

Abstract
Motivation: Next-generation RNA sequencing offers an opportunity to investigate transcriptome in an unprecedented scale. Recent studies have revealed widespread alternative polyadenylation (polyA) in eukaryotes, leading to various mRNA isoforms differing in their 3′ untranslated regions (3′UTR), through which, the stability, localization and translation of mRNA can be regulated. However, very few, if any, methods and tools are available for directly analyzing this special alternative RNA processing event. Conventional methods rely on annotation of polyA sites; yet, such knowledge remains incomplete, and identification of polyA sites is still challenging. The goal of this article is to develop methods for detecting 3′UTR switching without any prior knowledge of polyA annotations.
Results: We propose a change-point model based on a likelihood ratio test for detecting 3′UTR switching. We develop a directional testing procedure for identifying dramatic shortening or lengthening events in 3′UTR, while controlling mixed directional false discovery rate at a nominal level. To our knowledge, this is the first approach to analyze 3′UTR switching directly without relying on any polyA annotations. Simulation studies and applications to two real datasets reveal that our proposed method is powerful, accurate and feasible for the analysis of next-generation RNA sequencing data.
Conclusions: The proposed method will fill a void among alternative RNA processing analysis tools for transcriptome studies. It can help to obtain additional insights from RNA sequencing data by understanding gene regulation mechanisms through the analysis of 3′UTR switching.
Availability and implementation: The software is implemented in Java and can be freely downloaded from http://utr.sourceforge.net/.
Contact: zhiwei@njit.edu or hongzhe@mail.med.upenn.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The past two decades have witnessed dramatic changes brought on by high-throughput technology in both statistics and the biological sciences. Hybridization-based microarray technology, which emerged in the late 1990s, had been widely applied by researchers for more than a decade and led to a myriad of seminal advances. During the past few years, next-generation sequencing (NGS) has matured as a more powerful and accurate tool. It is replacing the once dominant microarray technology in all areas of application because of its affordable cost and highly accurate digital resolution (Wang et al., 2009). For transcriptome study, the introduction of RNA-Seq technology along with new analytic methods makes it possible to address an increasing number of compelling biological questions that may not be possible using microarray technology. In particular, alternative RNA splicing and processing, common phenomena in eukaryotes, play so critical a role in gene function regulation that they receive much attention in RNA-Seq analysis (Keren et al., 2010) and motivate quite a few methodological developments. For example, Mixture of Isoforms (MISO) uses a probabilistic mixture model to quantify alternative splicing and processing, and it, then, tests the equality of transcript isoform ratios between samples (Katz et al., 2010); multivariate analysis of transcript splicing (MATS), by using a Bayesian statistical framework, offers the flexibility to identify differential alternative splicing and processing events that match a given user-defined pattern (Shen et al., 2012); DEXSeq uses generalized linear models to test for differential usage of exons and provides reliable control of false discoveries by taking biological variation into account (Anders et al., 2012); other developments include (Griffith et al., 2010; Rogers et al., 2012; Trapnell et al., 2013). Despite the success of these methods, detecting 3′ untranslated regions (3′UTR) switching remains challenging. Very few, if any, methods and tools are available for directly analyzing this special alternative RNA processing event.
The pre-mRNA 3′ end processing plays a crucial role in eukaryotic mRNA maturation (Colgan and Manley, 1997; Proudfoot, 2011). Through cis elements in the 3′UTR of mRNAs, post-transcriptional gene regulation frequently occurs and determines the stability, localization and translation of mRNA (Martin and Ephrussi, 2009; Moore, 2005). These roles are mediated by interactions with RNA-binding proteins and microRNAs (miRNAs) (Licatalosi and Darnell, 2010). Over half of mammalian genes contain alternative cleavage and polyadenylation (or polyA) sites, which lead to various mRNA isoforms differing in their 3′UTRs (Zhang et al., 2005). Alternative cleavage and PolyAdenylation (APA) in 3′UTR, including shortening and lengthening events, have recently been identified as global phenomena under different cell conditions (Flavell et al., 2008; Ji et al., 2009; Mayr and Bartel, 2009; Sandberg et al., 2008) and different species (Sherstnev et al., 2012; Smibert et al., 2012; Ulitsky et al., 2012). They have also drawn much attention in cancer studies (Fu et al., 2011; Lembo et al., 2012; Lin et al., 2012; Mayr and Bartel, 2009).
In contrast with the increasingly recognized importance of APA, computational methods and tools for the APA analysis using RNA-Seq are underdeveloped. In unraveling APA regulation, Ji and colleagues scored relative expressions by taking the ratio of short reads density in extended and common regions, as defined by distal and proximal polyA sites, respectively (Ji et al., 2011). A higher score, therefore, indicated higher abundance of long 3′UTR isoform. A similar approach was taken in a recent tandem 3′UTR analysis, where the statistical significance was assessed by Fisher’s exact test for the switch score under different conditions (Wang et al., 2008). The same group further improved the approach and implemented a new computational tool, MISO (Katz et al., 2010). Specifically, tandem 3′UTR was treated as special alternative processing, and thus the quantification of expression level for each isoform can be estimated by computing Percent Spliced Isoform. These existing methods, however, have one critical drawback, namely, they rely on prior knowledge of annotated polyA sites. For example, MISO constructs 3′UTR isoform based on polyA sites information collected from the PolyA site database (Lee et al., 2007; Zhang et al., 2005). It is noted that the polyA sites from the current database are computationally inferred from cDNA (complementary DNA) / EST (expressed sequence tag) sequences. It is far from complete and may also contain false-positive findings. Therefore, these approaches that depend on polyA information may not be precise or powerful because of incomplete information of all potential cleavage sites on 3′UTR.
In this article, we propose using a change-point model for identifying 3′UTR switching. To our knowledge, this is the first available method that allows investigators to directly analyze 3′UTR length changes without being dependent on polyA site information. To determine whether a 3′UTR is shortening or lengthening to a certain extent, we further develop an additional testing procedure to make directional decisions. We show that this directional procedure can control the mixed directional FDR (mdFDR) at a pre-specified nominal level. Simulation studies in various settings and applications to two real NGS datasets have demonstrated that our proposed change-point model and the testing framework are powerful and accurate. The methods developed in this article have been implemented using Java in a computationally efficient and user-friendly software package available from http://utr.sourceforge.net/. This tool will allow investigators to analyze next-generation RNA sequencing data in an effective and efficient way.
The rest of the article is organized as follows. First, we introduce a change-point model based on a likelihood ratio test, followed by an iterative procedure for computing P-values. We then present a new directional testing procedure for identifying dramatic shortening or lengthening events while controlling mdFDR at a nominal level. We perform simulation studies to investigate numerical performance of the proposed method. Moreover, we apply the proposed method to analyze two real RNA sequencing datasets for identifying genes with length changes in their 3′UTRs. Finally, we conclude and discuss the results and methods.
2 METHODS
2.1 A change-point model for 3′UTR switching
The 3′UTR switching problem and the change-point model are illustrated via a toy example in Figure 1. We assume there are two 3′UTR isoforms, isoform 1 and isoform 2, ending with a distal and proximal polyA site, respectively. These two polyA sites define common and extended regions. We consider expression ratio of the two isoforms across two conditions, treatment and control, which can be quantified by the percentage of read counts from the treatment condition (Fig. 1c). We expect a constant ratio throughout the whole 3′UTR (pi = C, for i = 1, … ,T) if the isoform usages are identical under these two conditions. A ratio change at a certain position τ implies a ratio change between the two isoforms, which is the so-called 3′UTR switching. We wish to test the null hypothesis Ho that the ratio pi is constant against the alternative hypothesis that, for some point τ in the 3′UTR, the ratio changes from to ,
When the change-point location τ is known, e.g. based on isoform knowledge if it is available, detecting the change is straightforward. However, , and, most importantly, τ are unknown in our problem.
Fig. 1.
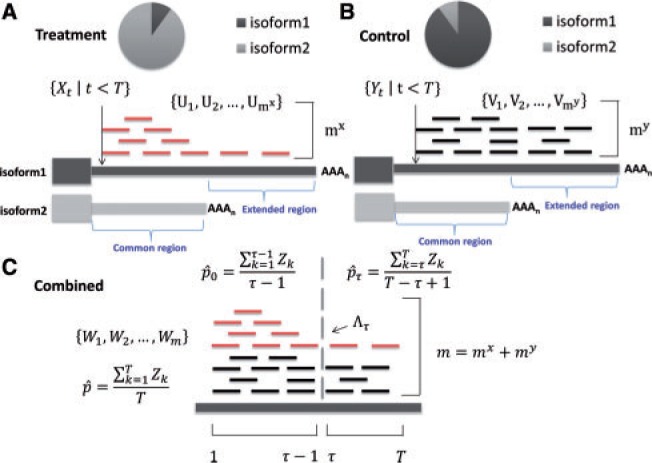
Illustration and notations of the change-point model for 3′UTR switching problem. (A) Treatment process; (B) Control process; (C) Combined process. Isoform 2 has a higher percentage expressed in the treatment condition, leading to a higher ratio of short reads density in common versus extended regions, as defined by the proximal and distal polyA sites, respectively
We start with a setup for the sequenced reads on 3′UTR with length T. Let be the number of reads whose first base maps to the left of base location t of a given 3′UTR under the treatment condition. Similarly, let be the number of such reads under the control condition. We denote and to be the total number of reads in the treatment and control conditions, respectively. Let and be the event locations for processes and , namely, U and V are the mapped positions of reads from the treatment and control samples. We let be the total number of reads combined from treatment and control samples, and then we obtain combined event locations . We define an indicator variable to denote whether an event is a realization of the treatment process or control process as follows:
For any short read i in the combined process, we use the term ‘success’ to refer to , that is, the read is from the treatment process. Hence, following Worsley (1983), we define a change-point model on the indices for read counts by the binomial log-likelihood function. Considering a candidate change point at τ, for , we have a generalized likelihood ratio statistic
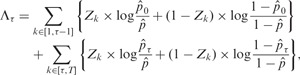 |
where , and are the maximum likelihood estimates of success probabilities:
This is an exact binomial generalized likelihood ratio statistic and can help to quantify the ratio change. Because the change-point location τ is unknown, we compute the statistic for all candidate loci τ = 2, … , T-1, and find the one yielding the maximal change. The solution is
2.2 A general iterative procedure for calculating P-value
We seek to compute the significance P-value for the maximum test statistic. To this end, following Worsley (1983), we use a general iterative procedure to calculate how likely the maximum likelihood ratio statistic L would be less than , denoted as , under the null hypothesis.
For the combined process in Section 2.1, let and be the total numbers of successes (from the treatment process) at intervals [1, k − 1] and [k, T], respectively, (k = 2, … ,T −1). The likelihood ratio test statistic depends only on and . Given that , and is fixed, depends only on .
Therefore, given and the test statistics, events of can be expressed as events of the form for suitable choices of and . For , we define so that the P-value can be derived as follows:
.
- by
where, for
A final iteration for k = T at v = S will produce = .
The desired probability will be .
The procedure is based on a dynamic programming approach. Its working logic is similar to the forward/backward algorithms used in hidden Markov models (Rabiner, 1989). Because the statistic depends only on Sk, as a naïve approach, we may enumerate all possible values of Sk for each position and consider all combinations S1S2 … ST, then pick the ones having and sum their likelihoods to obtain the final solution . Let D be the maximum number of possible values for Sk, k = 1,2 … ,T, then the computational complexity for this brute-force approach is O(DT), namely, exponential in terms of the problem size T.
In contrast, our algorithm solves this complex computation iteratively. Fk(v) represents the likelihood that no testing statistics can be found from position 1 to k, given Sk = v. So when k reaches the terminal point T and v = S, we can obtain the final solution . To compute Fk(v), we only need Fk−1(.), as illustrated in Supplementary Figure S1. Specifically, Fk(v) will assume that u successes are contributed by Fk−1(u) (namely, sampled from Mk − 1) and the remaining (v − u) successes come from mk, whose likelihood is then governed by the hypergeometric function hk(u,v). From the figure, we know that the computational complexity for one iteration, as determined by the number of edges, is O(D2). The total computational complexity is, therefore, O(TD2), namely, linear in terms of the problem size T.
2.3 A directional multiple testing procedure for identifying dramatic shortening or lengthening events
If the usage of the long isoform increases, we call it lengthening, and if it decreases, we call it shortening. Identifying shortening or lengthening events may be critical for downstream analyses, such as analyzing miRNA target sites. The significance we compute in the previous section is for a two-sided test. That is to say, when the null hypothesis is rejected, we can only state that there is a change, either lengthening () or shortening (). In practice, on rejecting the null H0, one may often conclude that the change is either lengthening or shortening based on the sign of (). There is a chance that this decision strategy will make a false statement about the sign, which is termed a directional error, or a type III error (Benjamini et al., 2005). It is desirable to control this error when making directional conclusions, which may not be negligible when a large number of tests are conducted simultaneously. In our applications, we often test for tens of thousands of genes at a time.
In the multiple-testing field, it is often argued that an exact null hypothesis is never true in reality; instead, more likely only significant differences matter (Benjamini et al., 2005; Williams et al., 1999). Here for our 3′UTR switching problem, small change may happen by chance and is irrelevant to the phenotype of interest. Dramatic change may be more robust and easier to replicate. Therefore, focusing on dramatic change is particularly meaningful, as we often have only one or few replicates in RNA-Seq experiments.
We propose to use the odds ratio (OR) at the estimated change-point to measure the change direction and magnitude, reasoning that the proposed method essentially chooses the location that gives the strongest association in a 2 × 2 contingency table among all possible locations. Thus, we perform Fisher’s exact test at the estimated change-point to make such directional decisions. We formulate this problem as controlling false discoveries within the multiple-testing framework. Using a similar definition as in Guo et al. (2010), we denote the mdFDR to be a combination of two parts. One is the false discovery rate (FDR), resulted from the change-point testing procedure. The other is the pure directional FDR (dFDR), derived from Fisher’s exact test,
where C is the number of falsely rejected true null hypotheses and R is the total number of rejected hypotheses among H1, … , Hm. F denotes the total number of false null hypotheses among H1, … , Hm that are correctly rejected while at least one directional error has been made when deciding on the signs of the components.
To control mdFDR, the expected proportion of Type I and directional errors among all the rejections, we propose a directional testing procedure as follows:
Apply the BH procedure (Benjamini and Hochberg, 1995) at level α to test whether there is a significant change among all the m hypotheses.
Let R denote the number of hypotheses rejected.
For every i = 1, … , R, perform one-sided Fisher’s exact test for testing OR > d (d ≥ 1).
If Fisher’s exact test has a P-value , then reject the null hypothesis.
It is shown that a similar BH procedure using the same two-sided P-value twice can control the mdFDR at level α (Benjamini et al., 2005). The directional testing procedure proposed here has its novel extension in comparison with the BH procedure in Benjamini et al. (2005). Specifically, the same significance P-values are reused in testing direction and controlling directional errors in (Benjamini et al., 2005); in contrast, our procedure uses an additional one-sided Fisher’s exact test for detecting dramatic change and the rejection is based on these new P-values. We show in our simulation studies that our new testing procedure can control mdFDR at the nominal level. It is noted that when d = 1, the one-sided test determines the direction of 3′UTR changes. The user may set d to be much larger than 1 to detect genes with more dramatic 3′UTR changes.
3 SIMULATION STUDIES
3.1 Power and FDR evaluation of the change-point model
We first present simulation results to demonstrate the performance of our change-point model. We assume there are two 3′UTR isoforms with different ending polyA sites as shown in Figure 1. The gene expression ratio before and after the change point (Fig. 1C) will critically influence how easily the change can be detected. So we generated the 3′UTR with different expression ratios under two conditions. Specifically, under condition 1, the entire 3′UTR has a constant expression level, whereas, under condition 2, the expression level was increased by K-fold in the common region and the extended region remained the same as in condition 1. Gene expression level was measured in reads per kilobase per million mapped reads (RPKM), Mortazavi et al., 2008). We simulated two constant expression levels RPKM = 1 and RPKM = 2 for condition 1. These two RPKM values are commonly used for determining expressed genes in RNA-Seq real data analyses (Ji et al., 2011; Zhang et al., 2013). We assumed that the total number of mapped reads was 100 million/sample and the 3′UTR length was 1000 bp. We considered three possible change points at 250, 500 and 750 bp of the 3′UTR. We varied the fold change K in the common region from 2 to 4 with increments of 0.5. The null distribution was simulated by setting K = 1 for estimating type I errors. We simulated 500 3′UTRs with change (K > 1) and 500 3′UTRs without change (K = 1) to estimate the power and FDR of our proposed method, respectively. We set FDR nominal level = 0.05. The simulation was repeated 50 times, and we reported the averaged power and FDR.
The simulation results are summarized in Figure 2. We see that FDR was controlled at the nominal level = 0.05 in all settings, suggesting that the proposed method is a valid testing procedure. Moreover, we find that the fold change, expression level and change-point position all influence 3′UTR switching detection. First, the power of our proposed method increases with the fold change from small to large. This is expected because the change is more likely to be detected when the signal becomes stronger. Second, the power increases when the gene expression level increases. Under the same fold change, the power of RPKM = 2 is always higher than that of RPKM = 1, suggesting that increasing the number of reads that are covered in the 3′UTR will also benefit change detection. Third, the position of the change point has an impact on the performance too. The change point in the middle yielded the highest power, compared with the change points close to the two ends.
Fig. 2.

Power and FDR evaluation of the change-point model at the nominal level FDR = 0.05. The FDR for the change-point model was controlled at the nominal level. Fold change, expression level and change-point position all have an influence on 3′UTR switching detection
3.2 Power and mdFDR evaluation of the proposed testing framework
We next evaluate the power and mdFDR for the proposed two-step testing framework. To simulate alternative hypotheses with mixed ORs, we used similar simulations as above but with the following modifications. For the 500 3′UTRs with fold change, we divided them into two groups with 250 each. The fold change for the first group is uniformly distributed from 1 to 3, and the second group is uniformly distributed from 3 to 5. We set d = 1 and d = 3 to test the changes with OR >1 and OR >3, respectively. We applied our proposed directional testing procedure at mdFDR level = 0.05.
As we can see from Figure 3, our proposed testing framework is able to control mdFDR at the nominal level = 0.05 for all the settings. Similarly, the power increases when the expression level doubles from RPKM = 1 to RPKM = 2, and the change point at the middle position is easier to detect than those closer to the two ends. It is noted that when the hypothesized OR is changed, the results change accordingly. For example, if we are interested in detecting the 3′UTRs with OR >3 by setting d = 3, the testing procedure then favors the second group of 3′UTRs with OR∼ and would not reject the 3′UTRs in the first group with OR∼. When we set d = 1 for testing the 3′UTR changes with OR >1, the 3′UTRs in the group with OR∼ are easier to detect because the signal is relatively stronger than that of d = 3. This explains the power difference between testing OR >1 and OR >3 as shown in Figure 3. In summary, it is easier to capture the switching events when the OR is higher, the expression level is higher or the change point is closer to the middle.
Fig. 3.

Power and mdFDR evaluation of the directional testing procedure at the nominal level mdFDR = 0.05. For all the settings, our proposed testing framework is capable of controlling mdFDR at the nominal level. It is easier to capture the 3′UTR switching events when the OR is higher, the expression level is higher or the change point is closer to the middle
4 REAL DATA APPLICATIONS
4.1 Application to regular RNA-Seq data
We first applied our proposed method to analyze regular RNA-Seq data that have been commonly produced to profile transcriptome changes. MYC is a notable transcriptional factor that has been frequently activated in many human cancers with profound cellular influence. Although MYC-binding sites and target genes have been documented extensively in the past decade, thanks to the widespread application of high-throughput technology, the role of MYC and MYC target genes in androgen-controlled breast cancer growth remains unclear. To elucidate MYC regulatory network in molecular apocrine breast cancers, Ni and colleagues used RNA-Seq to profile transcriptome changes before and after MYC knockdown by siRNA in MDA-MB-453 breast cancer cells with androgen stimulation (Ni et al., 2013). In summary, they transfected MDA-MB-453 breast cancer cells with control (siCtrl) or MYC siRNA (siMYC) for 48 h, followed by treatment with 10 nM DHT (DiHydroTestosterone, the most potent androgen) or vehicle (veh) for 6 h, resulting in three conditions: siCtrl-veh, siCtrl-DHT and siMYC-DHT. High-throughput 50 bp single-end sequencing was performed on Illumina HiSeq 2000 platform for each sample, generating total numbers of short reads ranging from ∼26 million to ∼39 million. Following the authors, we made two comparisons, siCtrl-DHT versus siCtrl-veh and siMYC-DHT versus siCtrl-DHT, but to detect 3′UTR shortening events instead of gene expression level changes.
We downloaded the dataset from NCBI (National Center for Biotechnology Information) Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under GSE45202. We aligned the raw reads to hg19 reference genomes using a conventional RNA-Seq aligner Tophat (Trapnell et al., 2009) v1.3.1 with default parameters. Coverage filter can help to reduce false-positive findings and is a heuristic strategy commonly used in existing RNA sequencing tools and analyses. Following MISO (Katz et al., 2010), we required that each 3′UTR should have at least 20 supporting reads in both samples, leading to 8052 and 7878 genes in the two comparisons, respectively, for further analysis. Our method was applied to detect shortening events with OR >2 at an mdFDR level of 0.05. We identified 947 shortening 3′UTRs in siCtrl-DHT versus siCtrl-veh and 1524 shortening 3′UTRs in siMYC-DHT versus siCtrl-DHT, respectively, with 461 genes in common. There are 1063 genes uniquely identified in the comparison of siMYC-DHT versus siCtrl-DHT but not in the comparison of siCtrl-DHT versus siCtrl-veh, which may be associated with MYC knockdown given the DHT treatment. We describe two examples of significant MYC-dependent shortening events, LDHA and OGDH, on the UCSC genome browser (Fig. 4), to demonstrate that our proposed method worked well in detecting such 3′UTR switching without relying on any polyA annotations. Because of space limitation, we only displayed the 3′UTR region despite that the actual reads spanned the whole gene body. We observed a highly non-uniform distribution of data in the 3′UTR, a common phenomenon in RNA-Seq data, which may be caused by polyA mRNA selection bias (Wang et al., 2009). We included the polyA track in the genome browser, which showed the annotated polyA sites (colored bars) from the PolyA_DB. We observed dramatic changes before and after the predicted change points. Clearly, the two genes LDHA and OGDH tend to use the proximal polyA site instead of the distal site in siMYC-DHT. These change points are also consistent and supported by the polyA sites annotated in the PolyA_DB. Together, these results suggest that our proposed method works well to detect 3′UTR switching without relying on any polyA annotations.
Fig. 4.

Examples of two MYC-dependent 3′UTR shortening events. The vertical lines indicate the estimated change points predicted by our proposed model. We observed dramatic changes before and after the predicted change points. Clearly, the two genes LDHA and OGDH tend to use the proximal polyA site instead of the distal site in siMYC-DHT. These change points are also consistent and supported by the polyA sites annotated in the PolyA_DB (colored bars in the PolyA_DB and Poly(A) tracks). Together, these results suggest that our proposed method works well to detect 3′UTR switching without relying on any polyA annotations
LDHA catalyzes the conversion of l-lactate and NAD to pyruvate and NADH in the final step of anaerobic glycolysis. It has been shown to be highly correlated with breast cancer growth (Wang et al., 2012). OGDH encodes one subunit of the 2-oxoglutarate dehydrogenase complex that catalyzes the overall conversion of 2-oxoglutarate (alpha-ketoglutarate) to succinyl-CoA and CO(2) during the Krebs cycle. It also plays an important role in breast cancer cells (Qattan et al., 2012). These shortening genes may be worthwhile for further biological study because the loss of distal region, if containing miRNA target sites, may help escape degradation destiny or translational repression.
We further conducted a gene set enrichment analysis (GSEA) of these 1063 MYC-dependent shortening genes using a hypergeometric test. The canonical pathways and Gene Ontology (GO) gene sets definitions were downloaded from the Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb/index.jsp). The results are summarized in Table 1 (Canonical pathways) and Supplementary Table S1 (GO gene sets). It has been suggested that MYC plays a crucial role in several aspects of cellular function, such as metabolism, growth, replication, differentiation and apoptosis (Ni et al., 2013). These pathway results suggest interesting transcription relevant functions of these 1063 MYC-dependent shortening genes, such as splicing, intron processing and transcript elongation. These genes are primarily associated with mRNA processing and gene expression, which are critical in cancer development (David and Manley, 2010; Sotiriou et al., 2006). The original studies (Ni et al., 2013) focused on conventional differential expression analysis. Capturing 3′UTR switching from the same RNA-Seq dataset using our proposed method would shed additional light on cancer transcriptome regulations and suggest new roles of MYC.
Table 1.
Significantly enriched canonical pathways in analysis of the breast cancer dataset of (Ni et al., 2013) at FDR = 0.05
| Canonical pathway | P-value |
|---|---|
| REACTOME_MRNA_SPLICING | 3.74E-05 |
| REACTOME_GENE_EXPRESSION | 4.99E-05 |
| REACTOME_PROCESSING_OF_CAPPED_ INTRONCONTAINING_PRE_MRNA | 7.74E-05 |
| BIOCARTA_PROTEASOME_PATHWAY | 1.06E-04 |
| REACTOME_FORMATION_AND_MATURATION_ OF_MRNA_TRANSCRIPT | 1.32E-04 |
| REACTOME_METABOLISM_OF_PROTEINS | 2.51E-04 |
| REACTOME_ELONGATION_AND_PROCESSING_ OF_CAPPED_TRANSCRIPTS | 2.55E-04 |
| KEGG_OXIDATIVE_PHOSPHORYLATION | 3.35E-04 |
| REACTOME_TRANSLATION | 6.15E-04 |
| KEGG_CARDIAC_MUSCLE_CONTRACTION | 7.68E-04 |
| REACTOME_INFLUENZA_LIFE_CYCLE | 9.18E-04 |
To compare with the existing methods that rely on polyA annotations for 3′UTR switching analysis, we also run MISO (Katz et al., 2010; version 0.4.1 with default parameters) to analyze this RNA-Seq dataset for identifying 3′UTR shortening events. We filtered tandem 3′UTR events following the MISO manual as follows: (i) at least one inclusion read, (ii) one exclusion read, such that (iii) the sum of inclusion and exclusion reads is at least 10 and (iv) the Δ Ψ is at least 0.2 and (v) the Bayes factor is at least 10, and (i)–(v) are true in one of the two samples. MISO did not output any tandem 3′UTR events, although it did report other alternative splicing events, such as skipped exons, intron retentions, etc. This shows that methods depending on polyA annotations may suffer from low power in 3′UTR switching analysis. The capability of our method for detecting 3′UTR switching will fill a void among current alternative splicing and processing analysis tools.
4.2 Application to special RNA-Seq data
We analyzed another breast cancer dataset (Fu et al., 2011) to highlight the flexibility of our proposed method to handle special RNA-sequencing data. To improve efficiency of capturing APA sites, Fu et al. (2011) developed a novel strategy to sequence only reads with poly(A) tails followed by a linear trend test method for analyzing APA site switching. Specifically, they modified oligo-d(T) tagged with sequencing primers after polymerase chain reaction (PCR) to sequence polyadenylated reads. They performed their SAPAS (Strategy of sequencing APA Sites) method to profile APA sites of human breast cancer lines and compared it with normal cell lines, generating in total ∼31 million short reads with 75 bp length from the Illumina platform. We downloaded the dataset from the NCBI sequence read archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi; accession number SRA023826). PolyA containing reads cannot be mapped to the genome directly. Therefore, we used Bowtie2 (Langmead and Salzberg, 2012) local model to align those polyA containing reads because this model does not require end-to-end mapping. We then applied our method to identify 3′UTR shortening events. The authors reported their results at FDR level = 0.01. To make a comparison, we reported shortening events at the same mdFDR level of 0.01 for OR >1. We identified 972 shortening events in the breast cancer cell line (MCF_7) in comparison with the control sample (MCF_10A). Their linear trend test method was conservative according to the authors and detected only 428 shortened 3′UTRs (Fu et al., 2011). We found that 85% of their shortening genes were also detected as shortening by our method. The larger numbers of 3′UTR shortening events we identified under the same significance level suggest the higher power of our method.
To demonstrate the accuracy of these findings, we first examined the four genes that were validated in their studies. All the four genes, DDX5, SEC61A1, HSBP1 and FAM134A, were detected to be shortening in MCF_7 by our method. The shortenings of these four genes were all experimentally confirmed (see the PCR results in the Supplementary Material of Fu et al., 2011). Moreover, visualization of the identified shortening events highlighted the accuracy of our prediction. Figure 5 shows two genes OAZ1 and SDC1 that were missed by the linear trend method but demonstrated clear shortening patterns. Both genes are known to be related with cancer (Kastl et al., 2010; Nikolova et al., 2009). Finally, we conducted the GSEA for these 972 shortening genes. The results are summarized in Table 2 (canonical pathways) and Supplementary Table S2 (GO gene sets). Interestingly, we also found the mRNA splicing pathway to be the most significantly enriched in this breast cancer dataset, as in the first breast cancer dataset we analyzed in the previous section. In particular, these genes are related to spliceosome, a large ribonucleoprotein complex that guides pre-mRNA splicing in eukaryotic cells. Recent studies have demonstrated the contribution of spliceosome as a core component in oncology (Quidville et al., 2013) and its role in determining 3′UTR length (Berg et al., 2012). Taken together, these results indicate the accuracy of our proposed method in capturing 3′UTR switching. Overall, this real data application highlights the flexibility of our method for analyzing NGS data that are specially generated to sequence and capture polyadenlyation cleavage sites.
Fig. 5.

Examples of two shortening events that were identified by our method but missed by the linear trend test. The vertical lines indicate the change points predicted by our proposed model. We observed a clear change before and after the predicted change points, suggesting that our proposed method work well to detect 3′UTR switching without relying on any polyA annotation information. The two genes OAZ1 and SDC1 tend to use the shorter isoform in the MCF_7 cancer cell line in comparison with the control sample MCF_10A, and demonstrated clear shortening patterns
Table 2.
Significantly enriched canonical pathways in analysis of the breast cancer dataset of (Fu et al., 2011) at FDR = 0.05
| Canonical pathway | P-value |
|---|---|
| REACTOME_MRNA_SPLICING | 3.83E-05 |
| REACTOME_ELONGATION_AND_PROCESSING_ OF_CAPPED_TRANSCRIPTS | 1.78E-04 |
| KEGG_CELL_ADHESION_MOLECULES_CAMS | 1.88E-04 |
| KEGG_SPLICEOSOME | 2.11E-04 |
| REACTOME_DIABETES_PATHWAYS | 2.25E-04 |
| BIOCARTA_EIF_PATHWAY | 4.07E-04 |
| REACTOME_PROCESSING_OF_CAPPED_INTRON_ CONTAINING_PRE_MRNA | 5.36E-04 |
| REACTOME_FURTHER_PLATELET_RELEASATE | 7.13E-04 |
5 CONCLUSION AND DISCUSSION
We propose a change-point model based on a generalized likelihood ratio statistic for identifying 3′UTR length change in the analysis of next-generation RNA sequencing data. We develop a directional multiple testing procedure for identifying dramatic shortening or lengthening events. The numerical performances of our approach are investigated using both simulated and real data. The results show that our proposed method is powerful, accurate and flexible for analyzing various types of next-generation RNA sequencing data.
Some experimental approaches may measure polyA sites explicitly, e.g. the one for the breast cancer dataset of (Fu et al., 2011) we analyzed. They can provide results serving directly to identify polyA sites. It is noted that detecting polyA sites and 3′UTR switching are two different problems, although related. Essentially, 3′UTR switching implies polyA site usage change. It will benefit from the discovery of polyA sites but does not require this information directly. As a simple strategy, one may first use an algorithm to identify polyA sites and then apply a simple approach such as Fisher's exact test to detect changes. However, the polyA site discovery step may not be trivial because of the heterogeneity of the cleavage sites at polyA sites and/or low coverage at some locations. For example, the authors of the Fu et al. (2011) dataset performed a modified snowball-like clustering (Tian et al., 2005) and then empirically took the cleavage clusters with more than one read as polyA sites. It may be complex and non-trivial to optimize such a two-step approach. In contrast, it is an advantage of our method over two-step strategies by providing a one-stop integrated solution.
The dynamic programming strategy has greatly reduced the computational complexity of our algorithm from exponential O(DT) to linear O(TD2). In practice, the typical 3′UTR size T is ∼1000–2000 bp. We observed that most legitimate regions [ak, bk] for Sk have size <150 for the conventional RNA-Seq data from Ni et al. (2013) we analyzed. We ran our program on a computing node with 800 MHz CPU and 4 GB memory. It took only ∼6 h to analyze the whole dataset of (Ni et al., 2013) for the two comparisons at the speed of a few seconds/gene. Therefore, our program is so efficient that it does not require high-performance computing facility and can run feasibly on most computers.
Many genes may have more than two polyA sites. Given more than two polyA sites, the alternative model (H1) in our method assuming two polyA sites will fit less well but still better than the null model H0 assuming one polyA site. The null remains the same. Thus, our testing procedure remains valid even though it may not be optimal under some circumstance. In other words, the actual FDR may be lower than the nominal FDR cutoff when more polyA sites are involved. Empirically, the user may use a more lenient cutoff if he feels our procedure is (over) conservative. Our method may not always be optimal when the signal is subtle. For instance, when there are multiple change points that are all subtle, a more powerful test can be designed by considering all change points. However, we do not expect many such instances. Moreover, if they are interested, the users may manually inspect further those identified genes to see whether there is more than one change point and where they are.
It is noted that the expression of 3′UTR and its regulation can be complex. For example, there are transcripts that are solely composed of 3′UTR (Carninci et al., 2006; Mercer et al., 2011). Although the development of our tool is motivated for the need to detect 3′UTR switching, it may also identify changes caused by such transcripts. Depending on applications, caution thus should be used with regard to the biology and the interpretation of the identified change points.
The proposed method can be improved in several ways. First, one limitation is that the current method cannot handle sample replicates. We may extend it by computing joint likelihood over multiple samples, assuming the same change point across samples but allowing , and to vary for different sample comparisons. Second, we assume there are only two isoforms with one change point. We may extend it for multiple isoforms with K > 1 change points. In principle, we may search similarly for the K points that yield the most significance with computational complexity of O(LK), where L is the whole UTR length. We may further assume K is unknown and determine it using model selection (Shen and Zhang, 2012). Third, statistical inference of confidence estimates is as important as point estimates. For example, the confidence intervals on the estimated change points could provide more information as needed for some downstream analyses, such as determining the loss/gain of miRNA target sites. This can be obtained based on the values accepted by a level α of likelihood ratio test (Worsley, 1986). Fourth, in addition to 3′UTR switching analysis, our proposed method can also be extended to other applications. For example, one can merge together the multiple 3′UTRs of a gene, if any, to perform alternative last exon analysis. Moreover, if input vector is the coverage of entire exon regions of a gene, our proposed method can also detect premature cleavage and polyA events, another set of interesting biological phenomena that has received much attention recently (Kaida et al., 2010). We are working on these extensions.
To our knowledge, the proposed approach is the first one to allow the analysis of 3′UTR switching without relying on any polyA annotations, one major limitation of existing methods. The closest existing approximate solution may be those requiring polyA annotation information, e.g. MISO compared in our analysis. These existing tools also have the same limitations as our method, such as not capable of handling sample replicates, not supporting multiple isoforms and no confidence interval estimates for the change point. These limitations warrant development of new bioinformatics methods.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Lili Wan for helping to review and improve the description of the biology. The authors thank Dr John Daye for proofreading and editorial assistance. The authors also thank all three referees for their constructive comments, which have greatly helped to improve the presentation of the article.
Funding: National Institutes of Health (grant number GM097505).
Conflict of Interest: none declared.
REFERENCES
- Anders S, et al. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Benjamini Y, et al. False discovery rate: adjusted multiple confidence intervals for selected parameters. J. Am. Stat. Assoc. 2005;100:71–93. [Google Scholar]
- Berg MG, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Colgan DF, Manley JL. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, et al. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011;21:741–747. doi: 10.1101/gr.115295.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, et al. Alternative expression analysis by RNA sequencing. Nat. Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- Guo W, et al. Controlling false discoveries in multidimensional directional decisions, with applications to gene expression data on ordered categories. Biometrics. 2010;66:485–492. doi: 10.1111/j.1541-0420.2009.01292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, et al. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl Acad. Sci. USA. 2009;106:7028–7033. doi: 10.1073/pnas.0900028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, et al. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol. Syst. Biol. 2011;7:534. doi: 10.1038/msb.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastl L, et al. Effects of decitabine on the expression of selected endogenous control genes in human breast cancer cells. Mol. Cell. Probes. 2010;24:87–92. doi: 10.1016/j.mcp.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Katz Y, et al. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, et al. Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 2010;11:345–355. doi: 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, et al. PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Res. 2007;35:D165–D168. doi: 10.1093/nar/gkl870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo A, et al. Shortening of 3′UTRs correlates with poor prognosis in breast and lung cancer. PLoS One. 2012;7:e31129. doi: 10.1371/journal.pone.0031129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012;40:8460–71. doi: 10.1093/nar/gks637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell. 2009;136:719–730. doi: 10.1016/j.cell.2009.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, et al. Expression of distinct RNAs from 3′ untranslated regions. Nucleic Acids Res. 2011;39:2393–2403. doi: 10.1093/nar/gkq1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Ni M, et al. Amplitude modulation of androgen signaling by c-MYC. Genes Dev. 2013;27:734–748. doi: 10.1101/gad.209569.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova V, et al. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis. 2009;30:397–407. doi: 10.1093/carcin/bgp001. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes Dev. 2011;25:1770–1782. doi: 10.1101/gad.17268411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qattan AT, et al. Spatial distribution of cellular function: the partitioning of proteins between mitochondria and the nucleus in MCF7 breast cancer cells. J. Proteome Res. 2012;11:6080–6101. doi: 10.1021/pr300736v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quidville V, et al. Targeting the deregulated spliceosome core machinery in cancer cells triggers mTOR blockade and autophagy. Cancer Res. 2013;73:2247–2258. doi: 10.1158/0008-5472.CAN-12-2501. [DOI] [PubMed] [Google Scholar]
- Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE. 1989;77:257–286. [Google Scholar]
- Rogers MF, et al. SpliceGrapher: detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol. 2012;13:R4. doi: 10.1186/gb-2012-13-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, et al. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JJ, Zhang NR. Change-point model on nonhomogeneous Poisson processes with application in copy number profiling by next-generation DNA sequencing. Ann. Appl. Stat. 2012;6:429–830. [Google Scholar]
- Shen S, et al. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherstnev A, et al. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 2012;19:845–852. doi: 10.1038/nsmb.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smibert P, et al. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep. 2012;1:277–289. doi: 10.1016/j.celrep.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J. Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- Tian B, et al. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, et al. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012;22:2054–2066. doi: 10.1101/gr.139733.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZY, et al. LDH-A silencing suppresses breast cancer tumorigenicity through induction of oxidative stress mediated mitochondrial pathway apoptosis. Breast Cancer Res. Treat. 2012;131:791–800. doi: 10.1007/s10549-011-1466-6. [DOI] [PubMed] [Google Scholar]
- Williams VS, et al. Controlling error in multiple comparisons, with examples from state-to-state differences in educational achievement. J. Educ. Behav. Stat. 1999;24:42–69. [Google Scholar]
- Worsley KJ. The power of likelihood ratio and cumulative sum tests for a change in a binomial probability. Biometrika. 1983;70:455–464. [Google Scholar]
- Worsley KJ. Confidence regions and tests for a change-point in a sequence of exponential family random variables. Biometrika. 1986;73:91–104. [Google Scholar]
- Zhang H, et al. PolyA_DB: a database for mammalian mRNA polyadenylation. Nucleic Acids Res. 2005;33:D116–D120. doi: 10.1093/nar/gki055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, et al. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl Acad. Sci. USA. 2013;110:19348–19353. doi: 10.1073/pnas.1319280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.