

Abstract
End-binding protein (EB1) is a microtubule protein that binds to the tumor suppressor adenomatous polyposis coli (APC). While EB1 is implicated as a potential oncogene, its role in cancer progression is unknown. Therefore, we analyzed EB1/APC expression at the earliest stages of colorectal carcinogenesis and in the uninvolved mucosa ("field effect") of human and animal tissue. We also performed siRNA-knockdown in colon cancer cell lines. EB1 is up-regulated in early and field carcinogenesis in the colon, and the cellular/nano-architectural effect of EB1 knockdown depended on the genetic context. Thus, dysregulation of EB1 is an important early event in colon carcinogenesis.
1. Introduction
The cytoskeleton plays a major role in cancer development and progression. Alterations in cytoskeletal regulatory pathways affect both the structure and function of the cytoskeleton, ultimately leading to tissue disruption, invasion, and genomic instability [1–3]. Dysregulation of cytoskeletal proteins are a critical early event of colon cancer initiation and progression [1,4,5]. Numerous studies have demonstrated that the adenomatous polyposis coli (APC) tumor suppressor gene is lost in more than 80% of all colorectal cancers and occurs during the earliest stages of carcinogenesis [6,7]. APC is a multi-functional protein responsible in regulating cellular β-catenin levels, cell differentiation, and maintaining microtubule stability, though the precise role of APC regulating the cytoskeleton during colon cancer remains unknown [8–10]. Microtubule end-binding protein 1 (EB1) was originally discovered as a binding partner of APC [11]. EB1 is encoded by the MAPRE1 gene and a member of the RP/EB family member involved the regulation of microtubule polymerization, cell polarity and chromosomal stability [12,13]. Together with APC, EB1 regulates chromosomal stability during mitosis [14]. Despite being an important binding partner of APC, the role of EB1 in colon carcinogenesis has not been well established. However, recent reports suggest that EB1 itself may play an important role in tumorigenesis.
Studies have demonstrated EB1 overexpression in hepatocellular carcinoma, gastric carcinoma, esophageal squamous cell carcinoma (ESCC) and breast cancer [15–18]. These studies indicate that EB1 promotes cellular proliferation and tumor growth, through the activation of β-catenin signaling [18,19]. However, MAPRE1 does not appear to be involved in somatic CRC and the role of EB1 regulation during CRC initiation and progression has not been extensively studied [20].
In the present study, we analyzed the expression of EB1 during early colorectal carcinogenesis and field carcinogenesis using the azoxymethane (AOM) rat model, polyposis in rat colon (Pirc) model and human samples. The AOM rat model is a chemically induced model of CRC, while the Pirc rat model is a genetic model of CRC through mutation of APC. We found that EB1 is markedly up-regulated at a pre-neoplastic time point in the AOM rat model. Similarly, we found that EB1 is also up-regulated in the histologically normal tissue in the Pirc rat model and AOM rat model. Given the potential clinical impact of EB1 dysregulation, we used low-coherence enhanced backscattering (LEBS) technique to study nano-architectural consequences of EB1 dysregulation. We knocked down EB1 in colon cancer cell lines with different APC status, HT-29 (APCmut/mut) and HCT-116 (APCwt/wt). Knockdown of EB1 resulted in different phenotypes based on the genetic context of the cell line, as seen by apoptosis, proliferation markers and LEBS spectral analysis. We report that EB1 is a proto-oncogene and propose that EB1 dysregulation is one of the earliest events in colon carcinogenesis.
2. Materials and Methods
2.1. Cell lines and tissues
HT-29 and HCT-116 cells were grown in McCoy’s 5A medium (ATCC, Manassas, VA) and supplemented with 10% fetal bovine serum + 50mg/mL penicillin/streptomycin under 5% CO2 environment at 37°C. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) for NorthShore University HealthSystem. Fisher 344 rats (Harlan, Madison, WI) on a standard AIN76a diet were treated with either 2 weekly injections (i.p.) of 15mg/kg Azoxymethane (AOM) or saline (Midwest Research Institute, Kansas City, MO). Rats were euthanized after 10 (pre-malignant time point) or 37 weeks (tumor bearing time point) post AOM injection. Genetic mutation of the Pirc rat model has been described [21]. For this study, male Pirc rats were obtained at 12 weeks of age (Taconic, Hudson, NY) and fed a standard AIN76a diet. Rats were euthanized at 24 weeks of age and adenocarcinomas in the colon were noted.
2.2. Immunohistochemistry
Human tissue microarrays (Collaborative Human Tissue Network, CHTN) were deparaffinized and then rehydrated with xylene and graded alcohol washes. Heat-induced epitope retrieval was performed using a pressure cooker. After quenching of endogenous peroxidase activity in 3% hydrogen peroxide, the slides were blocked with 5% horse serum. The slides were incubated overnight in EB1 (BD Biosciences, San Jose, CA) or APC (C-terminus) antibody (Santa Cruz Biotechnology, Santa Cruz, CA), followed by incubation with the appropriate biotinylated secondary antibody. Finally, the sections were developed using an avidin-biotin complex (ABC) kit (Vector Laboratories, Burlingame, CA). An observer blinded to the patient group scored the slides.
2.3 Transfection and cell viability assay
EB1 small-interfering RNAs (siRNAs) were transfected with lipofectamine 2000 reagent (Invitrogen Life Technologies, Grand Island, NY) in HT-29 and HCT-116 cells, following manufacturer’s instructions. Cells were then incubated in normal conditions for 72 hours in 96-well plates. At the end of the incubation, WST-1 reagent (Roche Diagnostics, Indianapolis, IN) was added. Following 30 min incubation with WST-1, the plate absorbance was read at 440nm and 600nm using the Spectramax Plus Spectrophotometer plate reader (Molecular Devices, Sunnyvale, CA).
2.4 Quantitative real time polymerase chain reaction (qRT-PCR)
RNA was isolated from samples using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH). Complementary DNA (cDNA) synthesis was performed using 5µg of RNA and Superscript RT (Invitrogen Life Technologies, Grand Island, NY), following standard protocol. Amplification of cyclinD1 and c-myc was performed using nested PCR protocols [22]. MAPRE1 PCR reactions were carried out using 80nM of the TaqMan probe and PCR Mastermix (Applied Biosystems, Carlsbad, CA) in a Cepheid Smart Cycle (Cepheid, Sunnyvale, CA). All samples were normalized to b-actin and average fold differences were calculated using the comparative Ct method [23]. Threshold of fold change significance was set as >1.5 (up-regulation) and <0.67 (down-regulation).
2.5 Western blotting
Proteins were resolved on SDS/PAGE gels and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Membranes were blocked in a 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20. Membranes were incubated overnight in EB1 antibody (BD Biosciences, San Jose, CA) at 4°C and then with the appropriate horseradish peroxidase-conjugated secondary antibody. Proteins were developed with enhanced chemiluminescence reagent (Santa Cruz Biotechnology, Santa Cruz, CA). The protein intensities were visualized using the UVP LabWorks system and software (UVP, Upland, CA).
2.6 Flow cytometry
HT-29 and HCT-116 transfected with EB1 siRNA and control cells were fixed 72 hours post-transfection. For apoptosis analysis, cells were fixed with ice-cold methanol and subsequently stained with M30 CytoDEATH-FITC, according to manufacturer’s protocol (Roche Applied Science, Indianapolis, IN). The CellQuest 3.1 software program was used to generate frequency histograms and data analysis.
2.7 LEBS instrumentation
The LEBS setup has been described in detail previously [24]. Briefly, LEBS enables simultaneous measurements of a scattering spectrum (400 – 700 nm) for a range of backscattering angles (−15° to 15°). The angular measurements were used to identify an enhanced backscattering peak and then the spectral properties of the enhanced backscattering were measured. The LEBS peak (Figure 4A) can be characterized by three parameters: width (W, average full width at half maximum), enhancement factor (E, average height), and spectral slope (S, linear coefficient from a linear regression). Cells were grown in 6-well plates, trypsinized, and pelleted. Nine separate measurements were obtained for each cell pellet. The variability between cell pellets was statistically non-significant (p >0.42). The LEBS markers from each experimental repeat were averaged.
Figure 4. EB1 knockdown caused alterations in sub-diffractional cell structure.
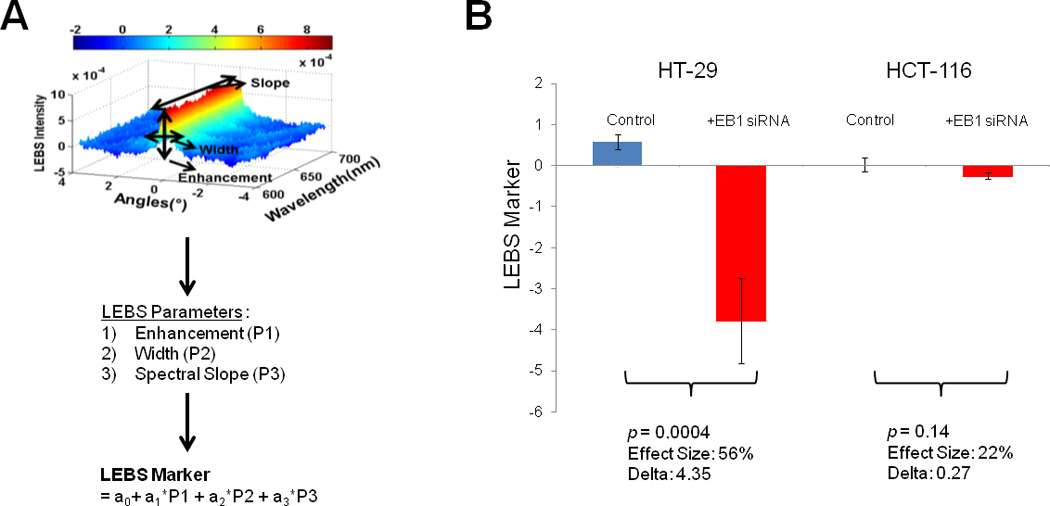
We used the novel low-coherence enhanced backscattering (LEBS) technique to measure nanoscale changes in cellular structure. A) The LEBS peak results from the backscattered intensity of a sample, as described in Material and Methods. B) Effect size percent between control and EB1 knockdown cells was calculated and averaged between experiments for each cell line. The effect size percent is greatly affected by EB1 knockdown in the HT-29 cells (p <0.001), but caused a more modest difference in HCT-116 cells (p =0.14). Error bars represent standard error.
2.8 LEBS Statistical Analysis
Previously, we have established a prediction rule based on LEBS spectral analysis for detection of field carcinogenesis in rectum and duodenum to predict risk of colon and pancreatic lesions elsewhere in the organ [24,25]. We used this binary logistic regression LEBS marker to evaluate and predict similar effect of EB1 knockdown in cells [26]. In short, the three LEBS parameters (E(P1), W(P2) , SS(P3)) were used as predictors by performing univariates analysis (ANOVA). To statistically construct a multivariable logistic model, all parameters with p <0.25 from univariate logistic regression were entered into the model and removed reversely, with the final model retaining parameters with p <0.05. The correlation coefficient was calculated for the selected parameters and verified to be non-significant. The final combined LEBS marker was built as linear combination of LEBS parameters as: LEBS Marker = a0 + a1*P1 + a2*P2 + a3*P3. The prediction rule development was carried out on HT-29 cells and then applied to HCT-116 cells. All p values were calculated using Student’s t-tests.
3. Results
3.1 EB1 is overexpressed in human colorectal adenoma and adenocarcinoma concomitant with APC reduction
A recent proteomic study showed that EB1 expression is increased in the tumor of CRC patients [27]. To determine whether EB1 is implicated in the development and progression of CRC, we first examined its expression in human colon tissue specimens by immunohistochemistry. Expression of EB1 was found at a low level in all normal colon tissues samples examined (Figure 1A). Approximately 50% of patients harboring an adenoma had high levels of EB1 expression. Compared to the normal colon, EB1 expression was significantly increased in adenoma cases (p <0.01; Figure 1B). Furthermore, 60% of adenocarcinoma patients also showed elevated levels of EB1 expression compared to the normal colon (p <0.01; Figure 1B). APC levels were significantly reduced in the adenoma and adenocarcinoma cases compared to the normal colon, as expected (Figure 1A). We next considered the adenoma tissue samples to assess a correlation between APC and EB1 expression. However, there was no significant correlation, possibly due to the relatively limited sample size (n=30 adenoma patients) (Figure 1C).
Figure 1. Immunohistochemical staining of EB1 and APC during human colon cancer progression.
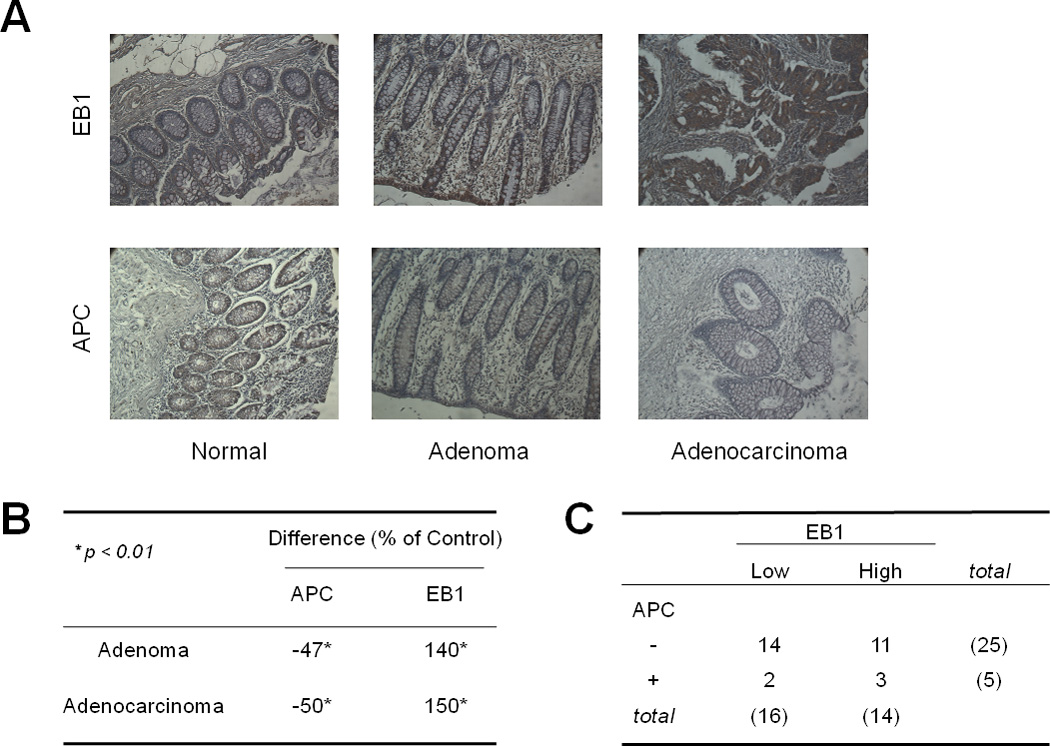
A) Expression of EB1 and APC (c-terminus) in human normal, adenoma, and adenocarcinoma colonic tissues. B) Quantification of the expression of EB1 and APC in adenoma and adenocarcinoma samples compared to normal tissue, *p <0.01. C) Correlation between EB1 and APC in adenoma tissue. High expression indicates an intensity score of 3 or higher.
3.2 EB1 is overexpressed in early and field colorectal carcinogenesis
While it has been shown that EB1 is overexpressed in tumor samples, the expression of EB1 in pre-malignant tissue has not yet been determined. Therefore, to study the involvement of EB1 in early colorectal carcinogenesis and field carcinogenesis, we analyzed MAPRE1 expression in different time points of the azoxymethane (AOM)–injected rat model using qRT-PCR methods. This model recapitulates many of the genetic and epigenetic features of human field carcinogenesis [28]. At a premalignant time point (10 weeks post-AOM injection), we found that MAPRE1 expression was 1.5-fold higher in the AOM-injected animals compared to their age-matched control counterparts (p <0.05; Figure 2A). Furthermore, at a cancerous time point of the AOM rat model (37 weeks post-AOM injection) MAPRE1 was more than 1.5-fold higher in the AOM uninvolved mucosa compared to control animals (p <0.01; Figure 2A). While these results demonstrate significant up-regulation of EB1 during early and field colon carcinogenesis, the AOM rat model cancer progression does not occur through APC mutation. We therefore analyzed MAPRE1 expression in the uninvolved mucosa of the Pirc (polyposis in rat colon) rat model for familial adenomatous polyposis (FAP), which develops colonic tumors in an APC mutant environment [21]. EB1 expression was 2–fold higher in Pirc rats containing a germline APC mutation compared to APC wildtype animals (p <0.05; Figure 2A), demonstrating EB1 up-regulation as a general event in early and field colorectal carcinogenesis. Next, immunohistochemical staining was performed for EB1 and APC. In early colon carcinogenesis (10 week AOM rat), EB1 was significantly up-regulated whereas APC was down-regulated (Figure 2B). Similarly, EB1 was over-expressed while APC was down-regulated in both the chemically-induced sporadic model (AOM) and genetic model (Pirc) for field carcinogenesis (Figure 2C). The relative protein expression quantification confirmed increased EB1 and reduced APC (p <0.1; Figure 2D). The animal results support the human data in which up-regulation of EB1 occurs in early colon carcinogenesis. Therefore, this is the first report demonstrating EB1 up-regulation in the pre-neoplastic tissue and field carcinogenesis.
Figure 2. MAPRE1 is increased in early carcinogenesis and the uninvolved mucosa.
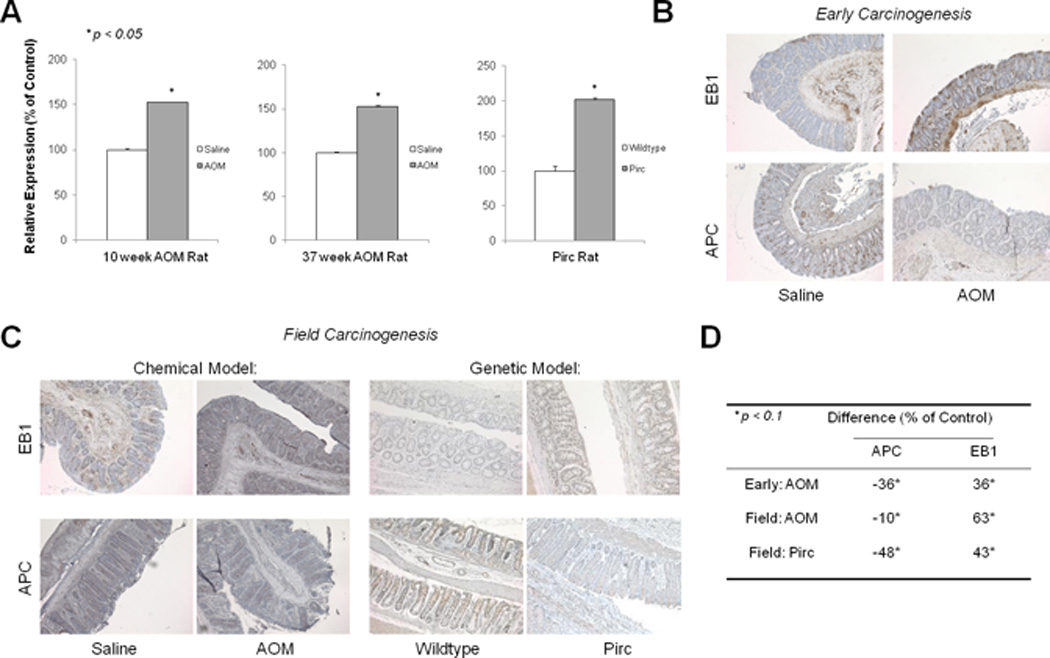
A) EB1 expression at a pre-neoplastic time point of the AOM rat model (10 weeks), and in the uninvolved mucosa (“field”) of the AOM rat model (tumor-bearing time point, 37 weeks) and the Pirc rat model using qRT-PCR. Standard error bars shown, *p <0.05. B) Protein expression of EB1 and APC (c-terminus) in the early colon carcinogenesis model. C) Protein expression of EB1 and APC (c-terminus) in the uninvolved mucosa in both a chemically induced rat model (AOM) and genetic Pirc rat model of colon carcinogenesis. D) Quantification of protein expression confirm reduced APC levels with increased EB1 levels, *p <0.1.
3.3 Loss of EB1 affects proliferation and apoptosis dependent on the cell line
To test the hypothesis that EB1 is a proto-oncogene in colon cancer, we studied the role of EB1 in proliferation and apoptosis by siRNA-mediated knockdown in an APC mutant (HT-29) and APC wildtype (HCT-116) colon cancer cell lines. Using the WST-1 assay, we found that EB1 knockdown decreased the rate of proliferation in HT-29 cells, but not in HCT-116 cells (Figure 3B). Previous reports have suggested that EB1 regulates proliferation through WNT/β-catenin pathways, showing alterations in cyclin D1 and c-myc [19]. We found that EB1 knockdown significantly decreased both cyclin D1 and c-myc expression in HT-29 cells, but not in HCT-116 cells (Figure 3C). Taken together, these results indicate that EB1 knockdown significantly reduced cell proliferation in APC mutant cells but has little effect in an APC wildtype environment.
Figure 3. EB1 knockdown in colon cancer cell lines induced changes in proliferation and apoptosis.
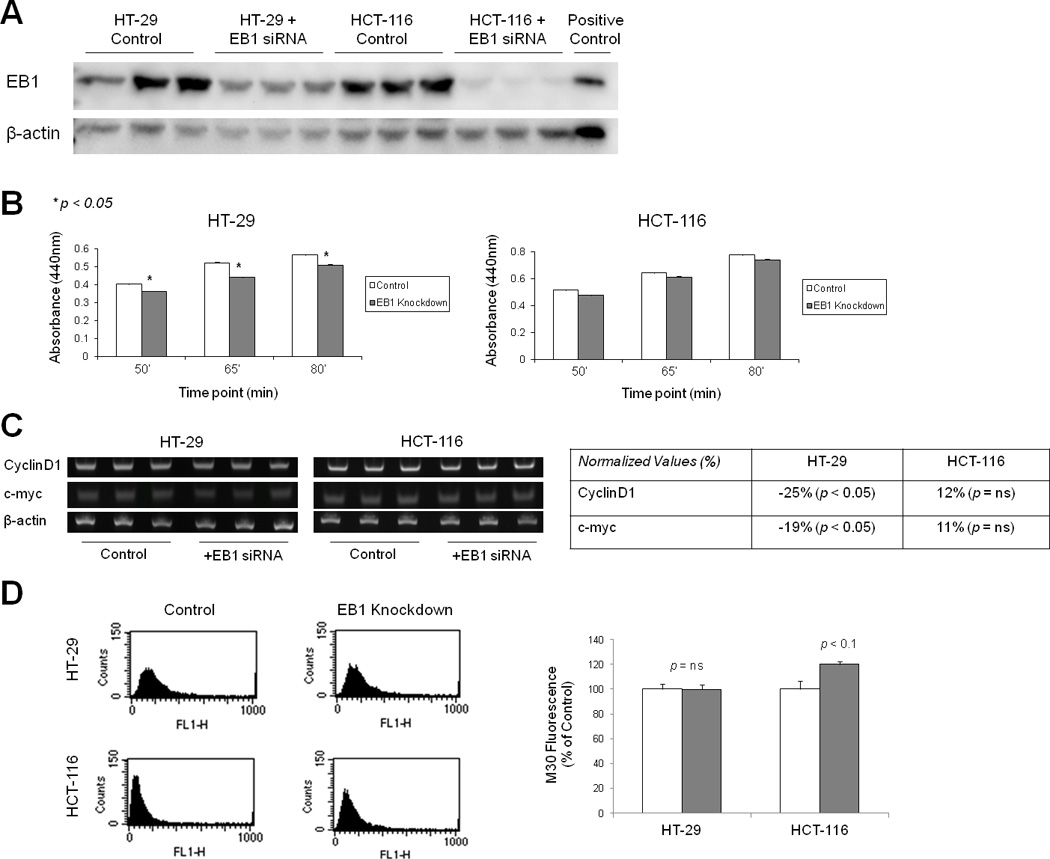
A) Western Blotting indicates 60–80% loss of EB1 in HT-29 and HCT-116 cell lines following siRNA-mediated knockdown. B) WST-1 assay showed that EB1 knockdown decreased HT-29 cell proliferation (p <0.05) but did not significantly affect HCT-116 cells. C) Knockdown of EB1 decreased cyclinD1 and c-myc expression in APCmut/mut HT-29 cells, but did not affect expression of these Wnt pathway members in APCwt/wt HCT-116 cells, using PCR. Expression was normalized to β-actin, *p <0.05. D) Control and EB1 knockdown cells were then subjected to flow cytometric analysis to measure M30 (apoptosis marker) levels. EB1 knockdown induced apoptosis in APCwt/wt HCT-116 cells (120% of control; p =0.06), it did not affect apoptosis in APCmut/mut HT-29 cells. Error bars represent standard error.
Considering the dissimilar proliferation results between HT-29 and HCT-116 cells, we next examined apoptosis. Control and EB1 knockdown cells were subjected to M30 CYTOdeath-FITC staining and flow cytometry analysis. The M30 antibody recognizes a caspase-cleaved epitope of the cytokeratin-18 protein to target cells undergoing apoptosis [29]. In HT-29 cells, there was no difference in M30 intensity in the EB1 knockdown compared to control cells. In contrast, we found a 20% increase in M30 intensity in the HCT-116 cells following EB1 knockdown compared to HCT-116 control cells (p = 0.06, Figure 3D). In view of the important interaction of APC and EB1, the anti-proliferative effect of EB1 knockdown may depend on the cell line, and particularly the APC status.
3.4 Low coherence enhanced backscattering (LEBS) measurements of cytoskeletal alterations
The LEBS marker is sensitive to micro-architectural alterations in both the AOM rat model and in human cancers [24,30]. Recently, our group has shown that cytoskeletal organization in epithelial cells is an important factor in determining differences in LEBS markers between control and pre-neoplastic mucosa [31]. In HT-29 cells, the LEBS marker was significantly decreased with EB1 knockdown compared to control cells (p <0.01; Fig. 2). However, in HCT-116 cells, the same LEBS marker was not as significantly affected by knockdown of the EB1 (p =0.14 Fig. 2). Similarly, the effect size difference between HT-29 and knockdown cell line was 56% whereas in the HCT-116 cell lines, it was only 21%. These results also demonstrate a different response to EB1 knockdown among cell lines, indicating distinct ultra-structural consequences between the cell lines.
4. Discussion
Early detection and intervention of cancer have been shown to greatly increase patient survival and provides a promising approach to combating cancer. Our group has developed novel optical techniques to target the earliest stages of carcinogenesis, such as LEBS. For the current study, we used the same LEBS prediction rule that previously has shown that ultra-structural alterations occur in very early stages in diffuse field of organs (colon and pancreas) at length scales as small as 40nm, which are irresolvable by conventional light microscopy [24,25]. Following this well-established capability of LEBS to detect ultra-structural manifestations and nanoscale, we used this technique to probe and understand cellular and proteomic processes resulting from these ultra-structural changes. Proper regulation of the cytoskeleton is a dynamic and crucial process for normal cell function, including proliferation, apoptosis and differentiation [10]. The microtubule-associate protein EB1 is a binding partner to APC, which is a key tumor suppressor frequently mutated in both sporadic and familial colorectal cancer (CRC). While it has been shown that EB1 is a potential oncogene in several cancers, the regulation of EB1 during CRC progression has not yet described. In this study, we demonstrate that EB1 is up-regulated in human CRC progression, consistent with APC down-regulation. Furthermore, we show that EB1 is significantly up-regulated at the message and protein level in both genetic and chemically induced models of CRC, also consistent with a decrease in APC. Therefore, we hypothesized that knockdown of EB1 in colon cancer cell lines would reduce cell tumorigenicity. We found that knockdown of EB1 induced apoptosis or decreased proliferation, depending on the genetic context of the cell line, which was also reflected by differential spectral analysis using LEBS.
The contrast in apoptotic induction between HT-29 and HCT-116 cells following EB1 knockdown may relate to the different p53 gene status, which is a critical regulator of apoptosis [32,33]. While there are a number of genetic differences between the cell lines, clearly the context plays an important role in the effect of EB1 knockdown and regulation. Thus, the potential role of EB1 and apoptosis in carcinogenesis requires closer attention. We also found that HT-29 cells following EB1 knockdown had decreased proliferation, cyclinD1, and c-myc expression but caused no change in the HCT-116 cell lines. As it was previously proposed that EB1 may disrupt APC regulation of the WNT signaling pathway, the altered phenotypes may be due to the different APC status of each cell line [18,19]. It has been shown that together APC and EB1 regulate the mitotic spindle, chromosome alignment and microtubule stabilization [14,34]. Dysregulation of the APC-EB1 interaction, through APC mutation or EB1 overexpression, may therefore promote cellular proliferation, spindle defects, and aberrant chromosomal segregation. Genetic instability, such as chromosomal instability (CIN), initiates cancer development, progression, and the multiplicity of mutations in tumors [35]. The dysregulation of the EB1-APC may therefore contribute to CIN in CRC initiation and progression, which is consistent with our results that EB1 plays an important role in early and field carcinogenesis.
Field carcinogenesis is the concept that the genetic/epigenetic and environmental milieu that results in a neoplastic lesion extends throughout the affected organ [36]. Thus, alterations in the diffuse field provide a mutational background and predisposition to carcinogenesis, while the initial neoplastic lesion occurred as a result of stochastic mutations [37]. Genomic instability is a common feature of early cancer development and field carcinogenesis in several cancers [36,38,39]. Therefore, our findings in the diffuse field of the organ and pre-malignant tissue suggest that EB1 up-regulation is one of the earliest events in colon carcinogenesis. To further examine the effect of EB1 loss in colon cancer cell lines, we also used the novel LEBS technology to assess the micro-architectural consequences in cells. LEBS markers are robustly sensitive to detecting the field effect in microscopically-normal tissue in human pancreatic and colorectal cancers [24,26,40]. Biologically, LEBS is capable of identifying nano- and micro-architectural alterations in cells, which in epithelial cells would correspond to structures such as the cytoskeleton [31]. Following EB1 knockdown, the LEBS marker was significantly altered in the HT-29 cells (56 % effect size change), but to a lesser degree in the HCT-116 cells (21% effect size change), indicating that EB1 loss induced significant, distinct alterations in nano-architecture between cell lines. Given that LEBS is well suited for morphological analysis of tissue architecture, this technique would be important for the assessment of in vivo effects of EB1 dysregulation on tissue micro-architecture.
Highlights.
EB1 expression is significantly increased in early and field carcinogenesis in the colon.
Knockdown of EB1 reduced the cancerous phenotype in colon cancer cell lines.
LEBS analysis of EB1 knockdown cells shows distinct changes in cell nanoarchitecture.
Acknowledgements
This work was supported by NIH grants U01CA111257, R01CA128641, R01CA165309, and R01CA156186.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Behrens J. Cadherins and catenins: role in signal transduction and tumor progression. Cancer Metastasis Rev. 1999;18:15–30. doi: 10.1023/a:1006200102166. [DOI] [PubMed] [Google Scholar]
- 2.Gavert N, Ben-Ze'ev A. beta-Catenin signaling in biological control and cancer. J Cell Biochem. 2007;102:820–828. doi: 10.1002/jcb.21505. [DOI] [PubMed] [Google Scholar]
- 3.Hall A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009;28:5–14. doi: 10.1007/s10555-008-9166-3. [DOI] [PubMed] [Google Scholar]
- 4.Hao X, Palazzo JP, Ilyas M, Tomlinson I, Talbot IC. Reduced expression of molecules of the cadherin/catenin complex in the transition from colorectal adenoma to carcinoma. Anticancer Res. 1997;17:2241–2247. [PubMed] [Google Scholar]
- 5.Marotta A, et al. Dysregulation of integrin-linked kinase (ILK) signaling in colonic polyposis. Oncogene. 2001;20:6250–6257. doi: 10.1038/sj.onc.1204791. [DOI] [PubMed] [Google Scholar]
- 6.Kinzler KW, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 7.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 8.Kroboth K, Newton IP, Kita K, Dikovskaya D, Zumbrunn J, Waterman-Storer CM, Nathke IS. Lack of adenomatous polyposis coli protein correlates with a decrease in cell migration and overall changes in microtubule stability. Mol Biol Cell. 2007;18:910–918. doi: 10.1091/mbc.E06-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langford KJ, Askham JM, Lee T, Adams M, Morrison EE. Examination of actin and microtubule dependent APC localisations in living mammalian cells. BMC Cell Biol. 2006;7:3. doi: 10.1186/1471-2121-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer. 2006;6:967–974. doi: 10.1038/nrc2010. [DOI] [PubMed] [Google Scholar]
- 11.Su LK, et al. APC binds to the novel protein EB1. Cancer Res. 1995;55:2972–2977. [PubMed] [Google Scholar]
- 12.Berrueta L, Kraeft SK, Tirnauer JS, Schuyler SC, Chen LB, Hill DE, Pellman D, Bierer BE. The adenomatous polyposis coli-binding protein EB1 is associated with cytoplasmic and spindle microtubules. Proc Natl Acad Sci U S A. 1998;95:10596–10601. doi: 10.1073/pnas.95.18.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tirnauer JS, Bierer BE. EB1 proteins regulate microtubule dynamics, cell polarity, and chromosome stability. J Cell Biol. 2000;149:761–766. doi: 10.1083/jcb.149.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell. 2005;16:4609–4622. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong X, et al. Oncogenic function of microtubule end-binding protein 1 in breast cancer. J Pathol. 2010;220:361–369. doi: 10.1002/path.2662. [DOI] [PubMed] [Google Scholar]
- 16.Nishigaki R, et al. Proteomic identification of differentially-expressed genes in human gastric carcinomas. Proteomics. 2005;5:3205–3213. doi: 10.1002/pmic.200401307. [DOI] [PubMed] [Google Scholar]
- 17.Orimo T, et al. Proteomic profiling reveals the prognostic value of adenomatous polyposis coli-end-binding protein 1 in hepatocellular carcinoma. Hepatology. 2008;48:1851–1863. doi: 10.1002/hep.22552. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, et al. Overexpression of EB1 in human esophageal squamous cell carcinoma (ESCC) may promote cellular growth by activating beta-catenin/TCF pathway. Oncogene. 2005;24:6637–6645. doi: 10.1038/sj.onc.1208819. [DOI] [PubMed] [Google Scholar]
- 19.Liu M, et al. EB1 acts as an oncogene via activating beta-catenin/TCF pathway to promote cellular growth and inhibit apoptosis. Mol Carcinog. 2009;48:212–219. doi: 10.1002/mc.20471. [DOI] [PubMed] [Google Scholar]
- 20.Jais P, Sabourin JC, Bombled J, Rougier P, Lasser P, Duvillard P, Benard J, Bressac-de Paillerets B. Absence of somatic alterations of the EB1 gene adenomatous polyposis coli-associated protein in human sporadic colorectal cancers. Br J Cancer. 1998;78:1356–1360. doi: 10.1038/bjc.1998.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amos-Landgraf JM, et al. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci U S A. 2007;104:4036–4041. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanez-Mo M, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med. 2003;348:403–413. doi: 10.1056/NEJMoa020809. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Roy HK, et al. Association between rectal optical signatures and colonic neoplasia: potential applications for screening. Cancer Res. 2009;69:4476–4483. doi: 10.1158/0008-5472.CAN-08-4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turzhitsky V, Liu Y, Hasabou N, Goldberg M, Roy HK, Backman V, Brand R. Investigating population risk factors of pancreatic cancer by evaluation of optical markers in the duodenal mucosa. Dis Markers. 2008;25:313–321. doi: 10.1155/2008/958214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy HK, Kim YL, Liu Y, Wali RK, Goldberg MJ, Turzhitsky V, Horwitz J, Backman V. Risk stratification of colon carcinogenesis through enhanced backscattering spectroscopy analysis of the uninvolved colonic mucosa. Clin Cancer Res. 2006;12:961–968. doi: 10.1158/1078-0432.CCR-05-1605. [DOI] [PubMed] [Google Scholar]
- 27.Sugihara Y, et al. Proteomic-based identification of the APC-binding protein EB1 as a candidate of novel tissue biomarker and therapeutic target for colorectal cancer. J Proteomics. 2012 doi: 10.1016/j.jprot.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee A, Quirke P. Experimental models of colorectal cancer. Dis Colon Rectum. 1998;41:490–505. doi: 10.1007/BF02235764. [DOI] [PubMed] [Google Scholar]
- 29.Demir Weusten AY, Groothuis PG, Dunselman GA, de Goeij AF, Arends JW, Evers JL. Morphological changes in mesothelial cells induced by shed menstrual endometrium in vitro are not primarily due to apoptosis or necrosis. Hum Reprod. 2000;15:1462–1468. doi: 10.1093/humrep/15.7.1462. [DOI] [PubMed] [Google Scholar]
- 30.Kim YL, Liu Y, Wali RK, Roy HK, Backman V. Low-coherent backscattering spectroscopy for tissue characterization. Appl Opt. 2005;44:366–377. doi: 10.1364/ao.44.000366. [DOI] [PubMed] [Google Scholar]
- 31.Mutyal NN, Radosevich A, Tiwari AK, Stypula Y, Wali R, Kunte D, Roy HK, Backman V. Biological mechanisms underlying structural changes induced by colorectal field carcinogenesis measured with low-coherence enhanced backscattering (LEBS) spectroscopy. PLoS One. 2013;8:e57206. doi: 10.1371/journal.pone.0057206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.el-Deiry WS, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 33.Shen Y, White E. p53-dependent apoptosis pathways. Adv Cancer Res. 2001;82:55–84. doi: 10.1016/s0065-230x(01)82002-9. [DOI] [PubMed] [Google Scholar]
- 34.Zhang T, Zaal KJ, Sheridan J, Mehta A, Gundersen GG, Ralston E. Microtubule plus-end binding protein EB1 is necessary for muscle cell differentiation, elongation and fusion. J Cell Sci. 2009;122:1401–1409. doi: 10.1242/jcs.039255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alberici P, Fodde R. The role of the APC tumor suppressor in chromosomal instability. Genome Dyn. 2006;1:149–170. doi: 10.1159/000092506. [DOI] [PubMed] [Google Scholar]
- 36.Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Research. 2003;63:1727–1730. [PubMed] [Google Scholar]
- 37.Backman V, Roy HK. Light-scattering technologies for field carcinogenesis detection: a modality for endoscopic prescreening. Gastroenterology. 2011;140:35–41. doi: 10.1053/j.gastro.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voravud N, Shin DM, Ro JY, Lee JS, Hong WK, Hittelman WN. Increased polysomies of chromosomes 7 and 17 during head and neck multistage tumorigenesis. Cancer Res. 1993;53:2874–2883. [PubMed] [Google Scholar]
- 39.Zaky AH, et al. Clinicopathologic implications of genetic instability in intestinal-type gastric cancer and intestinal metaplasia as a precancerous lesion: proof of field cancerization in the stomach. Am J Clin Pathol. 2008;129:613–621. doi: 10.1309/DFLELPGPNV5LK6B1. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, et al. Optical markers in duodenal mucosa predict the presence of pancreatic cancer. Clin Cancer Res. 2007;13:4392–4399. doi: 10.1158/1078-0432.CCR-06-1648. [DOI] [PubMed] [Google Scholar]