

Abstract
The rise of the “Top Down” method in the field of mass spectrometry-based proteomics has ushered in a new age of promise and challenge for the characterization and identification of proteins. Injecting intact proteins into the mass spectrometer allows for better characterization of post-translational modifications and avoids several of the serious “inference” problems associated with peptide-based proteomics. However, successful implementation of a Top Down approach to endogenous or other biologically relevant samples often requires the use of one or more forms of separation prior to mass spectrometric analysis, which have only begun to mature for whole protein MS. Recent advances in instrumentation have been used in conjunction with new ion fragmentation using photons and electrons that allow for better (and often complete) protein characterization on cases simply not tractable even just a few years ago. Finally, the use of native electrospray mass spectrometry has shown great promise for the identification and characterization of whole protein complexes in the 100 kDa to 1 MDa regime, with prospects for complete compositional analysis for endogenous protein assemblies a viable goal over the coming few years.
Proteomics in a Post-Genomics World
The rise in genome sequencing has greatly propelled the understanding of the living world, but alone is insufficient for full description of a biological system [1]. Focusing on the protein level, proteomics has emerged as another large-scale platform for improving the understanding of biology. Proteomic experiments can be used for the annotation and correction of genome sequences, quantitation of protein abundance, detection of post-translational modifications (PTMs), and identification of protein-protein interactions [2]. In many ways proteomics can serve as an important complement to genomics and transcriptomics [1]. For example, while mRNA abundance differences between cellular states can be routinely monitored, these levels may not be indicative of protein levels due to controls over protein translation and degradation. In certain systems, including extracellular fluids or subcellular organelles, transcript levels are of significantly less interest than protein abundance. Additionally, protein activity, perhaps the most important factor in understanding biological pathways, may be precisely regulated by post-translational modifications [1].
Top Down Proteomics
While a variety of methods, including cell imaging and protein arrays, are capable of large-scale protein study, mass spectrometry-based approaches are uniquely well suited in terms of throughput and sensitivity to handle proteome-wide investigations [2]. Mass spectrometry-based proteomics has traditionally been carried out in a Bottom Up approach [3,4]. This entails the chemical or enzymatic digestion of proteins prior to their introduction to the mass spectrometer. The detection and typically fragmentation-based identification of the peptides allows for the inferred identification of the original protein. Immediately several disadvantages to this approach become clear: a peptide or even several peptides may not be specific to an individual protein or protein form, large regions of the protein may not be identified which can leave behind important information regarding PTMs or sequence variants, and modifications or sequence variations may occur on disparate peptides, causing their relation to one another to be lost following digestion. Top Down mass spectrometry seeks to eliminate these problems by introducing the intact protein into the mass spectrometer where both its intact and fragment ions masses are measured (Fig. 1). This approach routinely allows for 100% sequence coverage and full characterization of proteoforms, the specific molecular form of the protein resulting from combinations of genetic variation, alternative splicing, and post-translational modifications [5].
Fig. 1.

Comparison of Top Down and Bottom Up mass spectrometry [3]. In the traditional Bottom Up approach, intact proteins are digested into peptides prior to introduction into the mass spectrometer where they are then detected and fragmented. In Top Down mass spectrometry, the protein is ionized directly, allowing for improved sequence coverage and detection of PTMs.
The potential for achieving full protein characterization has made the Top Down mass spectrometry approach extremely useful for analysis of single proteins or simple mixtures of significant biological interest [6,7,8,9,10,11]. However, the technical difficulty of proteome-wide analysis at the intact protein level has caused Top Down proteomics to lag behind Bottom Up in terms of proteome coverage, sensitivity, and throughput. However, recent advances in separations, mass spectrometry instrumentation, and tailored bioinformatic tools have propelled the Top Down approach towards becoming a powerful complement and perhaps a viable alternative to digestion-based approaches.
Intact Protein Separation Methods
The great complexity within most proteomic samples requires that they be fractionated prior to introduction to the mass spectrometer [12]. Many separation strategies can be applied off-line, or independent of the mass spectrometer [13]. This entails collection of the eluted fractions followed by their infusion into the mass spectrometer. Using this approach, more instrument time can be spent collecting data on a single protein or simple mixture. Additionally, off-line separations are more flexible as the separation conditions do not need to be mass-spectrometry compatible. In comparison, on-line separations couple directly to mass spectrometry, allowing for increased throughput and reduced sample handling but with limitations to data acquisition and separation conditions. Given the complexity of most proteomics samples, multiple separations are often required to achieve sufficient separation, often using an off-line approach coupled to an on-line separation.
Liquid Chromatography
One of the most common methods for the separation of intact proteins, peptides, and small molecules is liquid chromatography (LC). This general separation approach relies on differential partitioning of analytes between a liquid mobile phase and a stationary phase. In many cases, liquid chromatography can often be coupled to electrospray ionization (ESI), proving an effective method for on-line analysis [14]. While a variety of liquid chromatography methods have been developed, reversed-phase liquid chromatography (RPLC), hydrophobic interaction liquid chromatography (HILIC), and ion exchange chromatography (IEX) are three of the most common liquid chromatography approaches applied to intact proteins [13].
Reversed-Phase Liquid Chromatography
RPLC uses a non-polar stationary phase and a polar mobile phase, allowing the most hydrophilic analytes to elute first. Alkyl chains (C4, C5, C8, C18) linked to porous silica particles are common stationary phases, where shorter chains are generally preferred for intact proteins as these phases are less retentive and offer higher recovery [13]. Additionally, many reports have been published using derivatized nonporous silica (NPS) particles, which offer increased speed and protein recovery, but suffer from limited loading capacity and high back pressure [15,16,17,18,19,20]. Use of superficially porous particles, which contain a nonporous silica core and a porous shell, has been reported for protein separations, offering similar efficiency to that of nonporous columns but with reduced back pressure and improved loading capacity [21]. Polymeric reversed-phase materials, offering increased mechanical strength, uniform hydrophobicity, and high recovery, have also been utilized for the separation of intact proteins [13,22,23,24].
For intact proteins, RPLC is typically employed as a second dimension of separation, but it has often been successfully applied as the sole separation strategy [13,15,22,25]. For example, a 4.6 mm inner diameter derivatized nonporous silica column was used to fractionate Methanococcus jannaschii lysates prior to off-line analysis to facilitate protein detection and automated fragmentation [15]. Another study utilized a 1.0 mm polymeric column to separate histone extracts from HeLa cells [22]. The column effluent was split such that 300 nL/min was electrosprayed directly in the mass spectrometer and the remaining 99.7 μL/min was collected for later automated off-line analysis.
Hydrophobic Interaction Liquid Chromatography
In contrast with RPLC, HILIC utilizes a polar stationary phase and gradients of increasing water content, resulting in the elution of more hydrophobic species first [26,27]. Analytes partition between the mobile phase and water-enriched region surrounding the stationary phase, differing from traditional normal phase chromatography where analytes are actually adsorbed to the hydrophilic stationary phase. Membrane proteins extracted from bovine heart mitochondria have been fractionated using HILIC.[28,29] HILIC has also been extensively applied for the separation of modified histone forms prior to Top Down mass spectrometry [30,31,32].
Ion Exchange
While separation in RPLC and HILIC rely primarily on differences in hydrophobicity to achieve separation, ion-exchange chromatography (IEX) uses differences in the charge of the analyte. Increasing the ionic strength of the mobile phase is used to elute analytes from the charged stationary phase. Opiteck et al. reported the use of cation exchange coupled to on-line RPLC for the two dimensional separation of the Escherichia coli proteome [33]. Besides increasing the fractionation power of the system, the use of RPLC following IEX allows for efficient desalting prior to electrospray ionization. Strong anion exchange (SAX) coupled to RPLC-MS for intact protein separations has also been reported for the study of E. coli [34]. In this work, the authors combined Top Down and Bottom Up proteomics by collecting intact protein spectra and also digesting a portion of the protein effluent, allowing for identification. Anion exchange-RPLC has been utilized for the Top Down study of Shewanella oneidensis [35], yeast [36], and human leukocytes [37]. Chromatofocusing, a variant of IEX which uses a change in pH rather than ionic strength to achieve elution, has been coupled to off-line RPLC for fractionation of intact proteins from breast cancer [17] and Methanosarcina acetivorans [38]. In contrast to traditional IEX, chromatofocusing elutes proteins as a function of their isoelectric point (pI), a physical parameter which can be useful for protein identification.
Electrophoresis
In addition to chromatography, electrophoresis, which relies on the differential migration of proteins in an applied electric field, is an extremely popular general approach for separating intact proteins [2,13,39]. The most common electrophoretic method is SDS-PAGE, in which SDS-coated protein molecules migrate through a polyacrylamide gel matrix in an electric field achieving separation based largely on molecular weight [40]. This is commonly utilized in Bottom Up proteomics by digesting the proteins out of the gel, then performing on-line LC-MS [41,42]. This approach can be extended to two-dimensional gel electrophoresis (2-DE) in which isoelectric focusing (IEF) is used as the first dimension of separation to separate proteins according to their pI, which can be useful for resolving modified proteins, and then SDS-PAGE is performed as the second dimension [39,43]. While these gel-based approaches separate intact proteins, recovery of the protein from the gel is often very difficult and offers poor recovery [39]. Proteins can be electroeluted from SDS-PAGE gels using an orthogonally applied electric field to the gel; however few studies have been reported, likely due to poor recovery [44].
Tube Gel Electrophoresis
While traditional gel-based approaches are generally not applicable to Top Down proteomics, similar separation strategies have been applied. Continuous-elution gel electrophoresis utilizes a tube gel column to separate proteins which are then collected as they elute from the end of the gel column [16]. This approach was applied to the fractionation of the S. cerevisiae proteome using an acid-labile surfactant (ALS) rather than SDS, as it could be degraded upon acidification, limiting downstream interferences. The fractions were further separated using off-line RPLC on a 4.6 mm C4 column before MS analysis. An improvement to this original separation used a mini prep-cell, featuring a 7 mm gel column, rather than a 37 mm column, coupled to a 320 μm C4 column allowing for 15 to 300-fold less sample loading as well some on-line analysis [45].
The use of tube gel electrophoresis for protein separation was further expanded with the invention of gel-eluted liquid fraction entrapment electrophoresis (GELFrEE), shown in Fig. 2 [46]. This device differs from the prep cell in its use of a sample collection chamber, in which fractions are manually collected. Fractionating this way ensures that higher molecular weight proteins are not continually diluted and dispersed across many fractions. Additionally, utilizing a short gel column, this device offers separation in ~75% less time. The GELFrEE device was first applied to a Top Down study in 2009, utilizing SDS which was then removed using methanol/chloroform/water precipitation [47] prior to online nano-LC-MS [23]. Using a Tris-tricine [48] variant of the original separation, the authors were able to identify 35 unique proteins from a single GELFrEE separation of HeLa cells. In the last few years, several papers have been published using GELFrEE for molecular-weight based fractionation for Top Down proteomics, allowing for increased throughput and number of identifications [24,49,50,51,52].
Fig. 2.
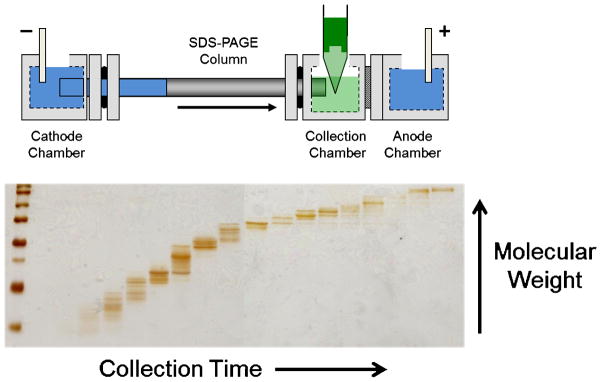
Diagram of the GELFrEE device [46]. A gel column is utilized to achieve electrophoretic separation of proteins, analogous to SDS-PAGE, which are then eluted into the liquid-phase for manual collection. The fractionation can then be visualized by running a portion of the fractions on a SDS-PAGE gel.
Isoelectric Focusing
Isoelectric focusing (IEF) for Top Down proteomics is generally considered more difficult as proteins tend to precipitate at their isoelectric point, significantly reducing their recovery from the gel media [18]. The Rotofor device uses an IEF separation but within an open channel, where the pH gradient is formed through the use of carrier ampholytes in solution between an acidic anode and a basic cathode [53]. While precipitation can still be problematic, especially for hydrophobic proteins, recovery can be increased by the use of 8 M urea and a nonionic detergent, such as CHAPS. A preparative-scale Rotofor, capable of separating ~1 g of protein within 55 mL of buffer, and the mini-Rotofor, a smaller version of the device which can hold ~15 mL of solution, have been used for the fractionation of the human erythroleukemia cell line proteome prior to NPS-RPLC-MS [18,19,20].
Another variant of solution isoelectric focusing (sIEF) for protein fractionation was reported in 2008 [54]. This device featured a separation channel divided into 8 chambers for fraction collection, where restriction channels and glass filter membranes were used to limit bulk flow between chambers. Recovery of precipitated proteins was achieved by washing the collection chambers with 0.1% TFA. It was shown that the device could be used as a first dimension of separation prior to a multiplexed version of GELFrEE, analogous to a two-dimensional gel separation [55]. This separation strategy was utilized for the MS2-based identification of over 1,000 human proteins using a Top Down approach, by far the most ever identified at that time [50].
Capillary Electrophoresis
Another electrophoretic technique used for the separation of intact proteins is capillary electrophoresis (CE). The small capillaries (<100 μm inner diameter) used within CE allow for high separation voltages (10–30 kV) without Joule heating, thereby reducing separation time and increasing peak capacity by limiting longitudinal diffusion [56,57,58]. Capillary zone electrophoresis (CZE), the simplest separation mode in CE, utilizes differences in the electrophoretic mobility of the analytes within an open capillary to achieve separation. CZE has been the most common CE mode applied to the mass spectrometry of intact proteins [59]. However, these studies have been largely limited to the analysis of a few target proteins. Notable examples include the detection of the α and β subunits of hemoglobin from a single erythrocyte [60] and the detection of various protein glycoforms of erythropoietin, fetuin, and α1-acid glycoprotein [61]. A larger scale study reported the detection of 55/56 ribosomal proteins from E. coli [62].
Capillary isoelectric focusing (cIEF), another common CE separation mode, provides a high resolution separation within an open capillary tube which has typically been coated with polyacrylamide to reduce electroosmotic flow and protein adsorption [63,64]. The use of cIEF-MS for the analysis of intact proteins was first reported in 1998, for the detection of protein standards, human hemoglobin variants, and E. coli cell lysate [65]. After focusing, proteins were mobilized into the mass spectrometer by applying potential to the inlet electrode and raising the inlet reservoir above the electrospray needle. Improvements to the cIEF separation and data acquisition, as well as the use of isotopically-depleted growth media for improved sensitivity, allowed for the detection of 1,000 polypeptides/proteins from ~300 ng of protein [66,67]. cIEF has also been coupled to RPLC-MS, which allowed for a second dimension of separation and the removal of ampholytes which can cause significant ionization suppression [68]. Using this platform, 1200 polypeptides/proteins from Chlorobium tepidum were detected over an eight hour separation [69].
Mass Spectrometry of Intact Proteins
The detection and identification of intact proteins, especially on a proteome-wide level, depends on high performance mass spectrometers [3]. High resolution and mass accuracy are critical to separate and accurately assign spectral peaks arising from complex precursor spectra containing multiple intact proteoforms or fragmentation spectra containing hundreds of fragment ions. Extremely high resolution may be required to distinguish disulfide bridges (Δm = 2 Da), deamidation (Δm = 1 Da), trimethylation versus acetylation (Δm = 39 mDa), and phosphorylation versus sulfation (Δm = 10 mDa) [70]. Sensitivity is also vital, as high molecular weight species such as proteins will have broad isotopic distributions, distributing the signal from a single protein across many peaks [71]. Additionally, in electrospray ionization (ESI) a population of protein ions will display a distribution of charge states. Combining these two effects, the signal arising from an individual proteoform may easily be split into hundreds of channels, reducing the signal at any mass-to-charge ratio (m/z). This becomes more significant at higher mass as the number of charge states and isotope peaks increase.
Before a protein can be detected or fragmented, it must first be ionized and desorbed into the gas phase. By far, the two most common techniques for protein ionization are matrix-assisted laser desorption ionization (MALDI) [72,73] and ESI [74,75]. MALDI primarily generates singly protonated protein ions, requiring a mass analyzer capable of detecting very high m/z species. This has largely limited MALDI analysis to the use of time-of-flight (TOF) instruments. One growing application of intact protein analysis using MALDI-TOF is tissue imaging [70,76]. However, the use MALDI-TOF in high-throughput proteomics has been limited due low resolution, poor fragmentation, the requirement of relatively purified samples, and difficulty coupling to separations [2,3]. ESI generates multiply charged ions and is the preferred method for the analysis of both peptides and intact proteins, especially on a proteomics wide-scale. Taking advantage of high resolution and mass accuracy, Top Down proteomics studies have largely been implemented using ESI coupled to either Fourier transform ion cyclotron resonance (FT-ICR) or Orbitrap mass analyzers and will be the focus of further discussion.
Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
Fourier transform ion cyclotron resonance mass spectrometry relies on the excitation of an ion at its cyclotron frequency within a strong magnetic field [77,78,79]. This excitation creates a spatially coherent packet of ions, which orbit at an increased radius, allowing for detection by monitoring the image current on a detection plate. The detected signal, also termed a transient, is converted from the time domain to the frequency domain through a Fourier transform, and then to m/z through mass calibration.
ESI-FT-ICR was first used for the analysis of intact proteins in 1989, with the detection of multiple charge states on a single protein, allowing for intact mass determination [80]. Further studies demonstrated isotopic resolution on proteins using a 2.8 Tesla (T) instrument, allowing for accurate mass determination [81,82]. The same instrument was also utilized to perform collision-induced dissociation (CID) and nozzle-skimmer dissociation (NSD) of ubiquitin, using the high resolving power of the instrument to determine charge state and identity of the fragment ions [83]. These fragmentation modes utilize collisions with gas molecules to fragment the protein backbone typically at the amide linkage, resulting in b- and y- type fragment ions (Fig. 3) [84]. Another technique, electron capture dissociation (ECD), has also been utilized for protein fragmentation [85]. A heated metal filament was used to introduce electrons into the ICR cell, where they are then captured in the Rydberg orbital of one of the protonated sites on the backbone of the protein, forming a radical which results in cleavage of the N-Cα bond to form c-and z•-type ions (Fig. 1.3) [84]. In the initial report ECD of several proteins resulted in an increase in the number of backbone cleavages compared with CID [85]. CID and other collisional methods are based upon vibrational excitation, where vibrational energy can be distributed throughout the molecule, often causing the fragmentation of the weakest bonds [86]. In ECD bond-cleavage occurs before energy can be redistributed throughout the molecule, often attributed to the process being “non-ergodic” and can be utilized to achieve significantly different fragmentation patterns than collisional methods [86,87].
Fig. 3.
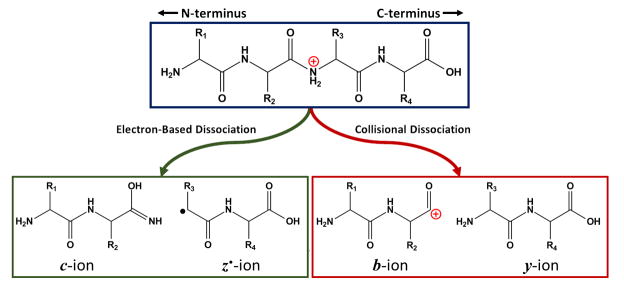
Comparison of collisional dissociation (CID/HCD) and electron-based dissociation (ECD/ETD) of peptides and proteins. Collisional dissociation results in cleavage of the amide-bond, resulting in b- and y-type ions. Electron-based methods cleave of the N-Cα bond, resulting in c- and z•- type ions.
Significant advances to FT-ICR technology, often focused on improving the analysis of intact proteins, included increases in magnetic field [88,89], addition of a resolving quadrupole for mass selection [90], and an accumulation octupole for ion storage before transmission to the ICR cell [91,92]. These modifications, allowing for increased sensitivity, dynamic range, and resolution were utilized on a 9.4 T instrument for the detection and identification of proteins from M. jannaschii and S. cerevisiae [93]. This instrument design has been utilized for a variety of Top Down proteomic studies [16,38,45,94].
The construction of a linear quadrupole ion trap/FT-ICR mass spectrometer was first reported in 2004 [95]. This instrument allowed for the storage and manipulation of ions from a continuous ion source in the linear trap before injecting them into the ICR cell. Mass accuracy was improved through the use of automatic gain control (AGC), accurately controlling the number of ions that are allowed to enter the ICR cell even from variable ion flux into the instrument which is typical of LC-MS. Fragmentation is also performed within the ion trap, with fragment ions able to be detected using the high resolution and mass accuracy of the ICR analyzer or the speed of the ion trap (mostly used for peptides). This instrument was commercialized utilizing a 7 T magnet, achieving 100,000 resolving power (m/z 400) with a one second transient as well as <2 ppm mass accuracy without internal calibration.
Use of 7 T LTQ-FT-ICR for Top Down proteomics includes the analysis of the S. cerevisiae proteome [36] and a variety of membrane proteins [96,97]. A 12 T version of the instrument has been utilized for increased throughput studies using the automated on-line/offline RPLC-MS platform for the study of HeLa nuclei [22], human leukocytes [37], and M. acetivorans [98] as well as the GELFrEE platform for the analysis of S. cerevisiae [49] and HeLa extracts [23,24,50]. A 14.5 T version of the LTQ-FT-ICR instrument has also been reported, featuring approximately 4-fold higher mass accuracy and twice the resolving power of the 7T instrument [99]. Besides the increase in field strength, a wired octupole following the ion trap allowed for the storage of an increased number of ions before detection in the ICR cell. This instrument was also utilized for analysis of GELFrEE fractions, using the improvement in sensitivity and resolution for the detection and identification of higher molecular weight proteins [23,100].
Orbitrap Mass Spectrometry
A new type of Fourier transform mass spectrometer was described in 2000, the Orbitrap mass analyzer [101]. This trap features a pair of axially symmetric electrodes: a central “spindle-like” electrode and an outer “barrel-like electrode”. In this electric field, ions rotate around the central electrode while oscillating down the length of the electrode. The frequency of these oscillations is proportional to (m/z)−1/2. Image current on the outer electrodes is monitored and the resulting time domain signal is converted to frequency and then to m/z as in FT-ICR. Also similar to FT-ICR, the Orbitrap mass analyzer has been coupled to a LTQ allowing for use of a continuous ion source (e.g., ESI), increasing mass accuracy with AGC, and enabling efficient fragmentation [102]. Coupling of the two analyzers was achieved by the use of a transfer octupole following the ion trap into a curved rf-only quadrupole (C-trap) used to eject ions axially towards the Orbitrap analyzer. This new instrument was capable of obtaining 60,000 resolving power (m/z 400) using a one second transient, achieving isotopic resolution of myoglobin and carbonic anhydrase.
The use of the LTQ-Orbitrap for more extensive examination of intact proteins was first reported in 2006 [103]. The authors demonstrated the ability of the instrument to consistently achieve <10 ppm mass accuracy on intact proteins and confidently identify the proteins using CID fragmentation (MS2 and MS3). The LTQ-Orbitrap was also utilized for the study of low molecular weight proteins from human blood including the quantitation of apolipoprotein proteoforms [104] and detection of transthyretin and hemoglobin variants [105]. The LTQ-Orbitrap was also used to distinguish several glycoforms from intact recombinant antibodies (~150 kDa) and fragment the reduced light and heavy chains using CID [106]. A similar study utilized higher-energy collisional dissociation (HCD) to obtain sequence information on subunits [107]. HCD is a collisional-based fragmentation approach similar CID, but allows for the detection of low m/z fragment ions and provides higher-energy collisions which can result in more informative fragmentation spectra [108]. In the original design, HCD was achieved by colliding oscillating ions within the C-trap with nitrogen gas, but an improved design relied on an additional higher pressure octupole collision cell following the C-trap.
Significant modifications were made to the design of the linear ion trap into a new instrument branded as the LTQ Velos [109]. Improved ion injection optics allowed for ~5-fold reduction in ion injection times and the use of two linear ion traps allowed for more efficient trapping and CID fragmentation in a higher pressure trap and higher resolution scanning in a lower pressure trap. While a stand-alone LTQ Velos was capable of achieving isotopic resolution of intact myoglobin, coupling the LTQ Velos to the Orbitrap (LTQ Orbitrap Velos) provided the routine high mass accuracy and resolving power needed to advance both Bottom Up and Top Down proteomics [110]. With similar coupling of the ion traps and Orbitrap as in the previous LTQ-Orbitrap, this instrument contained an improved integrated C-trap and collision cell enabling more efficient HCD. In the initial report, the LTQ Orbitrap Velos was able to achieve isotopic resolution of carbonic anhydrase along with its confident identification using HCD fragmentation. It should also be noted that the LTQ Velos has also been coupled to a 12 T FT-ICR mass spectrometer for Top Down analysis [52], however the instrument has not been commercialized and no other uses of such an instrument have been reported.
The LTQ Orbitrap Velos has been used for the analysis of disease causing hemoglobin variants from dried blood droplets for potential clinical use [111]. Another report utilized the instrument to identify 53 proteins, many of them containing modifications including pyroglutamates, disulfide bonds, and S-glutathiolation, from the periplasm of the bacterium Novosphingobium aromaticivorans [112]. Antibodies have also been analyzed using this instrument, allowing for improved sequence coverage through the use of electron transfer dissociation (ETD) of the disulfide intact species [113]. ETD is an electron-based fragmentation technique similar to ECD, but utilizes gaseous anions to transfer low-energy electrons to protonated analytes [114]. This approach allows for electron-based dissociation using a linear ion trap, which is not adequately capable of trapping free electrons used in ECD. ETD was originally coupled to a LTQ-Orbitrap through ESI of the precursor ETD reagent into the trap and then allowing for reaction [115], but an improved design transported the ions from a negative chemical ionization source to the ion trap [116].
A compact high-field Orbitrap, coupled with an improved Velos PRO dual ion trap mass spectrometer and advanced signal processing was recently reported, capable of a nearly four-fold increase in resolution [117]. This improved resolution was used to achieve isotopic resolution of carbonic anhydrase and enolase with a sub-second transient. The utility of this instrument for Top Down proteomics was demonstrated by the identification of 690 unique proteins from the H1299 human cancer cell line [51], followed by the identification of 1976 unique proteins from an H1299 cell line used to probe the proteome-wide difference in cellular senescence[118]. SID, CID, HCD, and ETD were each utilized to achieve complementary fragmentation and improved proteome coverage.
Data Processing for Top Down Proteomics
While powerful separation devices and mass spectrometers can be used together to generate data impressive in both quality and quantity, it must be adequately processed in order to identify and characterize proteoforms. As Top Down proteomics continues to increase in throughput and complexity of the samples analyzed, it is clear that a software platform must allow for fast, automated processing of raw data. ProSight PTM was the first search engine and web application designed for the identification of intact proteins [119,120]. In absolute mass searching (Fig. 1.4), the software uses the precursor mass and mass tolerance window to generate a possible list of candidates from a larger annotated database. The theoretical fragment ions from the candidates are then compared to the experimentally determined fragment ions within a fragment mass tolerance. A P-score is calculated for each hit, representing the probability that a random sequence could account for the matching ions [121]. Sequence tag searches can also be performed, allowing for identification of proteins based on amino acid mass differences from the fragmentation data (Fig. 1.4) [119,120]. An updated version, ProSight PTM 2.0, included the ability to include fixed modifications (e.g. alkylation of cysteine residues) and terminal modifications (e.g. N-terminal acetylation) [122]. ProsightPC, a desktop-based version of the software, has also been developed and commercialized. Additional features include error-tolerant searches, which applies the difference between observed and expected precursor masses to fragment masses, and biomarker searches, which searches against all possible protein fragments within the database (Fig. 4). A high-throughput mode allows for rapid processing of LC-MS data, utilizing the algorithms THRASH [123] or Extract to determine monoisotopic neutral precursor and fragment masses prior to database searching. ProSightPC has also been implemented on a computing cluster for processing large amount of Top Down data.[50] More recently, an online database search and retrieval has been paired with ProSight with a resulting 42 orders of magnitude gain in protein characterization[124].
Fig. 4.

Search modes utilized within ProSightPC. Absolute mass searching (a) uses an intact mass window to generate candidates, whose theoretical fragments are compared within experimental data. The hash marks indicate a matching fragment ion within a set fragment tolerance. In this case the experimental data was matched to a proteoform within the database featuring a cleaved initial methionine with 131.20 Da mass shift (outlined and boxed in orange). In sequence tag searching (b), a tag (green and boxed in orange) is generated from fragmentation data which is then used to search for candidates within the database displaying the same tag. Biomarker searching (c) is similar to absolute mass searching, but searches all possible sequences of proteins in the database, in this case resulting in identification of a proteoform containing a large truncation of the N-terminus (outlined and boxed in orange).
Alternatives to ProSight have been reported, but their uses have been limited.[125,126] BIG Mascot or MascotTD, utilizes the popular Bottom Up software platform Mascot,[127] but extends the precursor mass cutoff from the typical 16 kDa to 110 kDa.[125] The software was used for the identification of several standard proteins in addition to 13 variants of human superoxide dismutase. Additionally, the identification of a 669 kDa protein was achieved using funnel-skimmer dissociation without precursor detection. Another alternative platform, precursor ion independent Top Down algorithm (PIITA), searches tandem mass spectra against all possible tandem spectra from a sequence database [126]. This program was aimed primarily at protein identification, not full proteoform characterization. The use of the software for the analysis of Salmonella typhimurium outer membrane extracts resulted in 154 protein identifications.
Native Mass Spectrometry
Current high-throughput Top Down workflows have proven extremely successful at identifying a large number of the proteins present in human cells, yet the great majority of these studies have denatured the proteins prior to their introduction into the mass spectrometer[118]. While these conditions are gentle enough to preserve many covalent PTMs, the potentially biologically relevant non-covalent protein-protein and protein-ligand interactions are mostly destroyed. Native size-exclusion chromatography[128] and ion-exchange chromatography[129] have been reported for mapping out protein complexes from endogenous samples, however, these have mostly been followed by a bottom up approach; any stoichiometric information of subunit clustering or PTMs is lost in the digestion. While the top down study of intact protein complexes has been reported since the early 1990’s, [130,131] their characterization by gas-phase monomer ejection[132], and further by fragment ions have only recently been reported[133].
Interrogation of whole protein assemblies is a rapidly expanding sub-area of biological mass spectrometry. While most work has been on homomeric assemblies in the 60–700 kDa regime, the next major step will be to target heteromeric complexes like the proteasome, trying to find ways to eject each monomeric subunit for characterization by top down tandem MS. Many groups are active in this area [134,135,136,137,138] which really has shown that complexes present in the condensed phase can be transmitted into the gas phase with high fidelity. In addition to maintaining many of the non-covalent interactions crucial to biological processes, native mass spectrometry offers the additional benefit of a lower distribution of charge states. A 2011 study found that the large distribution of charge states observed for denatured proteins >30 kDa is the major cause of the decreased signal (and corresponding decrease in identification) for these large proteins[71]. However, ESI of these proteins under native conditions significantly reduces the number of charge states observed (coupled with a corresponding decrease in overall charge) in the mass spectrum; a recent study[139] of intact norovirus P particles (877 kDa) reported only 10 observable charge states, fewer than that for a denatured spectrum of cytochrome C (12 kDa)[140]. Fig. 5 shows the decrease in overall charge and number of different charge states for a protein when moving from denaturing to native solution conditions (by adjusting the solution pH).
Fig. 5.

The charge state distribution of electrosprayed ubiquitin ions from denaturing (top) and native (bottom) solution conditions.
Conclusions
Top Down proteomics offers an alternative to digestion-based approaches, with the promise of full protein characterization on a proteome-wide scale. While the measurement of intact proteins presents many technical challenges, the field has seen tremendous advances in separations tools, mass spectrometry instrumentation, and data processing. There has been a clear trend towards miniaturization of separations and increased use on-line and multidimensional separations. With increases in scanning speed, mass spectrometers have become more capable of handling on-line analysis and data acquisition has become focused largely on protein identification, rather than solely detection. Additionally, multiple modes of ion fragmentation have been exploited for increased sequence coverage. There has also been a shift towards commercial instrumentation, most recently the Orbitrap analyzer, which does not require expensive superconducting magnets. Software has become capable of performing various search types on complex databases while taking advantage of high mass accuracy. In general, there has been a trend towards analysis of more complex proteomes (e.g., human), with decreased sample amounts, resulting in both more identifications and improved proteoform characterization. Finally new advances in native separations and the direct detection of protein complexes offers many promising new directions of study for Top Down proteomics.
Top Down versus Bottom Up proteomics analysis
Separations methods for Top Down proteomics
Developments in mass spectrometrical instrumentation and fragmentation
Native mass spectrometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cox J, Mann M. Is proteomics the new genomics? Cell. 2007;130:395–398. doi: 10.1016/j.cell.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 3.Kelleher NL. Top-down proteomics. Anal Chem. 2004;76:196 A–203 A. [PubMed] [Google Scholar]
- 4.Chait BT. Mass spectrometry: Bottom-up or top-down? Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 5.Smith LM, Kelleher NL. Proteoform: A single term describing protein complexity. Nat Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siuti N, Roth MJ, Mizzen CA, Kelleher NL, Pesavento JJ. Gene-specific characterization of human histone H2B by electron capture dissociation. J Proteome Res. 2006;5:233–239. doi: 10.1021/pr050268v. [DOI] [PubMed] [Google Scholar]
- 7.Li M, Jiang L, Kelleher NL. Global histone profiling by LC-FTMS after inhibition and knockdown of deacetylases in human cells. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3885–3892. doi: 10.1016/j.jchromb.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Resemann A, Wunderlich D, Rothbauer U, Warscheid B, Leonhardt H, Fuchser J, Kuhlmann K, Suckau D. Top-down de Novo protein sequencing of a 13.6 kDa camelid single heavy chain antibody by matrix-assisted laser desorption ionization-time-of-flight/time-of-flight mass spectrometry. Anal Chem. 2010;82:3283–3292. doi: 10.1021/ac1000515. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, Guner H, Wang S, Kohmoto T, Young KH, Moss RL, Ge Y. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10:4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, Seeger MA, Miguel RN, Korkhov VM, van Veen HW, Venter H, Walmsley AR, Tate CG, Robinson CV. Mass spectrometry of membrane transporters reveals subunit stoichiometry and interactions. Nat Methods. 2009;6:585–587. doi: 10.1038/nmeth.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erales J, Gontero B, Whitelegge J, Halgand F. Mapping of a copper-binding site an the small CP12 chloroplastic protein of Chlamydomonas reinhardtii using top-down mass spectrometry and site-directed mutagenesis. Biochem J. 2009;419:75–82. doi: 10.1042/BJ20082004. [DOI] [PubMed] [Google Scholar]
- 12.Kellie JF, Tran JC, Lee JE, Ahlf DR, Thomas HM, Ntai I, Catherman AD, Durbin KR, Zamdborg L, Vellaichamy A, Thomas PM, Kelleher NL. The emerging process of Top Down mass spectrometry for protein analysis: biomarkers, protein-therapeutics, and achieving high throughput. Mol Biosyst. 2010;6:1532–1539. doi: 10.1039/c000896f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capriotti AL, Cavaliere C, Foglia P, Samperi R, Lagana A. Intact protein separation by chromatographic and/or electrophoretic techniques for top-down proteomics. J Chromatogr A. 2011;1218:8760–8776. doi: 10.1016/j.chroma.2011.05.094. [DOI] [PubMed] [Google Scholar]
- 14.Whitehouse CM, Dreyer RN, Yamashita M, Fenn JB. Electrospray interface for liquid chromatographs and mass spectrometers. Anal Chem. 1985;57:675–679. doi: 10.1021/ac00280a023. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JR, Meng FY, Forbes AJ, Cargile BJ, Kelleher NL. Fourier-transform mass spectrometry for automated fragmentation and identification of 5–20 kDa proteins in mixtures. Electrophoresis. 2002;23:3217–3223. doi: 10.1002/1522-2683(200209)23:18<3217::AID-ELPS3217>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Meng FY, Cargile BJ, Patrie SM, Johnson JR, McLoughlin SM, Kelleher NL. Processing complex mixtures of intact proteins for direct analysis by mass spectrometry. Anal Chem. 2002;74:2923–2929. doi: 10.1021/ac020049i. [DOI] [PubMed] [Google Scholar]
- 17.Chong BE, Yan F, Lubman DM, Miller FR. Chromatofocusing nonporous reversed-phase high-performance liquid chromatography/electrospray ionization time-of-flight mass spectrometry of proteins from human breast cancer whole cell lysates: a novel two-dimensional liquid chromatography/mass spectrometry method. Rapid Commun Mass Spectrom. 2001;15:291–296. doi: 10.1002/rcm.227. [DOI] [PubMed] [Google Scholar]
- 18.Wall DB, Kachman MT, Gong SY, Hinderer R, Parus S, Misek DE, Hanash SM, Lubman DM. Isoelectric focusing nonporous RP HPLC: A two-dimensional liquid-phase separation method for mapping of cellular proteins with identification using MALDI-TOF mass spectrometry. Anal Chem. 2000;72:1099–1111. doi: 10.1021/ac991332t. [DOI] [PubMed] [Google Scholar]
- 19.Wall DB, Kachman MT, Gong SYS, Parus SJ, Long MW, Lubman DM. Isoelectric focusing nonporous silica reversed-phase high-performance liquid chromatography/electrospray ionization time-of-flight mass spectrometry: a three-dimensional liquid-phase protein separation method as applied to the human erythroleukemia cell-line. Rapid Commun Mass Spectrom. 2001;15:1649–1661. doi: 10.1002/rcm.421. [DOI] [PubMed] [Google Scholar]
- 20.Wall DB, Parus SJ, Lubman DM. Comparison of the capabilities of liquid isoelectric focusing-one-dimensional nonporous silica reversed-phase liquid chromatography-electrospray ionization time-of-flight mass spectrometry and liquid isoelectric focusing-one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis mass mapping for the analysis of intact protein molecular masses. J Chromatogr B. 2001;763:139–148. doi: 10.1016/s0378-4347(01)00382-6. [DOI] [PubMed] [Google Scholar]
- 21.Roth MJ, Plymire DA, Chang AN, Kim J, Maresh EM, Larson SE, Patrie SM. Sensitive and reproducible intact mass analysis of complex protein mixtures with superficially porous capillary reversed-phase liquid chromatography mass spectrometry. Anal Chem. 2011;83:9586–9592. doi: 10.1021/ac202339x. [DOI] [PubMed] [Google Scholar]
- 22.Wenger CD, Boyne MT, II, Ferguson JT, Robinson DE, Kelleher NL. Versatile online-offline engine for automated acquisition of high-resolution tandem mass spectra. Anal Chem. 2008;80:8055–8063. doi: 10.1021/ac8010704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JE, Kellie JF, Tran JC, Tipton JD, Catherman AD, Thomas HM, Ahlf DR, Durbin KR, Vellaichamy A, Ntai I, Marshall AG, Kelleher NL. A robust two-dimensional separation for top-down tandem mass spectrometry of the low-mass proteome. J Am Soc Mass Spectrom. 2009;20:2183–2191. doi: 10.1016/j.jasms.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vellaichamy A, Tran JC, Catherman AD, Lee JE, Kellie JF, Sweet SMM, Zamdborg L, Thomas PM, Ahlf DR, Durbin KR, Valaskovic GA, Kelleher NL. Size-sorting combined with improved nanocapillary liquid chromatography-mass spectrometry for identification of intact proteins up to 80 kDa. Anal Chem. 2010;82:1234–1244. doi: 10.1021/ac9021083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SW, Berger SJ, Martinovic S, Pasa-Tolic L, Anderson GA, Shen YF, Zhao R, Smith RD. Direct mass spectrometric analysis of intact proteins of the yeast large ribosomal subunit using capillary LC/FTICR. Proc Natl Acad Sci U S A. 2002;99:5942–5947. doi: 10.1073/pnas.082119899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alpert AJ, Andrews PC. Cation-exchange chromatography of peptides on poly(2-sulfoethyl aspartamide)-silica. J Chromatogr. 1988;443:85–96. doi: 10.1016/s0021-9673(00)94785-x. [DOI] [PubMed] [Google Scholar]
- 27.Alpert AJ. Hydrophobic interaction chromatography of peptides as an alternative to reversed-phase chromatography. J Chromatogr. 1988;444:269–274. doi: 10.1016/s0021-9673(01)94030-0. [DOI] [PubMed] [Google Scholar]
- 28.Carroll J, Fearnley IM, Walker JE. Definition of the mitochondrial proteome by measurement of molecular masses of membrane proteins. Proc Natl Acad Sci U SA. 2006;103:16170–16175. doi: 10.1073/pnas.0607719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carroll J, Altman MC, Fearnley IM, Walker JE. Identification of membrane proteins by tandem mass spectrometry of protein ions. Proc Natl Acad Sci U S A. 2007;104:14330–14335. doi: 10.1073/pnas.0706817104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells. Nat Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 31.Pesavento JJ, Garcia BA, Streeky JA, Kelleher NL, Mizzen CA. Mild performic acid oxidation enhances chromatographic and top down mass spectrometric analyses of histones. Mol Cell Proteomics. 2007;6:1510–1526. doi: 10.1074/mcp.M600404-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Pesavento JJ, Bullock CR, Leduc RD, Mizzen CA, Kelleher NL. Combinatorial modification of human histone H4 quantitated by two-dimensional liquid chromatography coupled with top down mass spectrometry. J Biol Chem. 2008;283:14927–14937. doi: 10.1074/jbc.M709796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opiteck GJ, Lewis KC, Jorgenson JW, Anderegg RJ. Comprehensive on-line LC/LC/MS of proteins. Anal Chem. 1997;69:1518–1524. doi: 10.1021/ac961155l. [DOI] [PubMed] [Google Scholar]
- 34.Millea KM, Krull IS, Cohen SA, Gebler JC, Berger SJ. Integration of Multidimensional Chromatographic Protein Separations with a Combined “Top-Down” and “Bottom-Up” Proteomic Strategy. Journal of Proteome Research. 2005;5:135–146. doi: 10.1021/pr050278w. [DOI] [PubMed] [Google Scholar]
- 35.Sharma S, Simpson DC, Tolid N, Jaitly N, Mayampurath AM, Smith RD, Paša-Tolid L. Proteomic profiling of intact proteins using WAX-RPLC 2-D separations and FTICR mass spectrometry. J Proteome Res. 2006;6:602–610. doi: 10.1021/pr060354a. [DOI] [PubMed] [Google Scholar]
- 36.Parks BA, Jiang L, Thomas PM, Wenger CD, Roth MJ, Boyne MT, Burke PV, Kwast KE, Kelleher NL. Top-down proteomics on a chromatographic time scale using linear ion trap Fourier transform hybrid mass spectrometers. Anal Chem. 2007;79:7984–7991. doi: 10.1021/ac070553t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth MJ, Parks BA, Ferguson JT, Boyne MT, Kelleher NL. “Proteotyping”: Population proteomics of human leukocytes using top down mass spectrometry. Anal Chem. 2008;80:2857–2866. doi: 10.1021/ac800141g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patrie SM, Ferguson JT, Robinson DE, Whipple D, Rother M, Metcalf WW, Kelleher NL. Top down mass spectrometry of < 60-kDa proteins from Methanosarcina acetivorans using quadrupole FTMS with automated octopole collisionally activated dissociation. Mol Cell Proteomics. 2006;5:14–25. doi: 10.1074/mcp.M500219-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Rabilloud T. Two-dimensional gel electrophoresis in proteomics: Old, old fashioned, but it still climbs up the mountains. Proteomics. 2002;2:3–10. [PubMed] [Google Scholar]
- 40.Laemmli UK. Cleavage of structural proteins during assembly of head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 42.Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature. 1996;379:466–469. doi: 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- 43.Gygi SP, Corthals GL, Zhang Y, Rochon Y, Aebersold R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc Natl Acad Sci U S A. 2000;97:9390–9395. doi: 10.1073/pnas.160270797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davidsson P, Nilsson CL. Peptide mapping of proteins in cerebrospinal fluid utilizing a rapid preparative two-dimensional electrophoretic procedure and matrix-assisted laser desorption/ionization mass spectrometry. BBA - Gen Subjects. 1999;1473:391–399. doi: 10.1016/s0304-4165(99)00197-x. [DOI] [PubMed] [Google Scholar]
- 45.Du Y, Meng FY, Patrie SM, Miller LM, Kelleher NL. Improved molecular weight-based processing of intact proteins for interrogation by quadrupole-enhanced FT MS/MS. J Proteome Res. 2004;3:801–806. doi: 10.1021/pr0499489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tran JC, Doucette AA. Gel-eluted liquid fraction entrapment electrophoresis: An electrophoretic method for broad molecular weight range proteome separation. Anal Chem. 2008;80:1568–1573. doi: 10.1021/ac702197w. [DOI] [PubMed] [Google Scholar]
- 47.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 48.Schagger H, Vonjagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 49.Kellie JF, Catherman AD, Durbin KR, Tran JC, Tipton JD, Norris JL, Witkowski CE, Thomas PM, Kelleher NL. Robust analysis of the yeast proteome under 50 kDa by molecular-mass-based fractionation and top-down mass spectrometry. Anal Chem. 2012;84:209–215. doi: 10.1021/ac202384v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SMM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–U141. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL. Evaluation of the compact high-field Orbitrap for top-down proteomics of human cells. J Proteome Res. 2012;11:4308–4314. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Catherman AD, Li M, Tran JC, Durbin KR, Compton PD, Early BP, Thomas PM, Kelleher NL. Top down proteomics of human membrane proteins from enriched mitochondrial fractions. Anal Chem. 2013;85:1880–1888. doi: 10.1021/ac3031527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ayala A, Parrado J, Machado A. Use of Rotofor preparative isoelectrofocusing cell in protein purification procedure. Applied Biochemistry and Biotechnology. 1998;69:11–16. doi: 10.1007/BF02786017. [DOI] [PubMed] [Google Scholar]
- 54.Tran JC, Doucette AA. Rapid and effective focusing in a carrier ampholyte solution lsoelectric focusing system: a proteome prefractionation tool. J Proteome Res. 2008;7:1761–1766. doi: 10.1021/pr700677u. [DOI] [PubMed] [Google Scholar]
- 55.Tran JC, Doucette AA. Multiplexed size separation of intact proteins in solution phase for mass spectrometry. Anal Chem. 2009;81:6201–6209. doi: 10.1021/ac900729r. [DOI] [PubMed] [Google Scholar]
- 56.Hjerten S. Free zone electrophoresis. Chromatogr Rev. 1967;9:122–219. doi: 10.1016/0009-5907(67)80003-6. [DOI] [PubMed] [Google Scholar]
- 57.Jorgenson JW, Lukacs KD. Zone electrophoresis in open-tubular glass capillaries. Anal Chem. 1981;53:1298–1302. [PubMed] [Google Scholar]
- 58.Jorgenson JW, Lukacs KD. High-resolution separations based on electrophoresis and electroosmosis. J Chromatogr. 1981;218:209–216. [Google Scholar]
- 59.Haselberg R, de Jong GJ, Somsen GW. Capillary electrophoresis-mass spectrometry for the analysis of intact proteins. J Chromatogr A. 2007;1159:81–109. doi: 10.1016/j.chroma.2007.05.048. [DOI] [PubMed] [Google Scholar]
- 60.Hofstadler SA, Severs JC, Smith RD, Swanek FD, Ewing AG. High performance Fourier transform ion cyclotron resonance mass spectrometric detection for capillary electrophoresis. J High Resolut Chrom. 1996;19:617–621. [Google Scholar]
- 61.Balaguer E, Neususs C. Glycoprotein characterization combining intact protein and glycan analysis by capillary electrophoresis-electrospray ionization-mass spectrometry. Anal Chem. 2006;78:5384–5393. doi: 10.1021/ac060376g. [DOI] [PubMed] [Google Scholar]
- 62.Moini M, Huang HL. Application of capillary electrophoresis/electrospray ionization-mass spectrometry to subcellular proteomics of Escherichia coli ribosomal proteins. Electrophoresis. 2004;25:1981–1987. doi: 10.1002/elps.200305906. [DOI] [PubMed] [Google Scholar]
- 63.Hjerten S, Zhu MD. Adaptation of the equipment for high-performance electrophoresis to isoelectric-focusing. J Chromatogr. 1985;346:265–270. [Google Scholar]
- 64.Kilar F, Hjerten S. Fast and high resolution analysis of human serum transferrin by high performance isoelectric focusing in capillaries. Electrophoresis. 1989;10:23–29. doi: 10.1002/elps.1150100107. [DOI] [PubMed] [Google Scholar]
- 65.Yang LY, Lee CS, Hofstadler SA, Pasa-Tolid L, Smith RD. Capillary isoelectric focusing-electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry for protein characterization. Anal Chem. 1998;70:3235–3241. doi: 10.1021/ac980224o. [DOI] [PubMed] [Google Scholar]
- 66.Jensen PK, Pasa-Tolic L, Anderson GA, Horner JA, Lipton MS, Bruce JE, Smith RD. Probing proteomes using capillary isoelectric focusing-electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 1999;71:2076–2084. doi: 10.1021/ac990196p. [DOI] [PubMed] [Google Scholar]
- 67.Jensen PK, Pasa-Tolic L, Peden KK, Martinovic S, Lipton MS, Anderson GA, Tolic N, Wong KK, Smith RD. Mass spectrometic detection for capillary isoelectric focusing separations of complex protein mixtures. Electrophoresis. 2000;21:1372–1380. doi: 10.1002/(SICI)1522-2683(20000401)21:7<1372::AID-ELPS1372>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 68.Zhou F, Johnston MV. Protein characterization by on-line capillary isoelectric focusing, reversed-phase liquid chromatography, and mass spectrometry. Anal Chem. 2004;76:2734–2740. doi: 10.1021/ac035446n. [DOI] [PubMed] [Google Scholar]
- 69.Zhou F, Hanson TE, Johnston MV. Intact protein profiling of Chlorobium tepidum by capillary isoelectric focusing, reversed-phase liquid chromatography, and mass spectrometry. Anal Chem. 2007;79:7145–7153. doi: 10.1021/ac071147c. [DOI] [PubMed] [Google Scholar]
- 70.Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Kelleher NL. Analysis of intact protein isoforms by mass spectrometry. J Biol Chem. 2011;286:25451–25458. doi: 10.1074/jbc.R111.239442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. On the scalability and requirements of whole protein mass spectrometry. Anal Chem. 2011;83:6868–6874. doi: 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T, Matsuo T. Protein and polymer analyses up to m/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2:151–153. [Google Scholar]
- 74.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass-spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 75.Yamashita M, Fenn JB. Electrospray ion source. Another variation on the free-jet theme. J Phys Chem. 1984;88:4451–4459. [Google Scholar]
- 76.Seeley EH, Caprioli RM. MALDI imaging mass spectrometry of human tissue: method challenges and clinical perspectives. Trends Biotechnol. 2011;29:136–143. doi: 10.1016/j.tibtech.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Comisarow MB, Marshall AG. Fourier transform ion cyclotron resonance spectroscopy. Chem Phys Lett. 1974;25:282–283. [Google Scholar]
- 78.Comisarow MB, Marshall AG. Frequency-sweep fourier transform ion cyclotron resonance spectroscopy. Chem Phys Lett. 1974;26:489–490. [Google Scholar]
- 79.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 80.Henry KD, Williams ER, Wang BH, McLafferty FW, Shabanowitz J, Hunt DF. Fourier-transform mass spectrometry of large molecules by electrospray ionization. Proc Natl Acad Sci U S A. 1989;86:9075–9078. doi: 10.1073/pnas.86.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Henry KD, McLafferty FW. Electrospray ionization with Fourier-transform mass spectrometry. Charge state assignment from resolved isotopic peaks. Org Mass Spectrom. 1990;25:490–492. [Google Scholar]
- 82.Henry KD, Quinn JP, McLafferty FW. High-resolution electrospray mass spectra of large molecules. J Am Chem Soc. 1991;113:5447–5449. [Google Scholar]
- 83.Loo JA, Quinn JP, Ryu SI, Henry KD, Senko MW, McLafferty FW. High-resolution tandem mass spectrometry of large biomolecules. Proc Natl Acad Sci U S A. 1992;89:286–289. doi: 10.1073/pnas.89.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coon JJ. Collisions or electrons? Protein sequence analysis in the 21st century. Anal Chem. 2009;81:3208–3215. doi: 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 86.Zubarev RA, Haselmann KF, Budnik B, Kjeldsen F, Jensen F. Towards an understanding of the mechanism of electron-capture dissociation: a historical perspective and modern ideas. Eur J Mass Spectrom. 2002;8:337–350. [Google Scholar]
- 87.Sleno L, Volmer DA. Ion activation methods for tandem mass spectrometry. Journal of Mass Spectrom. 2004;39:1091–1112. doi: 10.1002/jms.703. [DOI] [PubMed] [Google Scholar]
- 88.Beu SC, Senko MW, Quinn JP, McLafferty FW. Improved Fourier-transform ion-cyclotron-resonance mass spectrometry of large biomolecules. Journal of the American Society for Mass Spectrometry. 1993;4:190–192. doi: 10.1016/1044-0305(93)85077-B. [DOI] [PubMed] [Google Scholar]
- 89.Senko MW, Hendrickson CL, Paša-Tolid L, Marto JA, White FM, Guan S, Marshall AG. Electrospray Ionization Fourier Transform Ion Cyclotron Resonance at 9.4 T. Rapid Communications in Mass Spectrometry. 1996;10:1824–1828. doi: 10.1002/(SICI)1097-0231(199611)10:14<1824::AID-RCM695>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 90.Belov ME, Nikolaev EN, Anderson GA, Auberry KJ, Harkewicz R, Smith RD. Electrospray ionization-fourier transform ion cyclotron mass spectrometry using ion preselection and external accumulation for ultrahigh sensitivity. Journal of the American Society for Mass Spectrometry. 2001;12:38–48. doi: 10.1016/S1044-0305(00)00198-7. [DOI] [PubMed] [Google Scholar]
- 91.Senko M, Hendrickson C, Emmett M, Shi SH, Marshall A. External accumulation of ions for enhanced electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Journal of the American Society for Mass Spectrometry. 1997;8:970–976. [Google Scholar]
- 92.Wilcox B, Hendrickson C, Marshall A. Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. Journal of the American Society for Mass Spectrometry. 2002;13:1304–1312. doi: 10.1016/S1044-0305(02)00622-0. [DOI] [PubMed] [Google Scholar]
- 93.Patrie SM, Charlebois JP, Whipple D, Kelleher NL, Hendrickson CL, Quinn JP, Marshall AG, Mukhopadhyay B. Construction of a hybrid quadrupole/Fourier transform ion cyclotron resonance mass spectrometer for versatile MS/MS above 10 kDa. Journal of the American Society for Mass Spectrometry. 2004;15:1099–1108. doi: 10.1016/j.jasms.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 94.Roth MJ, Forbes AJ, Boyne MT, Kim YB, Robinson DE, Kelleher NL. Precise and parallel characterization of coding polymorphisms, alternative splicing, and modifications in human proteins by mass spectrometry. Mol Cell Proteomics. 2005;4:1002–1008. doi: 10.1074/mcp.M500064-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Syka JEP, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T, Shabanowitz J, Hunt DF. Novel linear quadrupole ion trap/FT mass spectrometer: Performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J Proteome Res. 2004;3:621–626. doi: 10.1021/pr0499794. [DOI] [PubMed] [Google Scholar]
- 96.Ryan CM, Souda P, Bassilian S, Ujwal R, Zhang J, Abramson J, Ping PP, Durazo A, Bowie JU, Hasan SS, Baniulis D, Cramer WA, Faull KF, Whitelegge JP. Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation. Mol Cell Proteomics. 2010;9:791–803. doi: 10.1074/mcp.M900516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thangaraj B, Ryan CM, Souda P, Krause K, Faull KF, Weber APM, Fromme P, Whitelegge JP. Data-directed top-down Fourier-transform mass spectrometry of a large integral membrane protein complex: Photosystem II from Galdieria sulphuraria. Proteomics. 2010;10:3644–3656. doi: 10.1002/pmic.201000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ferguson JT, Wenger CD, Metcalf WW, Kelleher NL. Top-down proteomics reveals novel protein forms expressed in Methanosarcina acetivorans. Journal of the American Society for Mass Spectrometry. 2009;20:1743–1750. doi: 10.1016/j.jasms.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW, Marshall AG. High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 tesla. Anal Chem. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- 100.Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Lee JE, Kellie JF, Kelleher NL, Hendrickson CL, Marshall AG. Nano-LC FTICR tandem mass spectrometry for top-down proteomics: Routine baseline unit mass resolution of whole cell lysate proteins up to 72 kDa. Anal Chem. 2012;84:2111–2117. doi: 10.1021/ac202651v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Makarov A. Electrostatic axially harmonic orbital trapping: A high-performance technique of mass analysis. Anal Chem. 2000;72:1156–1162. doi: 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- 102.Makarov A, Denisov E, Kholomeev A, Baischun W, Lange O, Strupat K, Horning S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal Chem. 2006;78:2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 103.Macek B, Waanders LF, Olsen JV, Mann M. Top-down protein sequencing and MS3 on a hybrid linear quadrupole ion trap-Orbitrap mass spectrometer. Mol Cell Proteomics. 2006;5:949–958. doi: 10.1074/mcp.T500042-MCP200. [DOI] [PubMed] [Google Scholar]
- 104.Mazur MT, Cardasis HL, Spellman DS, Liaw A, Yates NA, Hendrickson RC. Quantitative analysis of intact apolipoproteins in human HDL by top-down differential mass spectrometry. Proc Natl Acad Sci U S A. 2010;107:7728–7733. doi: 10.1073/pnas.0910776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Théberge R, Infusini G, Tong W, McComb ME, Costello CE. Top-down analysis of small plasma proteins using an LTQ-Orbitrap. Potential for mass spectrometry-based clinical assays for transthyretin and hemoglobin. Int J Mass Spectrom. 2011;300:130–142. doi: 10.1016/j.ijms.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bondarenko P, Second T, Zabrouskov V, Makarov A, Zhang Z. Mass measurement and top-down HPLC/MS analysis of intact monoclonal antibodies on a hybrid linear quadrupole ion trap-orbitrap mass spectrometer. Journal of the American Society for Mass Spectrometry. 2009;20:1415–1424. doi: 10.1016/j.jasms.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 107.Zhang J, Liu H, Katta V. Structural characterization of intact antibodies by high-resolution LTQ Orbitrap mass spectrometry. Journal of Mass Spectrometry. 2010;45:112–120. doi: 10.1002/jms.1700. [DOI] [PubMed] [Google Scholar]
- 108.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 109.Pekar Second T, Blethrow JD, Schwartz JC, Merrihew GE, MacCoss MJ, Swaney DL, Russell JD, Coon JJ, Zabrouskov V. Dual-pressure linear ion trap mass spectrometer improving the analysis of complex protein mixtures. Anal Chem. 2009;81:7757–7765. doi: 10.1021/ac901278y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E, Denisov E, Lange O, Remes P, Taylor D, Splendore M, Wouters ER, Senko M, Makarov A, Mann M, Horning S. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics. 2009;8:2759–2769. doi: 10.1074/mcp.M900375-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Edwards R, Griffiths P, Bunch J, Cooper H. Top-down proteomics and direct surface sampling of neonatal dried blood spots: Diagnosis of unknown hemoglobin variants. Journal of The American Society for Mass Spectrometry. 2012;23:1921–1930. doi: 10.1007/s13361-012-0477-9. [DOI] [PubMed] [Google Scholar]
- 112.Wu S, Brown RN, Payne SH, Meng D, Zhao R, Tolid N, Cao L, Shukla A, Monroe ME, Moore RJ, Lipton MS, Paša-Tolid L. Top-down characterization of the post-translationally modified intact periplasmic proteome from the bacterium Novosphingobium aromaticivorans. Int J Proteomics. 2013;2013:10. doi: 10.1155/2013/279590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fornelli L, Damoc E, Thomas PM, Kelleher NL, Aizikov K, Denisov E, Makarov A, Tsybin YO. Analysis of intact monoclonal antibody IgG1 by electron transfer dissociation Orbitrap FTMS. Mol Cell Proteomics. 2012;11:1758–1767. doi: 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of electron-transfer dissociation on a hybrid linear ion trap Orbitrap mass spectrometer. Anal Chem. 2007;79:3525–3534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McAlister GC, Berggren WT, Griep-Raming J, Horning S, Makarov A, Phanstiel D, Stafford G, Swaney DL, Syka JEP, Zabrouskov V, Coon JJ. A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-orbitrap mass spectrometer. J Proteome Res. 2008;7:3127–3136. doi: 10.1021/pr800264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Mueller M, Viner R, Schwartz J, Remes P, Belford M, Dunyach JJ, Cox J, Horning S, Mann M, Makarov A. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics. 2012;11:1–11. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Catherman AD, Durbin KR, Ahlf DR, Early BP, Fellers RT, Tran JC, Thomas PM, Kelleher NL. Large-scale Top-down Proteomics of the Human Proteome: Membrane Proteins, Mitochondria, and Senescence. Mol Cell Proteomics. 2013;12:3465–3473. doi: 10.1074/mcp.M113.030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taylor GK, Kim YB, Forbes AJ, Meng F, McCarthy R, Kelleher NL. Web and database software for identification of intact proteins ssing “top down” mass spectrometry. Anal Chem. 2003;75:4081–4086. doi: 10.1021/ac0341721. [DOI] [PubMed] [Google Scholar]
- 120.LeDuc RD, Taylor GK, Kim YB, Januszyk TE, Bynum LH, Sola JV, Garavelli JS, Kelleher NL. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004;32:W340–W345. doi: 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Meng FY, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol. 2001;19:952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 122.Zamdborg L, LeDuc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res. 2007;35:W701–W706. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. Journal of the American Society for Mass Spectrometry. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 124.Durbin KR, Fellers RT, Ntai I, Kelleher NL, Compton PD. Autopilot: An Online Data Acquisition Control System for the Enhanced High-throughput Characterization of Intact Proteins. Analytical Chemistry. 2014 doi: 10.1021/ac402904h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Karabacak NM, Li L, Tiwari A, Hayward LJ, Hong PY, Easterling ML, Agar JN. Sensitive and specific identification of wild type and variant proteins from 8 to 669 kDa using top-down mass spectrometry. Mol Cell Proteomics. 2009;8:846–856. doi: 10.1074/mcp.M800099-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tsai Y, Scherl A, Shaw J, MacKay CL, Shaffer S, Langridge-Smith PR, Goodlett D. Precursor ion independent algorithm for top-down shotgun proteomics. Journal of the American Society for Mass Spectrometry. 2009;20:2154–2166. doi: 10.1016/j.jasms.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 127.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 128.Wessels HJCT, Vogel RO, Lightowlers RN, Spelbrink JN, Rodenburg RJ, van den Heuvel LP, van Gool AJ, Gloerich J, Smeitink JAM, Nijtmans LG. Analysis of 953 Human Proteins from a Mitochondrial HEK293 Fraction by Complexome Profiling. PLoS ONE. 2013;8:e68340. doi: 10.1371/journal.pone.0068340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Havugimana Pierre C, Hart GT, Nepusz T, Yang H, Turinsky Andrei L, Li Z, Wang Peggy I, Boutz Daniel R, Fong V, Phanse S, Babu M, Craig Stephanie A, Hu P, Wan C, Vlasblom J, Dar V-u-N, Bezginov A, Clark Gregory W, Wu Gabriel C, Wodak Shoshana J, Tillier Elisabeth RM, Paccanaro A, Marcotte Edward M, Emili A. A Census of Human Soluble Protein Complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ganem B, Li YT, Henion JD. Detection of noncovalent receptor-ligand complexes by mass spectrometry. Journal of the American Chemical Society. 1991;113:6294–6296. [Google Scholar]
- 131.Li YT, Hsieh YL, Henion JD, Senko MW, McLafferty FW, Ganem B. Mass spectrometric studies on noncovalent dimers of leucine zipper peptides. Journal of the American Chemical Society. 1993;115:8409–8413. [Google Scholar]
- 132.Jurchen JC, Williams ER. Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. Journal of the American Chemical Society. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL. From protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal Chem. 2013;85:11163–11173. doi: 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- 134.Jones CM, Beardsley RL, Galhena AS, Dagan S, Cheng G, Wysocki VH. Symmetrical Gas-Phase Dissociation of Noncovalent Protein Complexes via Surface Collisions. Journal of the American Chemical Society. 2006;128:15044–15045. doi: 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- 135.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Native electrospray and electron-capture dissociation FTICR mass spectrometry for top-down studies of protein assemblies. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hogan CJ, Ruotolo BT, Robinson CV, Fernandez de la Mora J. Tandem Differential Mobility Analysis-Mass Spectrometry Reveals Partial Gas-Phase Collapse of the GroEL Complex. The Journal of Physical Chemistry B. 2011;115:3614–3621. doi: 10.1021/jp109172k. [DOI] [PubMed] [Google Scholar]
- 137.Heck AJR. Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 138.Kaddis CS, Lomeli SH, Yin S, Berhane B, Apostol MI, Kickhoefer VA, Rome LH, Loo JA. Sizing Large Proteins and Protein Complexes by Electrospray Ionization Mass Spectrometry and Ion Mobility. Journal of the American Society for Mass Spectrometry. 2007;18:1206–1216. doi: 10.1016/j.jasms.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bereszczak JZ, Barbu IM, Tan M, Xia M, Jiang X, van Duijn E, Heck AJR. Structure, stability and dynamics of norovirus P domain derived protein complexes studied by native mass spectrometry. Journal of Structural Biology. 2012;177:273–282. doi: 10.1016/j.jsb.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 140.Konermann L, Douglas DJ. Acid-Induced Unfolding of Cytochrome c at Different Methanol Concentrations: Electrospray Ionization Mass Spectrometry Specifically Monitors Changes in the Tertiary Structure†. Biochemistry. 1997;36:12296–12302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]