the irradiated, untransplanted animals continued to lose weight, before dying between days 12 and 16.
Diarrhea
Despite the similar weight losses among fasudil-treated and untreated T cell recipients in the first 2 weeks, the 2 groups had different rates of diarrhea. Most untreated mice (>70%) had loose stools for 1 or more days between the second and third weeks post-transplantation, the period of highest mortality incidence. By comparison, less than 20% of the fasudil-treated mice developed clinically observable diarrhea during that time. Diarrhea resolved by the fourth week among survivors in both groups. Irradiated mice not receiving donor cells were not observed to have diarrhea before death.
Gut Histopathology
H & E staining of tissue sections from long-term survivors of fasudil-treated or untreated groups showed minimal or no inflammation of small intestines, liver, spleen, or tongue. Figure 3A shows the normal histology of gut crypts and villi seen in most sections among 5 fasudil-treated mice killed 90 days post-transplant. In 3 of 5 mice examined, moderate inflammatory cell infiltrates were seen at the base of the crypts in some sections, but crypt and villous architecture was intact (Figure 3B), giving an overall score of only 2/11 in the most severe cases. Of the 3 untreated long-term survivors killed for histopathology, none exhibited even mild inflammation of the sampled tissues in any of the examined sections (scoring 0/11).
Figure 3.
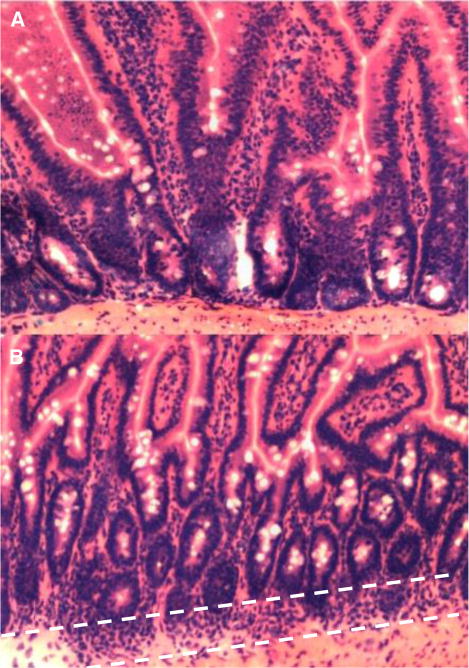
Long-term fasudil-treated and untreated survivors of GVHD have minimal inflammation of sampled tissues. Most small bowel sections from fasudil-treated animals showed normal architecture of crypts and villi, without evidence of inflammation (A). In 3 of 5 animals, scattered areas of lamina propria and basilar crypt inflammation were seen (dashed white lines), with no disruption of the crypt or villous architecture (B). No inflammation was observed in multiple samples taken from 3 untreated surviving animals (not shown).
Spleen Size
A suggestion of modest non-IT protection was the roughly 50% increase in day 90 spleen cellularity among fasudil-treated (3.6 × 106 per spleen) versus untreated (2.2 × 106 per spleen) mice receiving mature donor T cells. However, both groups had greatly reduced splenocyte counts (roughly 7% versus 4%, respectively) compared with ATBM-only recipients’ spleens, which contained approximately 5 × 107 cells per spleen.
Skin Involvement
Despite the better survival outcome and decreased diarrhea, all fasudil-treated mice lost hair and developed grossly observable skin inflammation and ulceration with similar frequency and time course as untreated animals within each experiment, starting at week 2 and resolving by week 8 (not shown). Again, as for weight recovery, skin lesion resolution by week 8 applied to the subset of animals from both groups surviving through week 8, comprised mostly of animals subsequently found to be long-term survivors.
Systemic Alloreactivity
Persistent skin involvement, weight loss, and only a very modest increase in spleen cellularity suggested that neither systemic host-specific allotolerance nor profound immunosuppression was the basis for fasudil’s beneficial impact on GVHD mortality. To directly address the issue of systemic alloreactivity to host MHC molecules, we harvested spleen cells 10 days after transplantation, the earliest time point at which donor T cells were recoverable in sufficient numbers from host spleens, and, at the other extreme, after the final week of observation (day 98 post-transplantation). Table 1 presents data for day 10 splenic lymphocytes, which reflect only donor T cells, because no stem or progenitor cell-derived T cells emigrating from the host thymus are present in host spleens at this time point. As shown, there was no evidence of fasudil-induced allotolerance to host MHC, as measured by IFN-γ ELISpot after 1-way donor C3H versus irradiated B6C3F1 stimulation of splenocytes in vitro. Early time point donor-derived T cells responded at least as robustly as splenocytes from untreated mice. Indeed, the greater response to B6 parental MHC alloantigens than to third party DBA/2 (H2d) alloantigens suggested possible in vivo priming within the B6C3F1 hosts. As expected, spleens from mice receiving no mature donor T cells (ATBM only) made no responses to any of the stimuli, because no newly formed T cells had yet emerged from the thymus and migrated to the spleen.
Table 1.
Day 10 IFN-γ Secreting Spleen Cell Frequencies after 5-Day One-Way Mixed Lymphocyte Reactions versus Self (C3H), Host B6C3F1, or Third-Party (DBA/2, H2d) Stimulators
| Irradiated Stimulators | Responding Splenocytes (per 2 × 105 cells) from Hosts Receiving:
|
||
|---|---|---|---|
| ATBM Only | ATBM + T Cells | ATBM + T Cells + Fasudil | |
| C3H | 0 | 4.6 | 2.3 |
| B6C3F1 | 0 | 82.3 | 115.6 |
| DBA/2 (H2d) | 0 | 16.0 | 18.0 |
| Con A | 1 | 61.3 | 120.0 |
At this early time point, only donor mature T cells are present and show no evidence of general immune suppression or tolerance for host B6 parental derived MHC alloantigens in fasudil-treated animals. Differences between ATBM-only and ATBM + T cell hosts, with or without fasudil treatment, were significant for all stimulations (P < .001).
Spleens from day 98 survivor mice had many more lymphocytes, allowing CFSE labeling of pooled spleen cells with flow-based assessment of cell division (dilutional peaks) in response to alloantigen recognition. Figure 4 shows that CFSE-labeled splenic lymphocytes from B6C3F1 recipients of ATBM cells alone had a relatively low proportion of cells undergoing multiple rounds of division after 5 days of stimulation with irradiated host cells (Figure 4A), compared with the proportion of cells undergoing multiple rounds of division in response to third-party allogeneic BALB/c stimulators (Figure 4B). The limited response to parental B6 MHC antigens is expected from C3H stem cell–derived T cells generated de novo within the F1 host thymic environment. By contrast, splenocytes from recipients of ATBM + donor T cells made a much stronger response to B6C3F1 cell stimulation (Figure 4C), very similar in magnitude to their response to BALB/c stimulation (Figure 4D). Fasudil-treated recipients of ATBM + T cells also contained splenocytes that made a strong response to B6C3F1 stimulation, with multiple rounds of cell division that yielded weakly fluorescent peaks (Figure 4E), compared with their response to BALB/c stimulation (Figure 4F).
Figure 4.
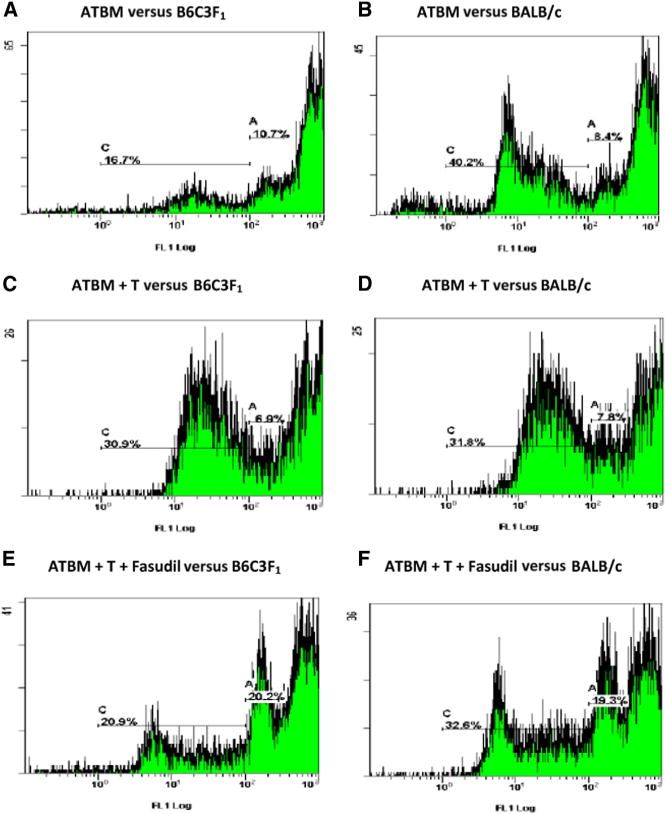
CFSE dilution peak determination of spleen cell alloreactivity in 1-way mixed lymphocyte reactions (responder versus irradiated stimulator). Pooled splenocytes from mice 98 days after receiving C3H donor anti-thy-1 treated bone marrow only (ATBM) made some proliferative response to host B6C3F1 cells (A). Roughly 11% of cells divided once, and 17% divided more than once, which is expected as a consequence of normal responses to self-restricted foreign environmental antigens. More cells (40% versus 17%, P < .001) responded with 2 or more cell divisions when third-party BALB/c cells were used as stimulators (B). By contrast, spleen cells from mice receiving mature donor T cells along with ATBM made a strong response to host B6C3F1 cell stimulation, with ~ 7% dividing once and ~31% more than once (C), not significantly different from their response (8% and 32%, respectively) to BALB/c (D), but significantly greater than (A) ATBM anti-F1 responses (P < .001). Mice receiving fasudil, along with donor T cells and ATBM (E), also made a stronger alloresponse to B6C3F1 host cells than mice receiving only ATBM (A), with 20% dividing once and 21 % dividing more than once. Although these proportions both differ significantly (P < .001) from the respective proportions of cells from the untreated mice (C), they are still greater in both cases than the corresponding peaks in ATBM-only spleen cultures (P < .001) and, compared with (C), also have a population of cells undergoing additional rounds of alloreactive division (lowest signal intensity peak). Fasudil-treated mice also retained strong anti-BALB/c MHC responses (F), with a peak profile roughly similar in distribution to (E). Data are representative of 2 replicate experiments and 4 culture time points.
DISCUSSION
Loss of alloreactive GVT effect is the main drawback of purging mature T cells from the donor graft to avoid GVHD development after HCT. In theory, protecting the most vulnerable target organs from lethal GVHD, while maintaining systemic antihost alloreactivity, at least during the early post-transplantation period, might reduce NRM and still allow efficient elimination of residual tumor by cell-mediated effector mechanisms directed at host alloantigens. Through the use of donor cells with genetically defective gut homing, previous mouse studies suggested the feasibility of such an approach [12–16]. Moreover, monoclonal antibodies to gut homing lymphocyte surface receptors ameliorated GVHD, and, in the case of anti-α4β7, GVT were shown to remain intact [12]. A less organ specific, systemic inhibition of CTL PKCα and PKCθ proteins also prevented GVHD and left some alloreactivity intact, with the GVT effect not detectably diminished [13].
In humans, a trial of the CCR5 antagonist, maraviroc, from −2 to +30 days post-HCT resulted in dramatically reduced acute GVHD and NRM for at least 1 year post-transplantation, compared with historical control subjects at the same institution [5]. In particular, there was no grade III or IV IT or liver acute GVHD. Thus, a relatively short course of treatment during the initial period of transplantation appeared sufficient to alter the subsequent course of GVHD for at least 1 year. Although the 20% increase in cancer relapse observed (56% versus 46% in recent historic control subjects from the same institution) did not reach statistical significance but did raise concerns regarding the possibility that inhibition of CCR5 might interfere with GVT effects [32].
On the other hand, the lack of a much more dramatic increase in relapse after early and brief CCR5 blockade is consistent with a retrospective analysis that found a strong inverse correlation of recurrent cancer with chronic GVHD but not acute GVHD [6]. Furthermore, CCR5 has been identified as a stronger marker of gut-homing alloreactive cells [33] than as a skin-homing alloreactive cells [34] during the acute phase of GVHD in humans, and IT GVHD is more lethal than skin GVHD. If these findings hold up in further studies, they would give impetus to the idea that inhibition of IT acute GVHD by early, short-term immunomodulation is beneficial for reducing NRM without eliminating a GVT effect. The addition of a second agent, such as fasudil, with protective effects against IT GVHD might allow for dose sparing or a shortened course of maraviroc, thereby maintaining suppression of lethal GVHD with even less chance of relapse.
The results presented herein support the concept of protecting GVHD targeted IT organs while maintaining systemic antihost alloreactivity and, consequently, a GVT effect. Amelioration of diarrhea and ongoing recovery of lost weight suggests IT protection by fasudil. The early death of more severely affected mice before their weights could fall sufficiently to influence group averages almost certainly masked a more dramatic treatment impact. It is likely that the p.o. and i.p. administration of fasudil contributed to targeting the IT for protection against the effects of inflammatory cytokines and cytotoxic alloreactive T cells. These results do not elucidate mechanisms, except to rule out general allotolerance or immune suppression. It is possible that IT vascular endothelia permeability is more sensitive to rho kinase inhibition than vascular endothelia in the spleen or skin, thereby allowing more effective inhibition of inflammatory cell egress into the gut mucosa as compared with skin. Localized generation and/or targeting of Tregs and myeloid-derived suppressor cells is also a possibility.
Fasudil inhibition of rho-associated coiled coil kinase targets several functions that are critical for IT mucosal inflammation: Leukocyte movement along endothelial receptors is reduced [28,35–37], as are cell polarization and uropod formation [21,35,38–41] and diapedesis [35,39–43]—all required for penetration of endothelial intercellular junctions. Fasudil’s protective effects on the endothelium are complimentary. Tight junction integrity and impermeability are increased [23,44–48], making it more difficult for cells to create intercellular gaps through which to cross the endothelium. Additionally, fasudil may improve oxygenation within the IT mucosa, because inhibition of rho-associated coiled coil kinase increases eNOS activity, leading to NO mediated dilation of the gut microvasculature [49,50]. This, along with the drug’s VEGF antagonism [39,51], could decrease hypoxia driven neovascularization [52–55], which has been found to be a hallmark of IT GVHD [13,14].
Hypoxia has been associated with Treg induction, especially in the context of tumor growth [56–69], so alleviation of hypoxia by fasudil would not be expected to directly favor the development of IT Tregs. On the other hand, immunotolerance is the default setting of the healthy IT, with a dominant effect of Tregs suppressing the response to most antigens of gut origin. Most of the body’s vitamin A is within fat-storing hepatic stellate cells that, in the presence of vitamin A–derived retinoic acid and TGF-β, induce Ag-specific Tregs directly or induce resident myeloid-derived suppressor cells to generate Tregs [70] that home back to liver and IT. To the extent that fasudil helps to maintain and restore a relatively normal IT and liver microenvironment, it may favor the default generation of Tregs.
Identifying the various mechanisms of action for fasudil protection against GVHD and demonstrating preservation of alloreactive GVT effect support a shift in the paradigm of GVHD treatment away from systemic immunosuppression and toward organ targeted protection. In combination with 1 or more agents discussed above, fasudil could become a mainstay of GVHD prophylaxis and treatment in the setting of allogeneic HCT. Clinical translation should be facilitated by the drug’s excellent safety profile during 2 decades of clinical use for prevention of poststroke cerebral spasm [29,30].
There is room for improvement with the fasudil approach. First, one-fourth of the treated animals still succumbed to GVHD, including some during initial i.p. treatment, with an incidence indistinguishable from the untreated group for the first 8 days. Second, treated surviving mice experienced severe weight loss for the initial 2 weeks, similar to that seen among untreated animals. Third, splenocytes from treated, long-term survivors were dramatically reduced in number compared with ATBM-only recipients. Fourth, the skin remained a major site of GVHD in animals receiving fasudil via the i.p. and p.o. routes, although all surviving mice resolved their skin lesions. Skin GVHD is a major cause of morbidity in patients. Topical fasudil may eventually be useful in treating persistent skin targeted GVHD.
Finally, it is worth noting that, other than improved weight gain, the grossly observable, histopathological, and immunological phenotype of fasudil-treated long-term survivors is largely indistinguishable from the much less frequent untreated long-term survivors. That is, fasudil appears to foster a series of events favoring organ selective tolerance or resistance, which also occurs naturally in some untreated animals. The details of this protective process remain to be elucidated, but the observation suggests that short-term treatments, with other agents having different proximate mechanisms of action, such as those mentioned above, may be combined with fasudil to generate potent, dose-sparing regimens.
Acknowledgments
We thank Dr. Jenny Zilberberg for expert technical advice with flow cytometry and ELISpot procedures. We thank Dr. Themba Nyirenda for expert statistical support, and Dr. David Lagunoff for histopathology staining and reading of slides.
Footnotes
Financial disclosure: ■■■.
Conflict of interest statement: ■■■.
References
- 1.Hill GR, Ferrara JL. The primacy of the gastrointestinal tract as a target organ of acute graft-versus-host disease: rationale for the use of cytokine shields in allogeneic bone marrow transplantation. Blood. 2000;95:2754–2759. [PubMed] [Google Scholar]
- 2.Chen X, Dodge J, Komorowski R, Drobyski WR. A critical role for the retinoic acid signaling pathway in the pathophysiology of gastrointestinal graft-versus-host disease. Blood. 2013;121:3970–3980. doi: 10.1182/blood-2012-08-445130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takatsuka H, Iwasaki T, Okamoto T, Kakishita E. Intestinal graft-versus-host disease: mechanisms and management. Drugs. 2003;63:1–15. doi: 10.2165/00003495-200363010-00001. [DOI] [PubMed] [Google Scholar]
- 4.Cho BS, Lee SE, Song HH, et al. Graft-versus-tumor effect according to type of graft-versus-host disease defined by National Institutes of Health consensus criteria and associated outcomes. Biol Blood Marrow Transplant. 2012;18:1136–1143. doi: 10.1016/j.bbmt.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Reshef R, Luger SM, Hexner EO, et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. 2012;367:135–145. doi: 10.1056/NEJMoa1201248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ringdén O, Shrestha S, da Silva GT, et al. Effect of acute and chronic GVHD on relapse and survival after reduced-intensity conditioning allogeneic transplantation for myeloma. Bone Marrow Transplant. 2012;47:831–837. doi: 10.1038/bmt.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee SJ, Klein JP, Barrett AJ, et al. Severity of chronic graft-versus-host disease: association with treatment-related mortality and relapse. Blood. 2002;100:406–414. doi: 10.1182/blood.v100.2.406. [DOI] [PubMed] [Google Scholar]
- 8.Signori A, Crocchiolo R, Oneto R, et al. Chronic GVHD is associated with lower relapse risk irrespective of stem cell source among patients receiving transplantation from unrelated donors. Bone Marrow Transplant. 2012;47:1474–1478. doi: 10.1038/bmt.2012.58. [DOI] [PubMed] [Google Scholar]
- 9.Patterson AE, Korngold R. Infusion of select leukemia-reactive TCR Vbeta+ T cells provides graft-versus-leukemia responses with minimization of graft-versus-host disease following murine hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2001;7:187–196. doi: 10.1053/bbmt.2001.v7.pm11349805. [DOI] [PubMed] [Google Scholar]
- 10.Fanning SL, Zilberberg J, Stein J, et al. Unraveling graft-versus-host disease and graft-versus-leukemia responses using TCR Vβ spectratype analysis in a murine bone marrow transplantation model. J Immunol. 2013;190:447–457. doi: 10.4049/jimmunol.1201641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadal E, Garin M, Kaeda J, et al. Increased frequencies of CD4(+) CD25(high) T(regs) correlate with disease relapse after allogeneic stem cell transplantation for chronic myeloid leukemia. Leukemia. 2007;21:472–479. doi: 10.1038/sj.leu.2404522. [DOI] [PubMed] [Google Scholar]
- 12.Petrovic A, Alpdogan O, Willis LM, et al. LPAM (alpha 4 beta 7 integrin) is an important homing integrin on alloreactive T cells in the development of intestinal graft-versus-host disease. Blood. 2004;103:1542–1547. doi: 10.1182/blood-2003-03-0957. [DOI] [PubMed] [Google Scholar]
- 13.Haarberg KM, Li J, Heinrichs J, et al. Pharmacologic inhibition of PKCα and PKCθ prevents GVHD while preserving GVL activity in mice. Blood. 2013;122:2500–2511. doi: 10.1182/blood-2012-12-471938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penack O, Henke E, Suh D, et al. Inhibition of neovascularization to simultaneously ameliorate graft-vs-host disease and decrease tumor growth. J Natl Cancer Inst. 2010;102:894–908. doi: 10.1093/jnci/djq172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penack O, Socié G, van den Brink MR. The importance of neovascularization and its inhibition for allogeneic hematopoietic stem cell transplantation. Blood. 2011;117:4181–4189. doi: 10.1182/blood-2010-10-312934. [DOI] [PubMed] [Google Scholar]
- 16.Koenecke C, Prinz I, Bubke A, et al. Shift of graft-versus-host-disease target organ tropism by dietary vitamin A. PLoS One. 2012;7:e38252. doi: 10.1371/journal.pone.0038252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer LA, Sale GE, Balogun JI, et al. Chemokine receptor CCR5 mediates alloimmune responses in graft-versus-host disease. Biol Blood Marrow Transplant. 2010;16:311–319. doi: 10.1016/j.bbmt.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murai M, Yoneyama H, Harada A, et al. Active participation of CCR5(+) CD8(+) T lymphocytes in the pathogenesis of liver injury in graft-versus-host disease. J Clin Invest. 1999;104:49–57. doi: 10.1172/JCI6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stirzaker RA, Biswas PS, Gupta S, et al. Administration of fasudil, a ROCK inhibitor, attenuates disease in lupus-prone NZB/W F1 female mice. Lupus. 2012;21:656–661. doi: 10.1177/0961203312436862. [DOI] [PubMed] [Google Scholar]
- 20.Wang DS, Dou KF, Li KZ, Song ZS. Enhancement of migration and invasion of hepatoma cells via a rho GTPase signaling pathway. World J Gastroenterol. 2004;10:299–302. doi: 10.3748/wjg.v10.i2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vishnubhotla R, Sun S, Huq J, et al. ROCK-II mediates colon cancer invasion via regulation of MMP-2 and MMP-13 at the site of invadopodia as revealed by multiphoton imaging. Lab Invest. 2007;87:1149–1158. doi: 10.1038/labinvest.3700674. [DOI] [PubMed] [Google Scholar]
- 22.Kamai T, Tsujii T, Arai K, et al. Significant association of rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9:2632–2641. [PubMed] [Google Scholar]
- 23.Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of rho. Nat Cell Biol. 2002;4:408–415. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- 24.Croft DR, Sahai E, Mavria G, et al. Conditional ROCK activation in vivo induces tumor cell dissemination and angiogenesis. Cancer Res. 2004;64:8994–9001. doi: 10.1158/0008-5472.CAN-04-2052. [DOI] [PubMed] [Google Scholar]
- 25.Somlyo AV, Bradshaw D, Ramos S, et al. Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem Biophys Res Commun. 2000;269:652–659. doi: 10.1006/bbrc.2000.2343. [DOI] [PubMed] [Google Scholar]
- 26.Ying H, Biroc SL, Li WW, et al. The rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther. 2006;5:2158–2164. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]
- 27.Bourguignon LY, Zhu H, Shao L, et al. Rho-kinase (ROK) promotes CD44v(3,8–10)-ankyrin interaction and tumor cell migration in metastatic breast cancer cells. Cell Motil Cytoskeleton. 1999;43:269–287. doi: 10.1002/(SICI)1097-0169(1999)43:4<269::AID-CM1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Barreiro O, Yanez-Mo M, Serrador JM, et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki Y, Shibuya M, Satoh S, et al. Safety and efficacy of fasudil monotherapy and fasudil-ozagrel combination therapy in patients with subarachnoid hemorrhage: sub-analysis of the post-marketing surveillance study. Neurol Med Chir (Tokyo) 2008;48:241–247. doi: 10.2176/nmc.48.241. discussion 247–248. [DOI] [PubMed] [Google Scholar]
- 30.Zhao J, Zhou D, Guo J, et al. Fasudil Aneurysmal Subarachnoid Hemorrhage Study Group. Efficacy and safety of fasudil in patients with subarachnoid hemorrhage: final results of a randomized trial of fasudil versus nimodipine. Neurol Med Chir (Tokyo) 2011;51:679–683. doi: 10.2176/nmc.51.679. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen HT, Dalmasso G, Torkvist L, et al. CD98 expression modulates intestinal homeostasis, inflammation, and colitis-associated cancer in mice. J Clin Invest. 2011;121:1733–1747. doi: 10.1172/JCI44631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies JK, Gribben JG. Blockade of chemotaxis in graft-versus-host disease. N Engl J Med. 2012;367:1667. doi: 10.1056/NEJMc1209665. author reply 1667–1668. [DOI] [PubMed] [Google Scholar]
- 33.Gomez A, Hammer S, Braun T, et al. A novel CD4+CD146+CCR5+ T-cell population is a biomarker of intestinal graft-versus-host disease; Presented at the 39th annual meeting of the European Group for Blood and Marrow Transplantation; April 10, 2013; London, UK. abstract 0390. [Google Scholar]
- 34.Morita NI, Matsumura Y, Morita K, Miyachi Y. Expression of CCR5 in graft-versus-host disease (GVHD) of the skin: immunohistochemical staining of 38 cases. J Dermatol. 2007;34:254–257. doi: 10.1111/j.1346-8138.2007.00263.x. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Harada T, Juang YT, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007;178:1938–1947. doi: 10.4049/jimmunol.178.3.1938. [DOI] [PubMed] [Google Scholar]
- 36.Smith A, Bracke M, Leitinger B, et al. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J Cell Sci. 2003;116(Pt 15):3123–3133. doi: 10.1242/jcs.00606. [DOI] [PubMed] [Google Scholar]
- 37.Noma K, Rikitake Y, Oyama N, et al. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–1644. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JH, Katakai T, Hara T, et al. Roles of p-ERM and rho-ROCK signaling in lymphocyte polarity and uropod formation. J Cell Biol. 2004;167:327–337. doi: 10.1083/jcb.200403091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu F, Zhang Z, Wu G, et al. Rho kinase inhibitor fasudil suppresses migration and invasion through down-regulating the expression of VEGF in lung cancer cell line A549. Med Oncol. 2011;28:565–571. doi: 10.1007/s12032-010-9468-5. [DOI] [PubMed] [Google Scholar]
- 40.Lämmermann T, Bader BL, Monkley SJ, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453:51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 41.Bardi G, Niggli V, Loetscher P. Rho kinase is required for CCR7-mediated polarization and chemotaxis of T lymphocytes. FEBS Lett. 2003;542:79–83. doi: 10.1016/s0014-5793(03)00351-x. [DOI] [PubMed] [Google Scholar]
- 42.van Buul JD, Hordijk PL. Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol. 2004;24:824–833. doi: 10.1161/01.ATV.0000122854.76267.5c. [DOI] [PubMed] [Google Scholar]
- 43.Li B, Zhao WD, Tan ZM, et al. Involvement of rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells. FEBS Lett. 2006;580:4252–4260. doi: 10.1016/j.febslet.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 44.Alevriadou BR. CAMs and rho small GTPases: gatekeepers for leukocyte transendothelial migration. Focus on “VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration”. Am J Physiol Cell Physiol. 2003;285:C250–C252. doi: 10.1152/ajpcell.00189.2003. [DOI] [PubMed] [Google Scholar]
- 45.Benais-Pont G, Punn A, Flores-Maldonado C, et al. Identification of a tight junction-associated guanine nucleotide exchange factor that activates rho and regulates paracellular permeability. J Cell Biol. 2003;160:729–740. doi: 10.1083/jcb.200211047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nohria A, Grunert ME, Rikitake Y, et al. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. 2006;99:1426–1432. doi: 10.1161/01.RES.0000251668.39526.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Persidsky Y, Heilman D, Haorah J, et al. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006;107:4770–4780. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto M, Ramirez SH, Sato S, et al. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am J Pathol. 2008;172:521–533. doi: 10.2353/ajpath.2008.070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bivalacqua TJ, Champion HC, Usta MF, et al. RhoA/rho-kinase suppresses endothelial nitric oxide synthase in the penis: a mechanism for diabetes-associated erectile dysfunction. Proc Natl Acad Sci USA. 2004;101:9121–9126. doi: 10.1073/pnas.0400520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ming XF, Viswambharan H, Barandier C, et al. 3Rho GTPase/rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002;22:8467–8477. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuno M, Takai S, Matsushima-Nishiwaki R, et al. Rho-kinase inhibitors decrease TGF-beta-stimulated VEGF synthesis through stress-activated protein kinase/c-Jun N-terminal kinase in osteoblasts. Biochem Pharmacol. 2009;77:196–203. doi: 10.1016/j.bcp.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 52.Takata K, Morishige K, Takahashi T, et al. Fasudil-induced hypoxia-inducible factor-1alpha degradation disrupts a hypoxia-driven vascular endothelial growth factor autocrine mechanism in endothelial cells. Mol Cancer Ther. 2008;7:1551–1561. doi: 10.1158/1535-7163.MCT-07-0428. [DOI] [PubMed] [Google Scholar]
- 53.Hata Y, Miura M, Nakao S, et al. Antiangiogenic properties of fasudil, a potent rho-Kinase inhibitor. Jpn J Ophthalmol. 2008;52:16–23. doi: 10.1007/s10384-007-0487-5. [DOI] [PubMed] [Google Scholar]
- 54.Washida N, Wakino S, Tonozuka Y, et al. Rho-kinase inhibition ameliorates peritoneal fibrosis and angiogenesis in a rat model of peritoneal sclerosis. Nephrol Dial Transplant. 2011;26:2770–2779. doi: 10.1093/ndt/gfr012. [DOI] [PubMed] [Google Scholar]
- 55.Nakabayashi H, Shimizu K. HA1077, a rho kinase inhibitor, suppresses glioma-induced angiogenesis by targeting the rho-ROCK and the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) signal pathways. Cancer Sci. 2011;102:393–399. doi: 10.1111/j.1349-7006.2010.01794.x. [DOI] [PubMed] [Google Scholar]
- 56.Clambey ET, McNamee EN, Westrich JA, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. 2012;109:E2784–E2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng B, Zhu JM, Wang Y, et al. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-β1 in gastric cancer. PLoS One. 2013;8:e63777. doi: 10.1371/journal.pone.0063777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fiegl M, Samudio I, Clise-Dwyer K, et al. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2009;113:1504–1512. doi: 10.1182/blood-2008-06-161539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan M, Jene N, Byrne D, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res. 2011;13:R47. doi: 10.1186/bcr2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 61.Synnestvedt K, Furuta GT, Comerford KM, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eltzschig HK, Köhler D, Eckle T, et al. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113:224–232. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor micro-environment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29:5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 65.Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. 2012;33:231–237. doi: 10.1016/j.it.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 66.Jin D, Fan J, Wang L, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245–2255. doi: 10.1158/0008-5472.CAN-09-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013;13:842–857. doi: 10.1038/nrc3613. [DOI] [PubMed] [Google Scholar]
- 68.Procaccini C, Galgani M, De Rosa V, Matarese G. Intracellular metabolic pathways control immune tolerance. Trends Immunol. 2012;33:1–7. doi: 10.1016/j.it.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Crecelius AR, Kirby BS, Richards JC, et al. Mechanisms of ATP-mediated vasodilation in humans: modest role for nitric oxide and vasodilating prostaglandins. Am J Physiol Heart Circ Physiol. 2011;301:H1302–H1310. doi: 10.1152/ajpheart.00469.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu TJ, Wang YC, Wu TH, et al. Inhibition of allogenic T-cell cytotoxicity by hepatic stellate cell via CD4+ CD25+ Foxp3+ regulatory T cells in vitro. Transplant Proc. 2012;44:1055–1059. doi: 10.1016/j.transproceed.2012.03.029. [DOI] [PubMed] [Google Scholar]