

Abstract
Motivation: Family-based designs are regaining popularity for genomic sequencing studies because they provide a way to test cosegregation with disease of variants that are too rare in the population to be tested individually in a conventional case–control study.
Results: Where only a few affected subjects per family are sequenced, the probability that any variant would be shared by all affected relatives—given it occurred in any one family member—provides evidence against the null hypothesis of a complete absence of linkage and association. A P-value can be obtained as the sum of the probabilities of sharing events as (or more) extreme in one or more families. We generalize an existing closed-form expression for exact sharing probabilities to more than two relatives per family. When pedigree founders are related, we show that an approximation of sharing probabilities based on empirical estimates of kinship among founders obtained from genome-wide marker data is accurate for low levels of kinship. We also propose a more generally applicable approach based on Monte Carlo simulations. We applied this method to a study of 55 multiplex families with apparent non-syndromic forms of oral clefts from four distinct populations, with whole exome sequences available for two or three affected members per family. The rare single nucleotide variant rs149253049 in ADAMTS9 shared by affected relatives in three Indian families achieved significance after correcting for multiple comparisons ().
Availability and implementation: Source code and binaries of the R package RVsharing are freely available for download at http://cran.r-project.org/web/packages/RVsharing/index.html.
Contact: alexandre.bureau@msp.ulaval.ca or ingo@jhu.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The advent of high-throughput sequencing of whole exomes and even whole genomes opens the possibility of detecting rare variants (RVs, including those unique to a family, and alleles up to a frequency of 1% in a population) impacting human health. The first successful applications of exome sequencing have been with rare Mendelian traits (Gilissen et al., 2012). A common study design to discover highly penetrant causal variants that are rare in families where previous genotyping has not been performed is to sequence the exome (or increasingly, the whole genome) of two or three affected subjects, and focus on novel variants predicted to be functional and shared by all sequenced family members as likely causal variants (Gilissen et al., 2012).
Contrary to monogenic Mendelian traits, considerable genetic heterogeneity must be expected with complex diseases. Familial forms of numerous common complex diseases are caused by RVs, supporting the hypothesis that RVs may explain a part of the so-called ‘missing heritability’ of these diseases, although the extent of the contribution of RVs to complex disease heritability is an ongoing debate (Gibson, 2012). In a family where cases cluster, there is a high probability that multiple affected members carry the same rare disease predisposing variant if such a variant exists and its penetrance is high (Cirulli and Goldstein, 2010; Wijsman, 2012). This gives an advantage to family samples over the samples of unrelated individuals, where disease-causing RVs may be seen only once or twice among tens of thousands of subjects.
As with Mendelian disorders, it has initially been proposed to use the RV sharing information to filter out RVs not shared in at least one family (Feng et al., 2011). For variants sufficiently rare so copies in the sequenced relatives are almost certainly identical by descent (IBD), the probability that an RV independent of the disease and detected in at least one sequenced subject would not be shared by other sequenced relatives who are affected was computed by Feng et al. (2011) to quantify the effectiveness of what they call the ‘concordance filter’ in discarding irrelevant RVs. We adopt the view that the probability that an RV would be shared by all affected relatives in a family—given it occurs in any one of them—computed disregarding the disease phenotype can be used to quantify evidence against the null hypothesis of absence of linkage and association to the disease and, therefore, establish that the RV may predispose to disease. We emphasize that RV sharing probabilities are not the same as IBD sharing probabilities. A parent–offspring pair carries DNA from three distinct chromosomes at any locus: one the parent and offspring share IBD, one in the parent and one in the offspring. The probability they share one allele IBD is hence equal to 1. However, a RV seen in one of the two relatives is present on only one of the three chromosomes. The probability that this chromosome is the one shared IBD is . So, parent–offspring pairs are informative for RV sharing analysis, whereas they are uninformative for IBD sharing-based linkage analysis. For more distant relationships, RV sharing probabilities remain smaller than IBD sharing probabilities as explained in Section 4. This information can be combined across all the families where the RV is seen, if more than one occurs. Mathematical expressions generalizing sharing probabilities to more than two relatives per family are given in the Section 2. This approach for calculating sharing probabilities does not require knowledge of the actual variant allele frequency in the population, and only assumes there is no identity by state (IBS) without IBD among sequenced family members. We illustrate that these calculated sharing probabilities are good approximations of the true IBS sharing probabilities for allele frequencies up to ∼1%, and also explore the power of the test based on these probabilities to detect disease susceptibility RVs using relative pairs under genetic heterogeneity models.
In addition to variant rarity, the known pedigree must be correct (in particular, all founders are unrelated) to insure a variant is introduced only once in the family. Cryptic relatedness can often be detected from dense marker genotype data. When founders of a known pedigree are related, an RV may be introduced more than once, leading to greater actual sharing probabilities than the value computed based on the known pedigree, and to an overstatement of the evidence against the null. We examine the impact of unknown relationships and propose to approximate the sharing probability using kinship coefficients among founders, estimated empirically from genome-wide marker data on family members. The validity of the approximation is evaluated in a simulation of small populations.
We then apply the RV sharing probability computation to a whole exome sequencing study of 55 multiplex families with apparent non-syndromic forms of oral clefts from four distinct populations. Oral clefts are the most common craniofacial malformations, representing a good example of a genetically heterogeneous disorder with at least a dozen different genes previously identified as genetic risk factors via genome-wide association studies (Beaty et al., 2013; Ludwig et al., 2012).
2 METHODS
Our goal is to compute the probability that a set of related subjects suffering from the same disease, whose DNA sequence has been obtained through sequencing (sequenced subjects), share an RV given that an RV has been observed at a site in the sequence in one of them, under the null hypothesis of no linkage and no association to any observed or unobserved disease susceptibility variant for the disease. In the basic setting, all founders are unrelated and we assume the variant is rare enough that a single copy exists among all the alleles present among the nf founders of the pedigree linking the sequenced subjects. In a generalization, we allow founders to be related, and allow for up to two copies of the RV to be introduced into the pedigree by related founders. We finally demonstrate how RV sharing probabilities computed in a single family can be combined across multiple families where the same variant is seen, and how to derive the P-value for the hypothesis test.
2.1 Rare variant sharing probability assuming unrelated founders
We define the following random variables:
Number of copies of the RV received by sequenced subject i,
Indicator variable that founder j introduced one copy of the RV into the pedigree,
Number of generations (meioses) between subject i and his or her ancestor j.
For a set of n sequenced subjects, we want to compute the probability
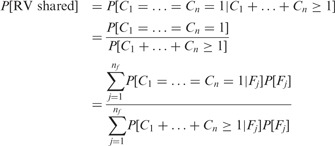 |
where the expression on the third line results from our assumption of a single copy of that RV among all alleles present in the nf founders. The probabilities cancel from the numerator and denominator. For the other terms, we first derive expressions for the special case where all the sequenced subjects descend from every founder among their ancestors through independent lines of descent. In that case,
and
where and d(j) is the subset of sequenced subjects who descend from founder j. The global expression is then
We note that this equation covers pedigrees with individuals marrying multiple times and marriage loops as in the family depicted in Figure 1B, provided all lines of descent are independent and there is no inbreeding. This is a generalization of the RV sharing probability for two relatives, (Feng et al., 2011) where D is the degree of relationship defined such that the expected proportion of alleles shared IBD equals . In the common situation where a single couple of founders is common to all sequenced subjects of a pedigree, the numerator simplifies and we obtain the following expression:
where f is any of the two founders forming the ancestral couple.
Fig. 1.
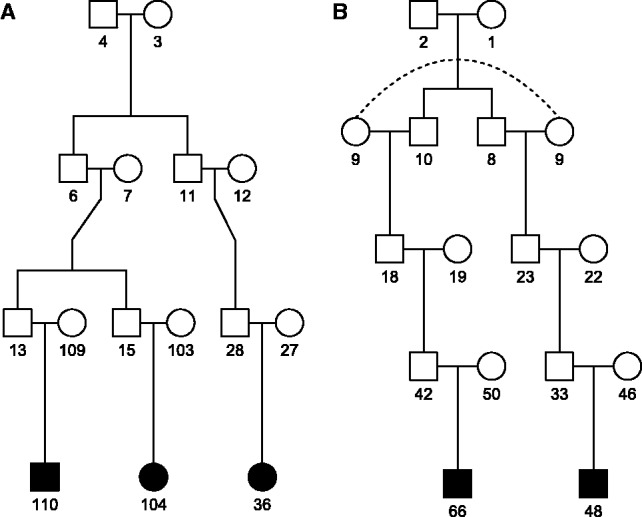
Syrian families sharing the T allele at rs117883393. Filled symbols represent affected members who have been sequenced. (A) Family with an affected first cousin pair and an affected second cousin. (B) Family with marriage loop
When the lineages of sequenced individuals ‘coalesce’ at a branching individual k who descended from founders of the pedigree (such as subject 6 in Fig. 1A), recursive computations described in the Supplementary Section A, and implemented in the RVsharing R package, are required.
The above approach for calculating sharing probabilities does not require knowledge of the actual variant frequency in the population, a distinct advantage when only few subjects are sequenced, possibly all of whom are affected. However, the variant has to be sufficiently rare so the probability of finding two copies IBS but not IBD among sequenced family members is negligible. For a fixed variant frequency, and assuming unrelated founders and Hardy–Weinberg equilibrium, exact IBS sharing probabilities for pedigree members can simply be derived using conditional probabilities under Mendel’s laws. We compare these true sharing probabilities with the sharing probabilities calculated under the ‘no IBS without IBD’ assumption for variant frequencies ranging from 0.001 to 0.05 in a variety of pedigrees, to assess when the above sharing probabilities are good approximations of the truth (see Section 3.1).
2.2 Computation allowing for relatedness among founders
We generalize our computation to the setting where founders are related, while still excluding the possibility the founders are themselves inbred (only their children will be). When the relatedness between specific pairs of founders is completely unknown and marker genotype data reveal IBD sharing in excess of the expectation based on the pedigree structure, we propose to approximate RV sharing probabilities between sequenced subjects using either a numerical approximation or Monte Carlo simulation, and show that either of the approaches gives a good approximation.
We assume all founders are related to the same extent, i.e. the kinship coefficient between all pairs of founders j and k is a (low, but positive) constant denoted , with the superscript f for ‘founders’. This assumption is required because there is a considerable variation in estimated kinship coefficients for pairs of subjects with the same degree of relatedness, even with perfect information on IBD sharing between subjects, because of variation in the length of genome shared from pair to pair (Manichaikul et al., 2010). Reliable inferences can thus only be obtained for the mean or another central tendency parameter. We also assume at most two founders introduced a copy of the RV considered in the computation.
Two situations can occur with respect to the marker genotype data available to estimate kinship among founders:
1. Polymorphic markers have been genotyped on the pedigree founders, typically from a genome-wide single nucleotide polymorphism (SNP) array. Then can be estimated for each founder pair j and k, and a global estimate obtained by averaging the over all founder pairs from the same population.
2. Genotype data are only available on the sequenced subjects (either from the sequencing data itself or from other genotyping). The common is estimated based on estimated kinship coefficients between sequenced subjects and the degrees of relationship between the sequenced subjects and all founders as described in the Supplementary Section B.
The numerical approximation consists in obtaining the probability every founder pair introduces the RV, and the probability every founder alone introduces the RV. Assuming only one or two founders introduce the RV, , and we only need to obtain PU. We obtain an approximation of PU from by expressing PU as an expectation over the distribution of the number A of alleles distinct by descent among founders. We show in the Supplementary Section C that
| (1) |
Among the a distinct alleles present, we then assume neither is present more than twice; of them are present twice; and the remaining are present only once. We further assume A only takes the values with positive probability, where d is a tuning parameter representing the maximum number of alleles present twice. We parameterize the probabilities to be proportional to
| (2) |
inspired from a truncated Poisson distribution. In the Supplementary Section C we explain in detail how θ is obtained from . We finally obtain the approximate RV sharing probability using the estimated value of PU:
| (3) |
where . The sharing probabilities conditional on the introduction of the RV by two of the founders and are computed exactly using the formulas in the Supplementary Section D.
When Monte Carlo sampling is used to approximate the RV sharing probability, we repeat the following steps for a large number R of replicates. We first sample an indicator variable of whether one or two copies of the RV were introduced into the family, with probability w and , respectively. In practice this is done by sampling the number of distinct alleles a from distribution (2), then sampling the RV among the a alleles. The RV is introduced twice if it is one of the first alleles, and introduced once otherwise. If it is introduced twice, the pair of founders introducing the RV is sampled with equal probability for all pairs. If it is introduced once, the sole founder introducing it is sampled instead. Then the transmission of the RV down the pedigree from the one or two founders introducing it is simulated according to Mendel’s laws. The events that the variant was observed in any of the sequenced subjects () and in all of them () are recorded. The proportions of these two events computed over R replicates estimate the numerator and denominator of Equation (3), respectively. The Monte Carlo approach is implemented in our R package RVsharing.
Monte Carlo simulation of transmission of an RV is also straightforward in pedigrees containing inbreeding loops. The simulation can be performed assuming pedigree founders are unrelated by forcing the introduction of only one copy of the RV (i.e. w = 1) or allowing for relatedness among founders as described earlier. Assuming unrelated founders, a method providing exact sharing probabilities with a single inbreeding loop and an approximation with two or more inbreeding loops is also presented in the Supplementary Section E.
2.3 Combining RV sharing probabilities across multiple families
For variants seen in only one family, the RV sharing probability can be interpreted directly as a P-value from a Bernoulli trial. For variants seen in M families and shared by affected relatives in a subset So of them, the P-value can be obtained as the sum of the probability of events as (or more) extreme as the observed sharing in the family subset So. If we denote pm as the sharing probability between the subjects in family m, the P-value is
where V is the subset of family sets Sv such that
2.4 Defining the set of rare variants tested
The lowest possible P-value for an RV seen in one or more families depends on family structure. The sharing probabilities between sequenced subjects in small or densely inbred families are high, and so is the potential P-value of an RV seen only in one such family (for instance, it is for an avuncular pair). We propose to test the null hypothesis of absence of linkage and association only among those variants with frequency less than a prespecified threshold (typically 1%) that could achieve a P-value below the level controlling the family-wise error rate, if shared by all affected subjects in the family (or families) in which they occur (i.e. potential P-value). These potential P-values are independent of the actual sharing pattern among affected subjects and, therefore, of the subsequent testing of RV sharing. We obtain this subset of RVs by ordering the potential P-values of RVs in decreasing order, stopping at the last potential P-value lower than the family-wise Type I error level α divided by the rank t of the P-value, yielding a threshold of .
3 RESULTS
3.1 Validation of the assumption that RVs are IBD
When all founders are unrelated, the assumption that only a single copy of the variant exists among the founders’ alleles (no IBS without IBD) provides good approximations of the true IBS sharing probabilities for a variety of relationship types between pairs and triplets of sequenced subjects (Fig. 2). The deviation from the actual IBS sharing probability remains <20% up to a frequency in the population slightly >1% for first cousin pairs and triples, and a frequency of about 0.5% for second cousin pairs and triples.
Fig. 2.
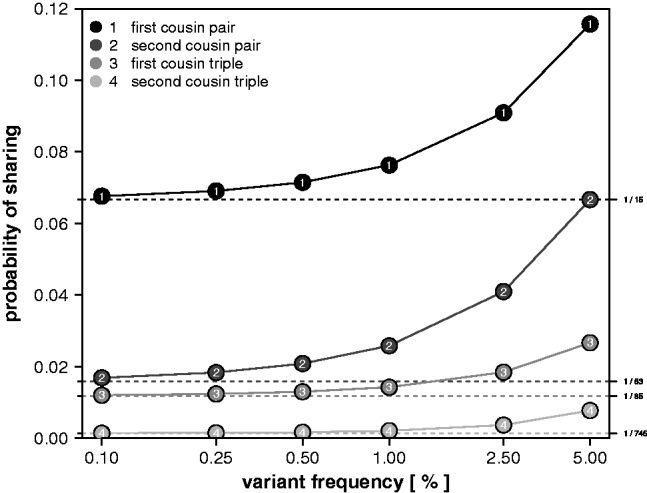
The exact sharing probabilities (Y-axis) as a function of variant allele frequency (x-axis), for a pair of first cousins (1), a pair of second cousins (2), three first cousins (3) and three second cousins (4). The sharing probabilities calculated under the assumption of ‘no IBS without IBD’ are 1/15, 1/63, 1/85 and 1/745, respectively, and indicated by the dashed horizontal lines
3.2 Validation of the approximation of the sharing probabilities with related founders
We simulated small populations as described in the Supplementary Section F from which we sampled founders of a pedigree to validate the quality of the approximation of sharing probabilities in presence of relatedness among founders. We used the pedigree for three second cousins shown in Supplementary Figure S1, with an RV sharing probability of when the founders are unrelated, as shown in Figure 2. This family structure was encountered in our oral cleft sample and was chosen for its three sequenced subjects with symmetric relationships. The simulation was repeated 100 times for each population size. Supplementary Table S1 shows the mean and SD of the mean kinship coefficient between pairs of subjects from the generation of the founders and of the mean number of copies of an RV in the eight subjects sampled to be the founders of the pedigree. With a population of 100 founders, the probability that the RV is introduced by more than two founders (given that it was seen in at least one founder) is too high to obtain a good approximation of the RV sharing probability when assuming the RV can only be introduced once or twice. The approximation was therefore computed only with 200 and 400 founders.
The first step in applying the approximation method was to estimate the parameter θ of the distribution of the number of distinct alleles among all founders. We used two different values for : the mean kinship coefficient among the eight sampled subjects and the mean kinship coefficient in the population. The former is a best case scenario, where a pedigree-specific is estimated without error, which is not possible in practice, whereas the latter can be approached with a sufficiently large sample from the population. Supplementary Figure S2 illustrates the approximations of the number of distinct alleles among founders are good, although the unlikely events of observing only 8–10 distinct alleles among the pedigree founders from the 200 founder population were not captured by the approximate distributions.
We approximated the RV sharing probability using formulas (1) and (3), plus those in the Supplementary Section D, and also alternatively by Monte Carlo, sampling 100 000 realizations of RV transmission down the pedigree of Supplementary Figure S1 in each replicate. The approximation of the probability that two founders introduced an RV was on average slightly lower than the value in the simulated populations, in particular when the number of founders was low (Supplementary Table S1). To evaluate the actual quality of the RV sharing approximation, we estimated the root mean squared error (RMSE) and bias over the simulation replicates, and observed that the RV sharing approximation was accurate and precise unless the number of population founders was low (Table 1). The RV sharing probability approximation was accurate for the population with 400 founders (negligible bias), where the mean kinship coefficient was approximately equal to second cousins once removed (). The RV sharing probability was on average underestimated (negative bias) for the population with 200 founders, where the mean kinship is between first cousins once removed () and second cousins (), suggesting limits for approximation methods restricted to two founders introducing an RV. The loss of precision and accuracy from using the population average instead of the average of the sampled subjects was smaller in the population with 400 founders than in the population with 200 founders, both in absolute and relative terms. Sampling 100 000 realizations of RV transmission in the Monte Carlo simulation was sufficient to achieve the same level of error as the numerical approximation with a relatively larger RV sharing probability in populations of 200 founders, but the Monte Carlo error remained slightly higher when approximating the smaller RV sharing probability in populations with 400 founders.
Table 1.
Approximation of rare variant sharing probabilities for three second cousins in small populations
| Number of founders | 200 |
400 |
|||
|---|---|---|---|---|---|
| Sample a | Population | Sample | Population | ||
| Analytical approximation | |||||
| RMSEb | Absolute | 0.0015 | 0.0026 | 0.0006 | 0.0007 |
| Relative | 0.27 | 0.34 | 0.24 | 0.28 | |
| Biasc | Absolute | −0.0009 | −0.0012 | −0.0002 | −0.0002 |
| Relative | −0.18 | −0.18 | −0.02 | 0.01 | |
| Monte Carlo approximation | |||||
| RMSE | Absolute | 0.0015 | 0.0026 | 0.0006 | 0.0008 |
| Relative | 0.27 | 0.35 | 0.26 | 0.32 | |
| Bias | Absolute | −0.0009 | −0.0012 | −0.0002 | −0.0001 |
| Relative | −0.17 | −0.17 | −0.01 | 0.02 | |
aMean kinship coefficient among founders from the sample or in the population. bRMSE: Root mean square error. If we denote the RV sharing probability at the rth replicate by and its approximation by , then the absolute RMSE is equal to , and the relative RMSE is equal to where . cThe absolute bias is equal to , and the relative bias is equal to with .
3.3 Power study
We assessed the probability of rejecting the null hypothesis when testing a causal RV (statistical power) using the proposed approach with parent–offspring, as well as first and second cousin pairs. We first determined with 4 and 10 pairs where the RV occurs, the number of pairs sharing an RV needed to reject the null at significance level , the level in our exome sequencing study of oral clefts. We then computed the power as the probability of equaling or exceeding these numbers of sharing pairs under alternative hypotheses where the probability of RV sharing given the event the two relatives are affected was computed assuming an RV had an effect on disease risk. For a dominant RV, the relationship between P1 and is well approximated by the expression where r is the marginal relative risk of the RV, for the classes of multilocus additive and genetic heterogeneity models [Risch (1990); the expression is exact when the RV is the single genetic cause of disease]. Under these model classes, the marginal relative risk of an RV is approximately the ratio of penetrance over disease prevalence. Figure 3 shows power as a function of penetrance and marginal relative risk of the RV for a genetic heterogeneity model detailed in the legend. For a fixed marginal relative risk, the power is roughly constant with respect to disease prevalence (results not shown). The penetrance required to keep the marginal relative risk fixed, however, varies and is obviously bounded above by one.
Fig. 3.
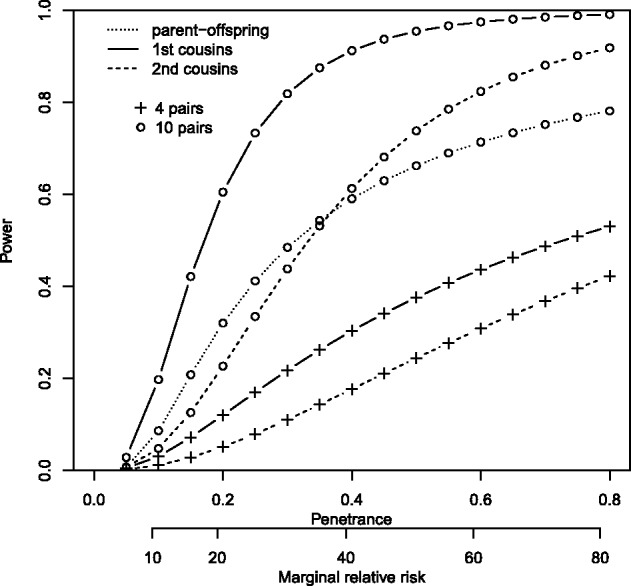
Power of the test based on sharing probabilities. Power was computed for a dominant RV with frequency of . The disease had a population prevalence of 1% and a recurrence risk to offspring of 5, to first cousins of 2 and to second cousins of 1.25. The significance level of was achieved when 10/10 parent–offspring pairs, 4/4 or 6/10 first cousins pairs and 3/4 or 4/10 second cousin pairs shared the RV (4 parent–offspring pairs are insufficient to achieve the significance level)
3.4 Whole exome sequencing study of non-syndromic oral clefts
We computed the sharing probability for all rare single nucleotide variants (SNVs) detected in exons and splice junctions in a whole exome sequencing study of affected relative pairs and triples drawn from 55 multiplex non-syndromic oral cleft families from diverse sites (Germany, Philippines, India, the Syrian Arab Republic, plus two Chinese families and one European–American family). One family was excluded from the present analysis because there was no known ancestor common to the three sequenced subjects, resulting in a sharing probability of 0 based on the pedigree structure. The sample for this analysis was composed of 51 families providing two affected subjects and 3 families providing three affected subjects for a total of 111 sequenced subjects. There were 60 038 exonic and splice site SNVs with frequency in the autosomal genome of these 111 sequenced subjects. Further details on the sequencing study are given in the Supplementary Section G.
We computed the RV sharing probabilities based on the known pedigree structures exactly in the 50 non-inbred families and using Monte Carlo simulation in the 4 inbred families. We then computed potential P-values of the rare SNVs (see Section 2.3) and obtained 2355 values below the threshold for a family-wise Type I error rate of 0.05. Supplementary Figure S3 shows the distribution of P-values for the selected SNVs. The SNV rs149253049 in ADAMTS9 had a . The G allele, the rarest of the three nucleotides A, C and G at this SNV, was shared by affected relatives in three families from India (Supplementary Table S2) and was not seen in any other family, neither in the ESP nor in the 1000 Genomes databases. For the Indian families, kinship estimates between affected subjects from genome-wide SNP genotypes based on the estimator of Manichaikul et al. (2010) did not produce any evidence of excess IBD sharing given the known degree of relatedness, nor of relatedness between subjects from distinct Indian families.
In addition to rs149253049, another SNV, rs117883393 in OR2A2, had a P-value below the Bonferroni-corrected significance threshold (). The T allele at this SNV was shared in a heterozygous state by all sequenced subjects from three families (the two Syrian families depicted in Figure 1 and an avuncular German pair) and was present in a heterozygous state in one of two sequenced first cousins once removed from another German family. Its frequency in the ESP database is 0.0063 for the whole sample, and 0.0081 for the European–American subsample. We suspected sharing probabilities may be underestimated in the Syrian families, where cultural and demographic factors make relationships between founders more likely. We obtained , close to the kinship coefficient of second cousins (). Application of the approximation method to the two Syrian families whose sequenced members shared the rare allele at rs117883393 using that value of reduced the evidence against the null hypothesis. A sensitivity analysis further revealed that allele frequencies as low as 0.5% in the Syrian population would render this finding non-significant. Additional details on the detected signals are presented in the Supplementary Section G, where we also report that the standard filtering consisting in keeping novel non-synonymous or truncating RVs predicted to be damaging and shared by affected relatives left us with a much greater number of variants to follow-up (656).
4 DISCUSSION
In this article, we propose using the probability of sharing of an RV by affected subjects under the null hypothesis of complete absence of linkage and association between an RV and disease status to build evidence against this null hypothesis in the context of exome sequencing studies of complex diseases in family samples. This approach will be successful at finding RVs with high penetrance for diseases where such variants are involved. We have presented formulas to compute exact probabilities of sharing of an RV by any number of affected subjects in arbitrary non-inbred pedigrees under the assumption the variant is sufficiently rare to be introduced only once in the pedigree, generalizing a previous formula applicable to two affected subjects.
It is important to stress that more information is extracted in this approach from each family than in the case of testing for linkage alone because we require the RV in question, and not any allele, to be shared. This is most easily illustrated with two relatives of degree D, for which the probability of sharing an allele IBD is while the RV sharing probability is . The ratio P[RV shared]/P[IBD] will tend to as D tends to infinity so RV sharing is more informative for making inference on a particular RV in families where the RV is seen. Our power study indicated moderate to good power to detect highly penetrant variants with a small number of families where the variant is observed. Power declines as the relative risk decreases, but if several RVs are involved in a heterogeneous disease, the probability of finding at least one would remain good even if power is low. First cousin pairs provided more power than second cousin and parent–offspring pairs under genetic heterogeneity and dominant effect of the variant, the model most compatible with RVs causing disease in unilineally related subjects.
The assumption that a RV is sufficiently rare for being almost certainly IBD among relatives is crucial to the validity of the RV sharing probabilities. We recommend performing an analysis of sensitivity to this assumption for any potential finding, as we have done for the two hits in the oral clefts study.
A potential pitfall with RV sharing probabilities based on a known pedigree structure is the possibility of cryptic relatedness among founders that would make the actual null sharing probability greater than the one computed here. We have developed an adjustment to RV sharing probabilities based on estimates of the kinship coefficients among founders of known pedigrees under the assumption of equal kinship coefficients for all pairs of founders. Our simulation study on a pedigree whose founders were drawn from larger pedigrees representing small populations showed the approximation is accurate when the mean kinship coefficient among founders of the known pedigree is no greater than the kinship coefficient of second cousins once removed (), but will underestimate RV sharing probabilities with closer relationships. The simulation study also revealed an accurate approximation can be achieved using Monte Carlo sampling with a reasonable number of draws for sharing probabilities of the order of .
An important aspect of our adjustment for unknown relationships is to be based solely on estimated kinship between founders, and not require an estimate of the RV frequency in the population from which the pedigree founders were drawn. We have proposed a formula to estimate mean kinship among founders based on the kinship estimates between sequenced subjects. A number of methods can be used to estimate kinship coefficients from genome-wide genotype data (Manichaikul et al., 2010; Speed et al., 2012; Thornton et al., 2012; Yang et al., 2011), and an appraisal of these methods is beyond the scope of this article. Because our approximation method requires only a mean kinship coefficient between founders, variation in the length of genome shared by pairs of subjects is smoothed by averaging. Using a population average instead of the true average over the founder pairs of the pedigree had a moderate impact on the error in our simulation study.
For this work, we have implemented the formulas to compute our numerical approximation of the RV sharing probability allowing for relatedness among founders and assess the sensitivity to allele frequency specifically for the family structures shown in Figure 1 and Supplementary Figure S1 and reported in Supplementary Table S2. Developing an implementation of these formulas to general pedigree structures remains challenging. However, these checks of the analysis assumptions can also be performed by Monte Carlo simulation, implemented in the RVsharing R package.
In our extension of the RV sharing probability to more than two subjects, we considered only the probability that all affected sequenced subjects share the RV. This is appropriate for three affected subjects sequenced in a pedigree as in the oral cleft study, where causal RVs not shared by all sequenced subjects are indistinguishable from benign RVs. However, it is too stringent a requirement when larger numbers of affected subjects from large multiplex families are sequenced, given the intrafamilial heterogeneity in disease causes typical of complex traits (Feng et al., 2011). At the same time, with sequenced subjects in a family, the event that n – 1 or n – 2 affected subjects out of n share an RV is itself evidence against the null hypothesis. The computation of the probability of such events in pedigrees of arbitrary structure will require further work.
Non-affected family members may also be included in future sequencing studies. While sequencing non-affected family members has been used to exclude private benign variation in studies of Mendelian traits (Gilissen et al., 2012), this risks excluding causal variants showing incomplete penetrance in studies of complex traits. An affected only analysis of RV sharing protects against unaffected carriers reducing evidence for a variant in the same way as it does in linkage analysis (McPeek, 1999). Sequence data on non-affected family members, in particular subjects marrying into the pedigree, will still be useful in narrowing down the number of founders that could have introduced a given RV in the pedigree and refine these RV sharing probabilities.
The methods and analyses presented are limited to considering a single RV at a time. Our results illustrate how with a few families it is possible to obtain substantial evidence of co-segregation between a RV and disease. Yet, rare causal variants found in a single family were not considered in our analysis of these multiplex cleft families because individual families provide limited information. A combined analysis of multiple RVs from the same functional unit, typically the same gene, will be needed to detect significant RV sharing at that level. Various issues still need to be resolved to implement such analysis, in particular, dealing with multiple RVs within the same family. This will be the object of future work.
Supplementary Material
ACKNOWLEDGEMENT
The authors thank J. Croteau (Centre de Recherche de l’Institut Universitaire en Santé Mentale de Québec) for his programming assistance.
Funding: This work was supported by NIH grants (R10-DE-014581, R01-DE016148, R01-DE009886, R37-DE008559 and X01-HG006177), which supported the whole exome sequencing at the Center for Inherited Disease Research. Recruitment of German families was supported by the Deutsche Forschungsgemeinschaft (FOR 423 and individual grants MA 2546/3-1, KR 1912/7-1, NO 246/6-1, WI 1555/5-1). Contacting patients and their families was supported by the German support group for individuals with cleft lip and/or palate (Deutsche Selbsthilfevereinigung für Lippen-Gaumen-Fehlbildungen e.V.). Recruitment of Syrian families was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, USA. A Bureau is supported by a research fellowship from the Fonds de recherche du Québec - Santé. I. Ruczinski was further supported by NIH (grant R01 GM083084).
Conflict of Interest: none declared.
REFERENCES
- Beaty TH, et al. Confirming genes influencing risk to cleft lip with/without cleft palate in a case-parent trio study. Hum. Genet. 2013;132:771–781. doi: 10.1007/s00439-013-1283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- Feng BJ, et al. Design considerations for massively parallel sequencing studies of complex human disease. PLoS One. 2011;6:e23221. doi: 10.1371/journal.pone.0023221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen C, et al. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012;20:490–497. doi: 10.1038/ejhg.2011.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig KU, et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 2012;44:968–971. doi: 10.1038/ng.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, et al. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPeek MS. Optimal allele-sharing statistics for genetic mapping using affected relatives. Genet. Epidemiol. 1999;16:225–249. doi: 10.1002/(SICI)1098-2272(1999)16:3<225::AID-GEPI1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Risch N. Linkage strategies for genetically complex traits I. Multilocus models. Am. J. Hum. Genet. 1990;46:222–228. [PMC free article] [PubMed] [Google Scholar]
- Speed D, et al. Improved heritability estimation from genome-wide SNPs. Am. J. Hum. Genet. 2012;91:1011–1021. doi: 10.1016/j.ajhg.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton T, et al. Estimating kinship in admixed populations. Am. J. Hum. Genet. 2012;91:122–138. doi: 10.1016/j.ajhg.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman EM. The role of large pedigrees in an era of high-throughput sequencing. Hum. Genet. 2012;131:1555–1563. doi: 10.1007/s00439-012-1190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, et al. Gcta: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.