

Abstract
Griscelli syndrome 2 is a rare autosomal recessive disorder of pigmentary dilution of hair, skin, splenohepatomegaly, pancytopenia, immune and neurologic dysfunction. Clinical course is characterized by recurrent infection triggered by uncontrolled T-lymphocyte and macrophage activation, called hemophagocytic syndrome. Since the primary presentation is with depigmented hair, we attempt to highlight diagnostic difficulties in such cases in developing countries like ours where pigmentary changes in hair and skin are commonly attributed to severe malnutrition. We also evaluated phenotype of all 10 cases of genotype (c.C550T; p.R184X), collected from published literature worldwide and emphasize the potential role of above mutation as hotspot in Southeast Asian region.
Keywords: Griscelli syndrome, Kala Azar, mutation
Introduction
What was known?
Phenotypic spectrum of Griscelli syndrome in all 10 patients described from India.
Griscelli syndrome (GS) is multisystem disorder with three subtypes (GS1, GS2, GS3), based on genetic loci (Myosin VA, Ras related protein Rab-27A, melanophilin). GS1 presents with primarily neurologic impairment with no immunologic involvement while GS2 presents with immunological dysfunction and multisystem involvement, whereas GS3 have only hypomelanosis. GS2 is the most common among three types with 10 cases reported from Indian literature. We attempt to discuss the difficulties in the diagnosis of these cases in developing countries like ours where hypopigmentation and splenomegaly are more commonly encountered in Kala Azar, thalassemia, leukemia, lymphoma, and tropical splenomegaly syndrome. We have summarized the clinical, laboratory, histopathology, and genotype of all 10 cases, compared our case with reported phenotypes and brought out the most consistent findings to direct the treating clinician. We also reviewed the phenotype of previously reported genotype (c.C550T; p.R184X) worldwide, and found (c.C550T; p.R184X) to be most prevalent in Southeast Asian subcontinent with potential to be founder mutation in this region.
Case Report
A two-and-a-half-year-old male born of non-consanguineous parentage was referred to with complaints of progressive abdominal distension since birth, progressive pallor and recurrent episodes of fever since 1 year of age and on and off ear discharge for last 2 months. There was history of four prior blood transfusions. There was no history of jaundice, vomiting, urinary or bowel complaints, bleeding from any site or neurological complaints.
His birth weight was not known. The mother had conceived three times, resulting in a normal 5-year-old female child and one female sibling who had the same clinical features as the proband but could not be evaluated as she had expired at around 9 months of age. The sibling had history of repeated episodes of fever and ear discharge and light colored hair. There was no history of any relatives affected by similar clinical presentation.
On examination his vital parameters were normal. He was grossly malnourished with a weight of 59.7% of expected (Grade III protein energy malnutrition according to IAP), height was 84.7% (severe stunting according to WHO) and Weight for height was 78% (wasting according to WHO). He had pallor, silvery gray scalp hair, white eyelashes, sparse eyebrows, and pedal edema [Figure 1]. Abdomen was distended, with firm hepatomegaly. The spleen was palpable 7 cm below left costal margin and was firm in consistency. There was no free fluid. The skin, iris and retina had normal pigmentation. Rest of the physical examination was unremarkable.
Figure 1.
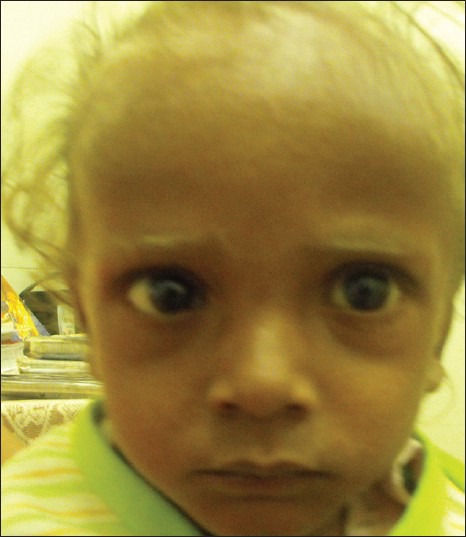
An Indian boy showing silvery hairs, eyelashes, and eyebrows
Investigations revealed pancytopenia with hemoglobin of 5.9 g/dL, a total leukocyte count 2310/mm3 N48%, L52% (ANC of 1100), platelet count of 46,000/mm3 and 0.8% reticulocytes. The peripheral smear showed moderate anisopoikilocytosis, predominantly normocytic hypochromic RBC with fair number of microcytes, ovalocytes and few ellipitocytes. Platelets and leucocytes were reduced. There were no giant cytoplasmic granules in leucocytes. The liver function tests were normal expect low albumin (2.1 mg/dL) and increased alkaline phosphatase (1196 mg/dL). Serum triglycerides and cholesterol were elevated. Hemoglobin electrophoresis, Blood culture, urine examination, prothrombin time, partial thromboplastin time, Immune profile, malarial serology, Kala Azar serology were normal. Bone marrow aspiration and biopsy were done during the accelerated phase of the disease (GS) for suspected hemo-phagocytosis. It showed erythroid hyperplasia, moderate megaloblastic erythropoiesis and depleted iron stores but did not reveal hemophagocytosis or giant granules in any of the cell line series.
Ultrasound of the abdomen was suggestive of hepatosplenomegaly with fatty liver. Magnetic resonance imaging of the brain revealed no pathology. Microscopic examination of the hair shaft of the proband showed pigmentation in outer hair sheath and hair shaft [Figure 2]. Dermis and appendages were unremarkable. Light microscopy of skin showed large coarse aggregates of melanin in the basal layer of the epidermis showing reactivity with Masson-Fontana stain [Figure 3]. Genetic study of RAB27A gene revealed a homozygous nonsense mutation in exon 6 (c.C550T; p.R184X).
Figure 2.
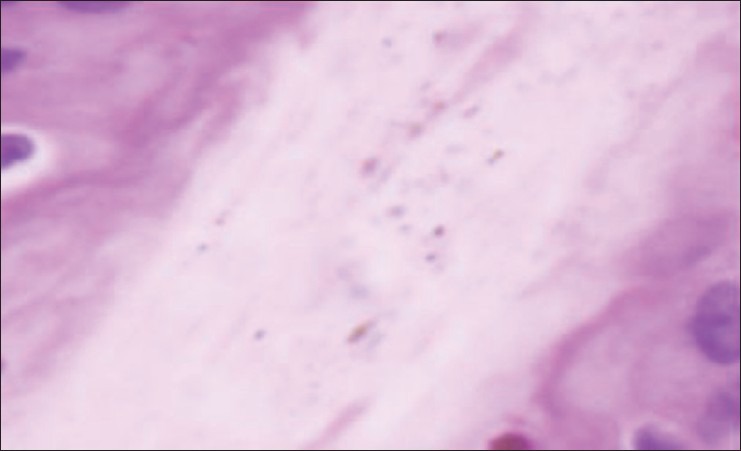
Hair shaft with irregular coarse melanin pigment (H and E, ×1000)
Figure 3.
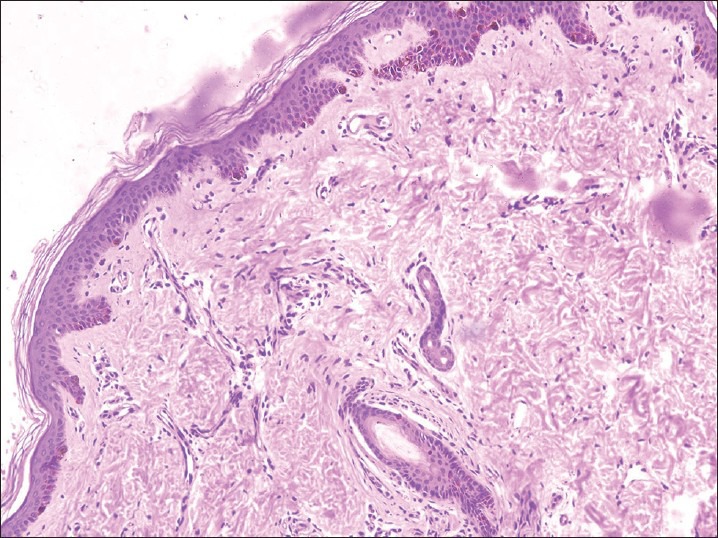
Irregular granular coarse melanin pigmentation of the basal layer of epidermis and hair bulb (H and E, ×100)
Discussion
GS is an autosomal recessive disorder with varied clinical manifestation. It was first reported by Griscelli et al. in two unrelated patients in 1978.[1] GS presents with variable phenotype and is categorized in to 3 types. GS1 patients primarily present with neurological involvement without immune dysfunction. Hepatosplenomegaly, recurrent infection, hypomelanosis and silvery gray hair are consistent features of GS2 patients. GS3 is categorized by hypomelanosis with no immunologic or neurologic involvement. Presence of grayish hair is a hallmark of all the three types of GS patients. Genetic loci for three different phenotypes (GS1, GS2, and GS3) are MYO5A, RAB27A, and MLPH respectively.[2,3] GS2 is the most common of all three disorders with GS3, being least common. There are only 10 case reports from India of GS2, with report of no cases of GS1 and GS3.[4,5,6,7,8,9,10,11,12] Although, history of consanguineous marriage was not forthcoming in our case. It cannot be ruled out as parents were married in same caste and same gotra of highly inbred society (teli or bania) of Hindu religion.
The constellation of hypopigmented hair, recurrent infection, severe malnutrition and hepatosplenomegaly pose a diagnostic challenge to treating pediatrician in a developing country where Kala Azar, thalassemia, leukemia, lymphoma, and tropical splenomegaly syndrome masquerade similarly. Possibility of Kala Azar was strong, as child came from endemic zone and the clinician attributed the pigmentary changes in hair to severe malnutrion. The work up for all these disorders came to be negative and child was referred to our facility for further workup. The most striking finding was presence of silvery gray hair, eyelashes, eyebrows which was attributed to severe malnutrition in developing countries like ours, where prevalence of malnutrition (under 5 years) is more than 40%.[13] We considered the differential diagnosis of silvery gray hair-GS and Chediak Higashi syndrome. The diagnosis of GS2 was made in presence of following features (silvery gray hair, splenohepatomegaly, recurrent infection) and absence of neurologic dysfunction and intracytoplasmic granules in leucocytes. Our diagnosis was further supported by hair shaft microscopy, skin biopsy [Figures 2 and 3] and molecular analysis.
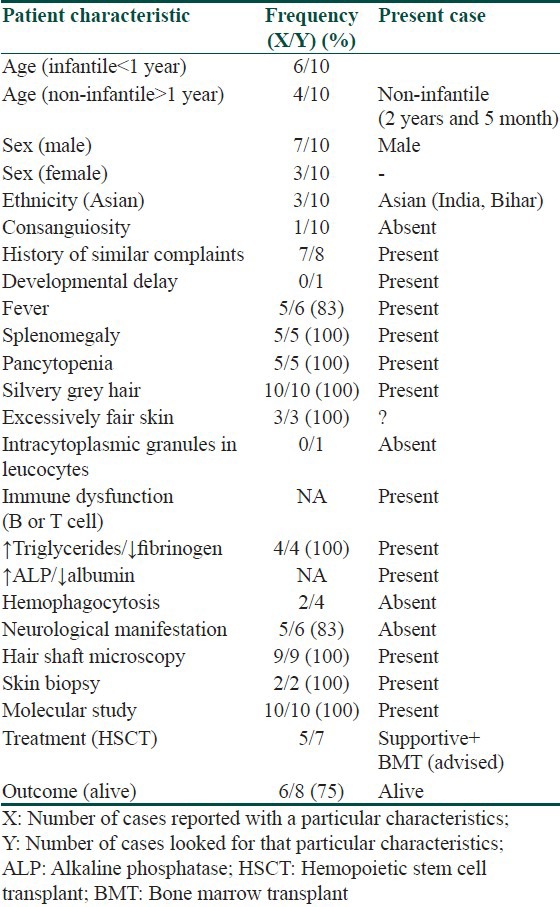
We reviewed the frequency of clinical, laboratory and mutational characteristics of Indian GS2 patients. We found following features to be most consistently present in affected cases including ours, i.e. fever, pallor, silvery gray hair, eyelashes and eyebrows, splenohepatomegaly, pancytopenia, immune dysfunction, uneven aggregation of melanin in hair shaft, large coarse aggregates of melanosomes in basal layer and genotype (C550T; R184X) [Table 1]. The pathogenesis of genotype had been previously reported in two Mauritian brothers in year 2000 (2). There have been 10 case reports of this genotype worldwide including one from India.[2,7,14,15,16] Ours is second case report from Indian subcontinent and eleventh worldwide. We tabulated frequency of phenotypic features of this genotype reported worldwide [Table 2]. Our case demonstrated all these features except neurological manifestations.
Table 1.
Frequency of clinical, laboratory, and mutational characteristic of Griscelli syndrome of Indian origin and comparison of our case with established phenotype
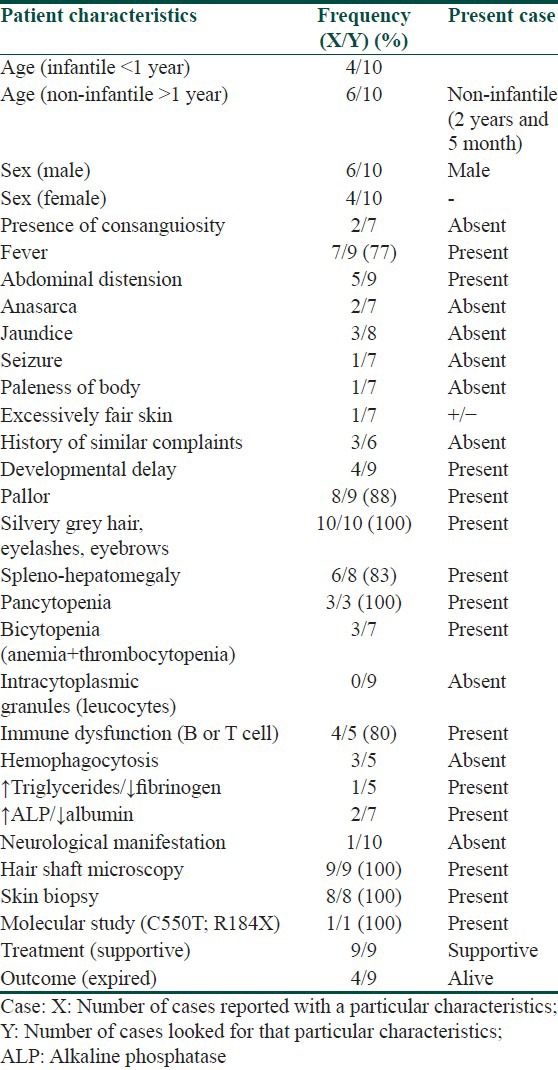
Table 2.
Spectrum of clinical and laboratory phenotype of RAB27A (c. 550C>T; p.Arg184X) mutation worldwide and comparison with our case
The diagnosis was delayed in present case due to following reasons: Attribution of hairy changes to malnutrition; triad of symptoms (fever, pancytopenia, and splenomegaly) to Kala Azar as child was from endemic region and low index of suspicion for GS2. This case highlights the difficulties in diagnosing a boy who presented to clinicians with fever, hairy changes, malnutrion and splenomegaly in an endemic region of Kala Azar. Cases of GS are under diagnosed in developing countries because of low index of suspicion and inadequate diagnostic facilities in primary and secondary care hospitals. Making a correct a diagnosis is of paramount importance as this will facilitate optimum treatment in cases with GS. GS1 patients require only palliative and supportive treatment, GS2 patients require bone marrow transplant whereas GS3 patients require no treatment. Genetic diagnosis further enables clinicians to offer prenatal genetic counselling.
What is new?
Problems in diagnosing such hypopigmentary cases in presence of Protein Energy Malnutrition (PEM) and in endemic region of Kala Azar
Compilation of all Indian patients with Griscelli syndrome with their phenotypic and genotypic spectrum
Ours is second genotypically proven case from India
Compilation of phenotypic features of genotype (C550T; R184X) from world literature
It helped us offer prenatal counseling to family in terms risk of recurrence in next pregnancy.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil.
References
- 1.Griscelli C, Durandy A, Guy-Grand D, Daguillard F, Herzog C, Prunieras M. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691–702. doi: 10.1016/0002-9343(78)90858-6. [DOI] [PubMed] [Google Scholar]
- 2.Ménasché G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25:173–6. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 3.Pastural E, Ersoy F, Yalman N, Wulffraat N, Grillo E, Ozkinay F, et al. Two genes are responsible for Griscelli syndrome at the same 15q21 locus. Genomics. 2000;63:299–306. doi: 10.1006/geno.1999.6081. [DOI] [PubMed] [Google Scholar]
- 4.Dinakar C, Lewin S, Kumar KR, Harshad SR. Partial albinism, immunodeficiency, hypergammaglobulinemia and Dandy-Walker cyst: A Griscelli syndrome variant. Indian Pediatr. 2003;40:1005–8. [PubMed] [Google Scholar]
- 5.Rath S, Jain V, Marwaha RK, Trehan A, Rajesh LS, Kumar V. Griscelli syndrome. Indian J Pediatr. 2004;71:173–5. doi: 10.1007/BF02723104. [DOI] [PubMed] [Google Scholar]
- 6.Manglani M, Adhvaryu K, Seth B. Griscelli syndrome: A case report. Indian Pediatr. 2004;41:734–7. [PubMed] [Google Scholar]
- 7.Sheela SR, Latha M, Injody SJ. Griscelli syndrome: Rab 27a mutation. Indian Pediatr. 2004;41:944–7. [PubMed] [Google Scholar]
- 8.Malhotra AK, Bhaskar G, Nanda M, Kabra M, Singh MK, Ramam M. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337–40. doi: 10.1016/j.jaad.2005.11.1056. [DOI] [PubMed] [Google Scholar]
- 9.Sori T, Nath AK, Thappa DM, Jaisankar TJ. Hypopigmentary disorders in children in South India. Indian J Dermatol. 2011;56:546–9. doi: 10.4103/0019-5154.87152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy RR, Babu BM, Venkateshwaramma B, Hymavathi Ch. Silvery hair syndrome in two cousins: Chediak-Higashi syndrome vs Griscelli syndrome, with rare associations. Int J Trichology. 2011;3:107–11. doi: 10.4103/0974-7753.90825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sahana M, Sacchidanand S, Hiremagalore R, Asha G. Silvery grey hair: Clue to diagnose immunodeficiency. Int J Trichology. 2012;4:83–5. doi: 10.4103/0974-7753.96910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar TS, Ebenazar S, Moses PD. Griscelli syndrome. Indian J Dermatol. 2006;5:269–71. [Google Scholar]
- 13.Srivastava A, Mahmood SE, Srivastava PM, Shrotriya VP, Kumar B. Nutritional status of school-age children: A scenario of urban slums in India. Arch Public Health. 2012;70:8. doi: 10.1186/0778-7367-70-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meschede IP, Santos TO, Izidoro-Toledo TC, Gurgel-Gianetti J, Espreafico EM. Griscelli syndrome-type 2 in twin siblings: Case report and update on RAB27A human mutations and gene structure. Braz J Med Biol Res. 2008;41:839–48. doi: 10.1590/s0100-879x2008001000002. [DOI] [PubMed] [Google Scholar]
- 15.Westbroek W, Tuchman M, Tinloy B, De Wever O, Vilboux T, Hertz JM, et al. A novel missense mutation (G43S) in the switch I region of Rab27A causing Griscelli syndrome. Mol Genet Metab. 2008;94:248–54. doi: 10.1016/j.ymgme.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meeths M, Bryceson YT, Rudd E, Zheng C, Wood SM, Ramme K, et al. Clinical presentation of Griscelli syndrome type 2 and spectrum of RAB27A mutations. Pediatr Blood Cancer. 2010;54:563–72. doi: 10.1002/pbc.22357. [DOI] [PubMed] [Google Scholar]