

Abstract
The relative influence of amyloid burden, neuronal structure and function, and prior cognitive performance on prospective memory decline among asymptomatic late middle-aged individuals at risk for Alzheimer’s disease (AD) is currently unknown. We investigated this using longitudinal cognitive data from 122 middle-aged adults (21 “Decliners” and 101 “Stables”) enrolled in the Wisconsin Registry for Alzheimer’s Prevention who underwent multimodality neuroimaging (11C-Pittsburgh Compound B (PiB), 18F-fluorodeoxyglucose (FDG), and structural/functional MRI) 5.7±1.4 years (range=2.9–8.9) after their baseline cognitive assessment. Covariate-adjusted regression analyses revealed that the only imaging measure that significantly distinguished Decliners from Stables (p=.027) was a Neuronal Function composite derived from FDG and fMRI. In contrast, several cognitive measures, especially those that tap episodic memory, significantly distinguished the groups (p’s < .05). Complementary receiver operating characteristic curve analyses identified the Brief Visuospatial Memory Test-Revised (BVMT-R) Total (.82±.05, p<.001), the BVMT-R Delayed Recall (.73±.06, p=.001), and the Reading subtest from the Wide-Range Achievement Test-III (.72±.06, p=.002) as the top three measures that best discriminated the groups. These findings suggest that early memory test performance might serve a more clinically-pivotal role in forecasting future cognitive course than is currently presumed.
Keywords: Longitudinal cognitive decline, amyloid, FDG, fMRI, family history of AD, preclinical AD
INTRODUCTION
The prevailing view of the temporal evolution of Alzheimer’s disease (AD) pathophysiological changes posits that inceptive alterations in β-amyloid trigger a cascade of events that sequentially leads to neuronal dysfunction, neurodegeneration and, ultimately, cognitive/functional impairments (Jack et al., 2010; Sperling et al., 2011). Converging support for this model has come from studies that simultaneously examined biological markers of these brain lesions—using positron emission tomography (PET) tracers such as 11C-Pittsburgh Compound B (PiB) and 18F-fluorodeoxyglucose (FDG)—among individuals spanning the AD spectrum from cognitively healthy to frank dementia (Landau et al., 2012; Lo et al., 2011; Villemagne et al., 2013). Even so, a number of studies now suggest that this cascade might be differentially expressed in certain risk-enriched cohorts such as cognitively-healthy persons with a parental family history (FH) of AD and/or carrying copies of the apolipoprotein E4 (APOE4) allele (Okonkwo, Xu, Dowling, et al., 2012; Sheline et al., 2010; Sperling et al., 2011). Furthermore, no studies have conducted integrative, multimodality examinations of the effect of various AD pathophysiological markers on cognitive decline in late midlife, a period when these markers are arguably just starting to become dynamic.
Accordingly, our aim in this study was to use an ensemble of imaging and neuropsychological measures to assess the relative contributions of β-amyloid deposition, neuronal dysfunction, neurodegeneration, and prior cognitive performance to cognitive decline among cognitively-healthy middle-aged adults who have been longitudinally followed since 2001 through the Wisconsin Registry for Alzheimer’s Prevention (WRAP; Sager, Hermann, & La Rue, 2005). Because baseline cognitive assessments in this cohort occurred several years prior to the imaging exams, the analyses presented here cannot directly address the hypothesized AD cascade (Jack et al., 2010; Sperling et al., 2011). However, these data may provide initial insights into associations between imaging markers of brain health and longitudinal cognitive course in a cohort that is very well-characterized and enriched with at-risk middle-aged adults.
METHODS
Participants
The WRAP was initiated in 2001 as a longitudinal registry of cognitively-normal, middle-aged adults (ages 40–65 at entry) with and without FH of AD (Sager et al., 2005). Participants were recruited on a rolling basis and the cohort currently consists of >1500 individuals. The procedures for verifying presence or absence of AD in the parent has been described previously (Okonkwo, Xu, Dowling, et al., 2012; Sager et al., 2005). The WRAP study protocol targeted a 4-year interval between baseline and wave 2 visits, and 2-year intervals between subsequent waves.
Between May 2010 and April 2012, 178 WRAP participants were recruited into a multimodality neuroimaging study consisting of PiB, FDG, T1 volume, and functional magnetic resonance imaging (fMRI) scans. The analyses reported in this article include those individuals (n=122) who had complete data across all imaging modalities and were classified as either “Stable” or “Decliner” based on longitudinal neuropsychological data (see Longitudinal Cognitive Status section below). These 122 individuals with complete data did not differ from the other 56 participants on age, gender, race, education, FH status, or APOE4 status. On average, participants completed the imaging exams 5.7±1.4 years (range=2.9–8.9) after their baseline WRAP study visit. The baseline WRAP visit was when cognition was first assessed, with the exception of a few tests that were added to the WRAP battery subsequently (see Neuropsychological Assessment section below).
The University of Wisconsin Institutional Review Board approved all study procedures and each subject provided signed informed consent before participation. The study was carried out in accordance with the Helsinki Declaration.
Neuroimaging Protocol
Details on the acquisition and post-processing of the PET and MRI exams are fully described in the Supplementary Material. In brief, both FDG and PiB images were acquired in 3D on a Siemens EXACT HR+ scanner. PiB imaging consisted of a 6-minute transmission scan and a 70-minute dynamic scan upon bolus injection. FDG imaging was done per ADNI protocol (Jagust et al., 2010) and involved a 30-minute scan acquired 30 minutes after bolus injection. Post-processing for both PiB and FDG was based on an in-house automated pipeline (Christian, Vandehey, Floberg, & Mistretta, 2010; Floberg et al., 2012). Distribution volume ratios (DVRs) were derived from the PiB images using the Logan method, with a cerebellar gray matter reference (Price et al., 2005). The MRI images were acquired on a GE x750 3.0T scanner. The anatomical imaging featured a T1-weighted inversion recovery-prepared SPGR volume. The fMRI paradigm, similar to that reported in our prior publication (Xu et al., 2009), consisted of an event-related task involving episodic recognition of neutral faces. The task required participants to discriminate previously viewed faces from novel ones. This paradigm reliably evokes activation in aspects of the precuneus/posterior cingulate cortex (PCC), regions known to be both vulnerable to early AD pathological changes and involved in episodic memory (Buckner et al., 2005; Huijbers et al., 2012; Johnson et al., 2006; Leech & Sharp, 2013; Rami et al., 2012; Vannini et al., 2011; Xu et al., 2009). Taken together, the imaging modalities implemented in this study target 3 core pathophysiological features of the hypothesized AD cascade viz. β-amyloid deposition (PiB imaging), neuronal dysfunction (FDG and fMRI scans), and neurodegeneration (T1 volume) (Jack et al., 2010; Sperling et al., 2011). As noted above, participants completed the imaging exams 5.7±1.4 years after their initial WRAP study visit.
Region-of-interest data sampling
To focus our analyses on the cortical midline structures (precuneus and PCC) known to be predilection sites for β-amyloid aggregation and neurometabolic alterations in preclinical AD (Buckner et al., 2005; Sperling et al., 2011), we used a series of binary masks to sample data from these regions of interest (ROI). For PiB, we sampled data from the precuneus using the Automated Anatomical Labeling atlas implemented in the WFU PickAtlas toolbox (Maldjian, Laurienti, Kraft, & Burdette, 2003). We sampled FDG using the PCC mask from the ADNI FDG Meta-ROI suite (Landau et al., 2011). Our fMRI mask was generated empirically by first performing a voxel-wise ANCOVA (adjusting for age, gender, APOE4, and FH) between an independent set of Stables (n=90) and Decliners (n=34) who had completed this task as part of enrollment in another imaging study. Interrogation of the results map from this analysis—at Pvoxel <.005, cluster extent ≥50 voxels—was restricted to a precuneus/PCC a priori search region (Xu et al., 2009). Significant clusters were then written out as a binary mask and used to sample fMRI data from the participants included in the present analyses. Of note, this ANCOVA for defining our fMRI ROI revealed that the Decliners had increased brain activation compared to the Stables. Analogous findings have been reported by other groups (O’Brien et al., 2010). Hippocampal volume was measured with the FreeSurfer image analysis suite (http://surfer.nmr.mgh.harvard.edu/). The masks used for sampling PiB, FDG, and fMRI data are shown in Figure 1.
Figure 1. Regions of interest for PET and fMRI data sampling.
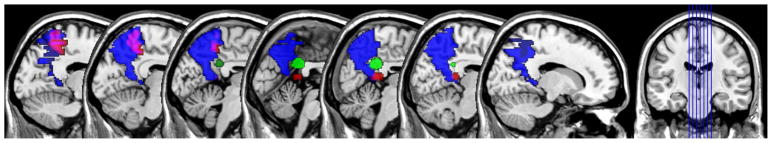
Binary masks with which PiB (blue), FDG (green), and fMRI (red, purple where intersects with PiB mask) were sampled. The masks are overlaid on the SPM single subject atlas conformed to MNI space at y-plane locations −15 −10 −5 0 5 10 15. The PiB mask was derived using the precuneus label in the WFU_PickAtlas toolbox (Maldjian et al., 2003); the FDG mask was taken from the PCC template in the Meta-ROI suite developed by ADNI;15 and the fMRI mask was empirically generated using data from an independent set of Stables (n=90) and Decliners (n=34) who had completed the fMRI task as part of enrolment in another imaging study.
PiB= Pittsburgh Compound B; FDG= fluorodeoxyglucose; fMRI=functional magnetic resonance imaging; SPM=statistical parametric mapping; MNI=Montreal Neurological Institute; WFU=Wake Forest University; PCC=posterior cingulate cortex; ROI=region of interest; ADNI=Alzheimer’s Disease Neuroimaging Initiative.
Neuropsychological Assessment
WRAP participants undergo a comprehensive neuropsychological battery (Sager et al., 2005). In addition to global measures such as the Clinical Dementia Rating Scale (CDR; Morris, 1993) and the Mini-Mental State Exam (MMSE; Folstein, Folstein, & McHugh, 1975), specific cognitive domains—and component psychometric measures assessed—include: Episodic Memory: Rey Auditory Verbal Learning Test (RAVLT; Schmidt, 1996), Brief Visuospatial Memory Test-Revised (BVMT-R; Benedict, 1997), Logical Memory subtest from the Wechsler Memory Scale, Revised edition (Wechsler, 1987); Attention/Working Memory: Digit Span, Arithmetic, and Letter-Number Sequencing subtests from the Wechsler Adult Intelligence Scale, 3rd edition (WAIS-III; Wechsler, 1997); Executive Function: Clock Drawing Test (Strauss, Sherman, & Spreen, 2006), Trail Making Test (Reitan & Wolfson, 1993), WAIS-III Digit Symbol, Wisconsin Card Sorting Test (Heaton, Chelune, Talley, Kay, & Curtiss, 1993), Stroop Test (Trenerry, Crosson, DeBoe, & Leber, 1989); Visuospatial ability: Block Design and Matrix Reasoning subtests from the Wechsler Abbreviated Scale of Intelligence (WASI; Wechsler, 1999), Judgment of Line Orientation Test (Benton, 1994); and Language: Wide-Range Achievement Test, 3rd edition (WRAT-III Reading, Wilkinson, 1993), Vocabulary and Similarities subtests from the WASI, Boston Naming Test (Kaplan, Goodglass, & Weintraub, 1983), Controlled Oral Word Association Test (Benton, Hamsher, & Sivan, 1976). The MMSE, BVMT-R, and Logical Memory tests were added to the WRAP study protocol at wave 2. The CDR was administered at the time of brain imaging. All tests were administered in a standardized fashion (Strauss et al., 2006).
Vascular Risk Factors
Participants underwent a clinic visit at the UW NIH-funded Institute for Clinical and Translational Research that included anthropometric measurements, blood pressure readings, and fasting blood draw for assaying a panel of laboratory tests implicated in vascular disease. These tests included total cholesterol, high density lipoprotein, insulin, glucose, interleukin-6, and high sensitivity C-reactive protein.
Longitudinal Cognitive Status
Adjudication of longitudinal cognitive status (Stable vs. Decliner) was based on RAVLT Total Learning and Long Delay scores, across all testing occasions, as early deficits in list learning/retention have been shown to be particularly prognostic of future clinical decline (Blacker et al., 2007; Elias et al., 2000). Specifically, we first derived age-, gender-, and education-adjusted RAVLT norms using baseline data from WRAP participants (n=408) who were FH- and also free from all major neurological and psychiatric conditions that could compromise cognition across all their study visits (spanning November 2001 to April 2012). Using these norms, an individual was labeled a Decliner if their score on the RAVLT Total Learning or Long Delay fell ≥ 1.5 SD below expected intra- or inter-individual levels on any visit other than baseline, and remained within this range on subsequent testing occasions. Conversely, persons were considered Stable if their RAVLT Total Learning and Long Delay scores never dipped below −1 SD (whether intra- or inter-individually) across all visits.
All participants had at least two cognitive assessments (median=3, range=2–4) and the median time interval between baseline and last cognitive assessment was 5.8 years (range=3.7–9.8). Mean±SD RAVLT Total Learning scores for the Stables (z-transformed and adjusted for age, gender, and education) were 0.22±0.79 at baseline and 0.44±0.81 at the last visit (t(100)=3.10, p=.003). For the Decliners, corresponding baseline and last visit scores were −0.69±0.81 and −1.22±0.62, respectively, (t(20)= −2.42, p=.025). Similarly, z-transformed and demographic-adjusted RAVLT Long Delay scores for the Stables were 0.28±0.79 at baseline and 0.45±0.79 at the last visit (t(100)=2.50, p=.014) whereas Decliners obtained scores of −0.58±0.98 and −1.14±0.66, respectively, (t(20)= −2.48, p=.022). These test scores reveal that whereas the Stables experienced some increment in test scores over longitudinal follow up perhaps due to practice effects, the Decliners experienced a worsening in test performance. Figure 2 plots mean±SE group scores on the RAVLT Total and Long Delay at each study visit.
Figure 2. Longitudinal performance on RAVLT Total and Long Delay among Stables and Decliners.


RAVLT=Rey Auditory Verbal Learning Test. Data points represent mean score on the RAVLT Total Learning (top panel) and RAVLT Long Delay (bottom panel) among Stables (blue diamonds) and Decliners (red squares). Note: Because the WRAP cohort was recruited on a rolling basis, not all subjects have had all 4 study visits (for example, of the 122 subjects who contributed data to the analyses, only six—4 Stables and 2 Decliners—have had a visit 4). Therefore, Visit 4 on this figure is not necessarily synonymous with the “last visit” described in the Longitudinal Cognitive Status section. This is why there is some discrepancy between the RAVLT values for Visit 4 on this plot and the values reported for “last visit” in the Longitudinal Cognitive Status section.
The reason that Decliners’ mean scores on the RAVLT Total Learning and Long Delay at the last visit did not quite hit the −1.5 SD mark was because we defined decline both inter- and intra-individually. Therefore, the Decliner group included persons whose absolute RAVLT Total Learning/Long Delay test scores at the last visit did not cross the −1.5 SD threshold (i.e., inter-individual rule) but who, nonetheless, experienced a longitudinal drop in test score that was ≥1.5 SD (i.e., intra-individual rule), such as the participant who had an RAVLT Total Learning score (z-transformed and demographic-adjusted) of 0.57 at baseline but dropped to a score of −1.31 at their last visit—an intra-individual worsening in performance of 1.88 SDs.
Statistical Analysis
Group differences on baseline demographic, clinical, and vascular measures were tested using independent samples t-test or χ2 analyses as appropriate. To assess the relationship between our imaging measures and prospective cognitive decline, we fitted a series of linear regressions with cognitive status (Stable vs. Decliner) as predictor and the imaging measures as outcome. This approach (versus a logistic or Cox regression, with cognitive status as the outcome) allowed us to assess the relationship between the imaging measures and cognitive decline, while simultaneously investigating the potential influence of other relevant covariates—e.g., age, gender, APOE4, and FH—on these imaging measures within our cohort. The association between baseline cognitive performance and prospective cognitive decline was assessed similarly using an ANCOVA design with age, gender, and education as covariates. Finally the predictive value of the imaging and cognitive measures vis-à-vis longitudinal cognitive status (i.e., Stable vs. Decliner) was evaluated using receiver operating characteristic (ROC) analyses. The input for each ROC analysis was a residualized version of the original imaging/cognitive measure derived by fitting a linear regression to the imaging/cognitive measure (y), using relevant covariates (x1 … xn), and computing the standardized residual. Because there are no unequivocal cut-points for adjudicating “abnormality” on these imaging/cognitive measures, the area under the ROC curve (AUC) and 95% confidence interval (CI) was adopted as the most representative summary statistic since it denotes a measure’s average sensitivity for all possible values of specificity (and vice-versa), thus capturing the measure’s overall performance (Zhou, Obuchowski, & McClish, 2011). All analyses were performed using SPSS 20.0 (IBM Corp., Armonk, NY) and Medcalc 12.7 (Ostend, Belgium). Only findings with a 2-tailed p value ≤ .05 were considered significant.
RESULTS
Background Characteristics
Table 1 details the background characteristics of the study participants. The Stable and Decliner groups did not differ significantly on any of the demographic, clinical, or vascular measures examined.
Table 1.
Background characteristics of study participants†
| Variable | Stable, n=101 | Decliner, n=21 | p value |
|---|---|---|---|
| Demographics | |||
| FH positive, % | 68.3 | 66.7 | .883 |
| APOE4 positive, % | 38.6 | 42.9 | .717 |
| Female, % | 73.3 | 57.1 | .140 |
| Caucasian, % | 99.0 | 100.0 | .647 |
| Age | 54.84 (5.97) | 55.12 (5.92) | .843 |
| Education | 15.87 (2.54) | 16.05 (2.29) | .769 |
| Clinical Measures | |||
| MMSE | 29.32 (0.97) | 29.31 (0.97) | .974 |
| CDR Global=0, % | 98.0 | 100.0 | .513 |
| CDR Sum of Boxes | .02 (0.11) | .00 (0.00) | .540 |
| CES-D | 6.59 (7.19) | 4.80 (6.64) | .304 |
| IQCODE | 47.52 (4.84) | 48.53 (3.17) | .304 |
| Vascular Indices | |||
| Body mass index (kg/m2) | 27.80 (5.08) | 28.74 (4.81) | .441 |
| Waist-to-hip ratio | .85 (.10) | .86 (.11) | .720 |
| Hypertension, % | 19.8 | 14.3 | .556 |
| Diabetes, % | 4.0 | 0.0 | .582 |
| Smoker (ever), % | 46.5 | 28.6 | .131 |
| Smoker (current), % | 5.0 | 4.8 | .971 |
| Systolic blood pressure (mmHg) | 123.69 (14.33) | 126.71 (13.22) | .375 |
| Diastolic blood pressure (mmHg) | 74.02 (8.96) | 77.43 (9.30) | .118 |
| Total cholesterol (mg/dL) | 202.56 (34.28) | 207.19 (30.51) | .568 |
| HDL cholesterol (mg/dL) | 63.00 (19.36) | 55.38 (16.27) | .095 |
| Insulin (μ/mL) | 8.81 (6.66) | 8.24 (4.13) | .705 |
| Glucose (mg/dL) | 95.61 (9.59) | 96.57 (8.40) | .672 |
| HOMA-IR | 2.14 (1.83) | 1.98 (1.14) | .702 |
| Interleukin-6 (pg/mL) | 2.27 (7.01) | 2.29 (2.37) | .993 |
| hs C-Reactive Protein (mg/L) | 1.94 (2.12) | 2.00 (2.00) | .900 |
Values are mean (SD) except where otherwise indicated. All measures were obtained at wave 1 with the following exceptions (1) MMSE, IQCODE, and waist-hip ratio were added to the WRAP protocol at wave 2, (2) the vascular laboratory measures were assayed from fasting blood samples drawn at wave 2, and (3) subjects completed the CDR as part of their brain imaging visit.
FH=family history of Alzheimer’s disease; APOE4=the varepsilon 4 allele of the apolipoprotein E gene; MMSE=Mini-Mental State Exam; CDR= Clinical Dementia Rating Scale; CES-D= Center for Epidemiologic Studies Depression Scale; IQCODE=Informant Questionnaire on Cognitive Decline in the Elderly; HDL=high-density lipoprotein; HOMA-IR=Homeostasis Model Assessment of Insulin Resistance.
Imaging Findings
Our imaging findings are displayed in Table 2 and summarized here.
Table 2.
Neuroimaging measures and cognitive decline
| Neuroimaging measure | Standardized β | T statistic | p value |
|---|---|---|---|
| PiB | |||
| Decliner | −.01 | −.08 | .939 |
| Age | .35 | 3.83 | <.001 |
| Female | .21 | 2.40 | .018 |
| APOE4 positive | .10 | 1.07 | .286 |
| FH positive | .04 | .42 | .674 |
| FDG | |||
| Decliner | −.04 | −1.69 | .093 |
| Age | −.06 | −2.32 | .022 |
| Female | −.10 | −3.71 | <.001 |
| APOE4 positive | −.01 | −.44 | .664 |
| FH positive | .01 | .41 | .684 |
| FDG global | 1.00 | 36.19 | <.001 |
| fMRI | |||
| Decliner | .15 | 1.65 | .100 |
| Age | .06 | .65 | .518 |
| Female | .12 | 1.32 | .189 |
| APOE4 positive | .01 | .03 | .976 |
| FH positive | .23 | 2.51 | .013 |
| Hippocampal volume | |||
| Decliner | .04 | .55 | .587 |
| Age | −.29 | −3.63 | <.001 |
| Female | −.05 | −.53 | .601 |
| APOE4 positive | .01 | .08 | .94 |
| FH positive | .09 | 1.10 | .273 |
| ICV | .44 | 4.44 | <.001 |
For each imaging measure, all predictors were entered in the regression model simultaneously.
PiB= Pittsburgh Compound B; FDG= fluorodeoxyglucose; fMRI=functional magnetic resonance imaging; APOE4=the varepsilon 4 allele of the apolipoprotein E gene; FH=family history of Alzheimer’s disease; ICV=intracranial volume.
PiB
Decliners did not differ from the Stables on β-amyloid deposition. Older age was associated with higher β-amyloid and women exhibited greater β-amyloid than men. Neither APOE4 nor FH was associated with increased β-amyloid in this model. Because amyloid does not aggregate uniformly across the entire precuneus, especially in earlier stages of AD, we re-ran the PiB analyses using data extracted from around the precuneus subregion with most overt β-amyloid in our sample (at MNI x,y,z coordinates [−8, −66, 52]). The results were unchanged.
FDG
There was a trend for Decliners to have lower glucose metabolism compared with Stables, but this was not significant at the .05 alpha threshold. As with β-amyloid, both older age and female sex were significantly associated with decreased glucose metabolism, and neither APOE4 nor FH status had an influence on glucose metabolism.
fMRI
Decliners tended to have increased brain activation compared with Stables. As with the FDG data, this did not meet the .05 alpha threshold. FH+ persons had significant hyperactivation compared with those who were FH−. No other demographic factor was associated with fMRI activation.
Hippocampal volume
Decliners and Stables were not distinguishable on hippocampal volume. Hippocampal volume was inversely related to age. There were no gender, APOE4, or FH effects on hippocampal volume.
Supplementary Imaging Analyses
Neuronal Function Marker
Given that (1) of the imaging measures, only FDG and fMRI showed trends for separating Decliners from Stables, and (2) both measures index complementary aspects of neuronal integrity, we combined them into a z-composite measure of Neuronal Function wherein higher scores reflect better neuronal integrity (Wirth, Madison, et al., 2013). The input that went into this composite measure was a residualized version of the original imaging measure derived by fitting a linear regression to the imaging measure using applicable covariates (see Table 3), and computing the standardized residual. Specifically, the FDG measure was residualized on age, sex, APOE4 status, FH status, and global FDG whereas the fMRI measure was residualized on age, sex, APOE4 status, and FH status. The test of between-group difference on this composite Neuronal Function marker revealed that Decliners had significantly lower scores (−.33±.74) compared with Stables (.07±.73), t(120)=2.23, p=.027.
Table 3.
Group differences on baseline neuropsychological measures†
| Variable | Stable, n=101 | Decliner, n=21 | p value |
|---|---|---|---|
| Episodic Memory | |||
| RAVLT Total | 53.01 (5.97) | 47.26 (5.90) | <.001 |
| RAVLT Short Delay | 10.98 (2.27) | 9.38 (2.24) | .004 |
| RAVLT Long Delay | 11.20 (2.21) | 9.13 (2.18) | <.001 |
| RAVLT Recognition | 14.30 (1.28) | 13.29 (1.26) | <.001 |
| BVMT-R Total | 25.40 (4.74) | 19.91 (4.69) | <.001 |
| BVMT-R Delayed Recall | 9.81 (1.93) | 8.34 (1.91) | .002 |
| BVMT-R Recognition | 5.85 (0.49) | 5.36 (0.49) | <.001 |
| WMS-R Logical Memory I | 30.72 (6.06) | 27.77 (5.99) | .044 |
| WMS-R Logical Memory II | 27.26 (6.51) | 23.91 (6.44) | .033 |
| Attention/Working Memory | |||
| WAIS-III Digit Span | 17.97 (3.97) | 17.10 (3.93) | .362 |
| WAIS-III Arithmetic | 15.22 (2.98) | 14.34 (3.00) | .225 |
| WAIS-III Letter Number Sequencing | 10.71 (2.23) | 10.20 (2.20) | .341 |
| Executive Function | |||
| Clock Drawing Test | 9.50 (0.78) | 9.58 (0.78) | .707 |
| Trail Making Test A, time | 27.27 (8.25) | 24.75 (8.17) | .203 |
| Trail Making Test B, time | 62.77 (19.81) | 57.06 (19.59) | .229 |
| WAIS-III Digit Symbol | 57.70 (8.40) | 56.50 (8.45) | .557 |
| WCST-64, perseverative responses | 8.59 (6.14) | 7.60 (6.07) | .500 |
| Stroop Color-Word, words named | 111.06 (19.17) | 106.06 (19.17) | .332 |
| Visuospatial Ability | |||
| WASI Block Design | 46.42 (10.96) | 40.52 (10.84) | .026 |
| WASI Matrix Reasoning | 25.83 (3.00) | 24.31 (2.97) | .036 |
| Judgment of Line Orientation | 25.23 (3.75) | 25.04 (3.77) | .832 |
| Language | |||
| WRAT-III Reading | 108.75 (8.42) | 102.71 (8.33) | .003 |
| WASI Vocabulary | 66.49 (6.47) | 64.81 (6.40) | .277 |
| WASI Similarities | 38.95 (3.81) | 37.70 (3.77) | .174 |
| Boston Naming Test, spontaneous | 56.43 (2.73) | 55.68 (2.74) | .248 |
| COWAT | 42.79 (10.62) | 41.38 (10.51) | .581 |
Values are raw scores statistically adjusted for age, gender, and education. All cognitive test scores were from wave 1 study visit, except the BVMT-R and Logical Memory which were added to the study protocol at wave 2.
WRAP= Wisconsin Registry for Alzheimer’s Prevention; RAVLT=Rey Auditory Verbal Learning Test; BVMT-R=Brief Visuospatial Memory Test-Revised; WMS-R=Wechsler Memory Scale, Revised edition; WAIS-III=Wechsler Adult Intelligence Scale, 3rd edition; WCST=Wisconsin Card Sorting Test; WASI=Wechsler Abbreviated Scale of Intelligence; WRAT III=Wide-Range Achievement Test, 3rd edition; COWAT= Controlled Oral Word Association Test, C-F-L version.
Posteromedial Cortex Volumetry
Because the precuneus/PCC regions are known to be vulnerable to AD pathology, are involved in episodic recollection, and were used to sample PiB, FDG, and fMRI data in our study, we also explored group differences in T1 gray matter volume sampled from these regions. The native T1 images were first preprocessed in SPM8 (www.fil.ion.ucl.ac.uk/spm) using standard voxel-based morphometric approaches—i.e., segmentation, normalization, modulation, and smoothing—as previously described (Okonkwo, Xu, Dowling, et al., 2012; Okonkwo, Xu, Oh, et al., 2012). We then applied the binary PCC and precuneus masks described above in “Region-of-interest data sampling” to the resulting gray matter probability maps in order to sample gray matter volumes from these regions. Covariate-adjusted regressions analyses revealed no significant gray matter volume differences between Decliners and Stables in either the PCC (p=.995) or the precuneus (p=.472).
Neuropsychological Findings
Table 3 shows the results of comparisons between Decliners and Stables on baseline cognitive measures. Decliners had poorer test scores than Stables on all study measures of episodic memory, spanning learning, free recall, and recognition memory metrics. They also had significantly lower literacy scores (WRAT-III Reading), and performed worse on two measures of visuospatial ability. In contrast, the groups did not differ on measures of executive function, attention/working memory, or language. Note that group comparisons on the RAVLT are presented in this table only for descriptive purposes given that serial performance on this measure was used in the adjudication of longitudinal cognitive status, i.e., Stable vs. Decliner.
We re-ran the cognitive analyses while adjusting for age, gender, and WRAT-III Reading instead of age, gender, and education given that, in some contexts, quality of education (as indexed by literacy measures such as the WRAT-III Reading) has been shown to be a more pertinent demographic confound than mere years of schooling (Manly, Schupf, Tang, & Stern, 2005). Our initial findings persisted, with the exception that Logical Memory I & II, Block Design, and Matrix Reasoning were no longer significantly different between the groups, with p values of .285, .302, .094, and .234 respectively. This variation in findings was driven by the fact that whereas longitudinal cognitive status was not significantly associated with education (r(120)=.03, p =.769; see also Table 1), it exhibited a significant, negative association with WRAT-III Reading (r(120)=−.23, p =.013). Thus, adjusting for WRAT-III Reading dampened some of the original findings, though the direction of each prior finding (i.e., Decliners having worse scores) persisted. Of note, education and WRAT-III Reading were moderately correlated in the sample, r(120)=.45, p <.001.
Predictive Value of Imaging and Cognitive Measures
The ROC analyses were performed for all imaging measures but only for those cognitive tests on which the groups differed significantly (see Table 3). AUCs (95% CI) from the ROC analyses are shown in Figure 3. Of the imaging measures, hippocampal volume had the lowest AUC (0.46 (0.32, 0.60)) whereas the composite Neuronal Function marker (0.66 (0.53, 0.79)) had the highest. Among cognitive measures, BVMT-R Total had the highest AUC (0.82 (0.72, 0.92)) whereas WASI Matrix Reasoning had the lowest (0.57 (0.43, 0.70)). With the exception of the WASI Matrix Reasoning, cognitive measures consistently exhibited better predictive utility than imaging measures. However, as can be seen from Figure 3, there is considerable overlap in the confidence bounds of observed AUCs, suggesting that while the measures may differ on point estimates, their effects may not be statistically distinguishable. Formal statistical comparisons of the ROCs confirmed this. The only significantly different AUCs were (1) PiB < BVMT-R Total (p=.001), WRAT-III Reading (p=.010), BVMT-R Delayed Recall (.040); (2) FDG < BVMT-R Total (p=.033); (3) fMRI < BVMT-R Total (p=.020); (4) Hippocampal volume < BVMT-R Total (p=.002), BVMT-R Delayed Recall (p=.049); (5) Matrix Reasoning < BVMT-R Total (p=.001). Note that AUCs from RAVLT indices are plotted in Figure 3 purely for descriptive purposes; hence, they are not featured in the foregoing statistical comparisons of ROCs.
Figure 3. ROC analyses for imaging and cognitive measures.

The dashed vertical line represents an AUC of 0.5, the AUC for a measure with no predictive utility.
AUC=area under the ROC curve; ROC=receiver operating characteristic curve; PiB= Pittsburgh Compound B; FDG= fluorodeoxyglucose; fMRI=functional magnetic resonance imaging; WRAT III=Wide-Range Achievement Test, 3rd edition; RAVLT=Rey Auditory Verbal Learning Test; BVMT-R=Brief Visuospatial Memory Test-Revised; WMS-R=Wechsler Memory Scale, Revised edition; WASI=Wechsler Abbreviated Scale of Intelligence.
DISCUSSION
In this study, we capitalized on the unique opportunity provided by a cohort of at-risk middle-aged adults who have just begun showing very mild memory changes—over several years of follow-up—to gain an aperture into the influence of amyloid burden, neuronal structure/function, and prior cognitive performance on longitudinal cognitive decline in midlife. While the temporal asynchrony between baseline cognitive assessment and the imaging exams in our study precludes a definitive evaluation of the hypothesized model of biomarker changes in AD in this cohort (Jack et al., 2010; Sperling et al., 2011), our results are nonetheless intriguing. We found that prior performance on several neuropsychological measures—particularly those in the domain of episodic memory—significantly discriminated the Decliners from the Stables. In contrast with these cognitive findings, no single imaging measure significantly differentiated Stables from the Decliners. However, a composite marker of Neuronal Function—synthesized from FDG and fMRI—did discriminate the groups successfully.
Studies on the impact of β-amyloid on longitudinal cognitive decline among cognitively-healthy adults are currently underway in diverse research groups, and initial reports have been variable (Chetelat, 2013; Chételat et al., 2013). While some of these investigations have shown increased β-amyloid deposition to be associated with prospective cognitive decline (Landau et al., 2012; Lim et al., 2012; Morris et al., 2009; Resnick et al., 2010; Snitz et al., 2013), others have failed to find such associations or have found it only when analyses are restricted to a subset of study subjects such as those with concomitant neuronal injury, low cognitive reserve, or significant decline (Cairns et al., 2009; Desikan et al., 2012; Ewers et al., 2012; Jack et al., 2009; Rentz et al., 2010). The age of the cohort is arguably a critical contributor to these inconsistencies given that age is a cardinal risk factor for both AD/cognitive decline and β-amyloid aggregation (Hedden, Oh, Younger, & Patel, 2013). Of note, our cohort’s mean age of 61 is 10–30 years younger than other cognitively-normal cohorts wherein associations between β-amyloid and cognitive decline have been detected (Landau et al., 2012; Lim et al., 2012; Morris et al., 2009; Resnick et al., 2010; Snitz et al., 2013). Other potential sources of variance in reported β-amyloid—cognitive decline findings include the algorithm for establishing cognitive decline, duration of follow up, and genetic characteristics (e.g., % APOE4+) of the sample.
Similar to results from the least-squares analyses, the ROC analyses indicated that measures of neurometabolic function (i.e., fMRI and FDG, singly and conjointly) exhibited greater predictive utility compared with measures of amyloidosis and neurodegeneration. Only a handful studies have integrated multimodality imaging in the prediction of cognitive decline among cognitively-healthy older adults. Landau et al. (2012) recently reported that, among healthy controls (mean age = 78 years), β-amyloid burden was associated with decline on a measure of global cognition whereas glucose metabolism was not. In another study (Wirth, Oh, et al., 2013) biomarkers of β-amyloid and neuronal injury exerted both independent and interactive effects on decline in memory and nonmemory composite measures in subjects with a mean age of 73 years. In contrast, Lo and colleagues (2011) found that baseline glucose metabolism explained a larger portion of the variance in global cognitive decline than amyloidosis in elderly controls at mean age 75 years. Our findings appear to be more in line with the latter report. It is also important to mention that the recent criteria for preclinical AD (Sperling et al., 2011) posited that biomarkers of synaptic dysfunction (e.g., fMRI and FDG) might become abnormal prior to detectable β-amyloid deposition in at-risk cohorts. Similarly, in their revised AD cascade model, Jack et al. (2013) raised the possibility that some measures of neural integrity (i.e., tau) might become abnormal prior to β-amyloid. There clearly is a need for further research into the predictive utility of β-amyloid vis-à-vis neuronal function in this pivotal stage of AD, including a thoughtful consideration of subgroups wherein one biomarker might be potentially more useful than the other.
To our knowledge, this is the first study to perform an integrated examination of the predictiveness of cognitive test scores and PET/MRI imaging data with respect to cognitive decline in cognitively healthy middle-aged adults. Although our design is less than ideal in that the imaging exams did not happen at the same time as the baseline cognitive assessment (there was a 5.7-year interval), our findings are nevertheless informative. For example, the observation that memory and neuronal function measures were comparatively more strongly associated with longitudinal cognitive decline are in accord with a recent multimodality study among persons with mild cognitive impairment (MCI) wherein glucose metabolism and episodic memory were, separately and jointly, the strongest predictors of conversion to AD (Landau et al., 2010). Episodic memory impairment is well-established as the chief cognitive feature of AD, and some prior studies have shown that early changes in episodic memory are detectable in cognitively-normal adults who go on to develop MCI or AD (Blacker et al., 2007; Elias et al., 2000). It was surprising, though, that a measure of visual memory was the best predictor of decline in our cohort. While the import of this is not immediately clear, a prior study (Kawas et al., 2003) from the Baltimore Longitudinal Study of Aging also found baseline visual memory to be predictive of AD, and a more recent study (Snitz et al., 2013) found that visual—but not verbal—memory declined precipitously among nondemented amyloid-positive subjects. Nonetheless, we cannot rule out the possibility that the BVMT finding was driven by its known shared variance with the RAVLT (our criterion measure) among cognitively-healthy subjects, even though the correlation between both measures within our sample was not particularly striking (e.g., r of .21 between BVMT Total and RAVLT total). We also noted with interest that the non-memory cognitive measure most predictive of decline in our sample was WRAT-III Reading, a measure of literacy. Literacy is considered a proxy for cognitive reserve (Stern, 2012), and several studies have now shown that higher cognitive reserve delays the onset of clinical impairments even in the context of marked neuropathological changes (Rentz et al., 2010). The relatively higher WRAT-III scores exhibited by our Stables suggest that this protective effect of cognitive reserve might be in effect within our sample.
Among the demographic factors examined for associations with our imaging biomarkers, the two most consistent findings were with age and sex. The age effect was expected given that age is the highest risk factor for AD, and that age-associated brain alterations are now well-documented (Drachman, 2006). The sex finding was surprising and perhaps intriguing. Although controversial, evidence from epidemiological and neuropathological studies suggests comparatively higher risk for AD in women versus men (Alzheimer’s Association, 2012; Corder et al., 2004). Relatedly, a growing number of publications (including some from our group) are providing evidence for a greater likelihood of AD-related brain changes among persons with a maternal FH of AD, even in midlife (Mosconi et al., 2009; Okonkwo, Xu, Oh, et al., 2012). While some reports have tied the disproportionate representation of women in AD case finding to estrogen-related mitochondrial toxicity (Vina & Lloret, 2010), there is scant consensus on these questions. Additional, well-controlled studies would be helpful in bringing clarity.
A key limitation of our study is the asynchrony between baseline cognitive assessment and the imaging exams. The temporal lag between these evaluations potentially created a situation wherein the baseline cognitive measures predicted decline prospectively whereas the imaging measures predicted decline retrospectively for most subjects. It is unclear the extent to which this asynchrony influenced our findings. As the WRAP cohort is ongoing, it will be of interest to repeat our analyses upon collection of additional longitudinal data such that “baseline” can be set at the same time point for all study measures. Another possible limitation of our study involves the use of cut-points (and the specific cut-point chosen) to define cognitive decline. No matter how carefully chosen, cut-points are often vulnerable to classification errors. It is arguable that a score of, say, −1.4 is substantively different than a score of −1.5. Still, cut-points are endemic to clinical research and practice. In addition, because the cut-point algorithm for determining decline was applied evenly across all subjects, it is expected that any resulting bias or classification errors would be stochastically—versus systematically—distributed in the sample. Lastly, we cannot rule out the possibility that the incipient cognitive decline we have observed in our sample is simply a marker for senescence rather than a signal for underlying AD especially given the observed associations with cognition/neuronal function and the lack thereof with amyloid (Jack et al., 2013). Additional follow up—anchored to clinical endpoints such as MCI—will allow us more definitively evaluate the prognostic utility of this early decline
In summary, this integrative, multimodality study found that, among cognitive measures, tests of episodic memory were the most sensitive to prospective cognitive decline in an at-risk middle-aged cohort whereas, among imaging measures, markers of neuronal function were more strongly associated with cognitive decline over a preceding epoch compared with β-amyloid deposition. Among several clinically-relevant indications, this finding suggests that early memory difficulties might be a useful harbinger of future decline. As the WRAP is an ongoing study, and prospective imaging and cognitive assessments are occurring, it will be of great interest to revisit the analyses presented here in the future, when all predictors of interest can be set to a uniform origin, thereby allowing a truly prospective comparison of imaging markers vis-à-vis cognitive measures with respect to future cognitive decline in this at-risk cohort.
Supplementary Material
Acknowledgments
This research was supported by NIA grants R01 AG021155 (SCJ), R01 AG027161 (MAS), P50 AG033514 (SA), P50 AG033514-S1 (OCO), and K23 AG045957 (OCO); by a Veterans Administration Merit Review Grant I01CX000165 (SCJ); and by a Clinical and Translational Science Award (UL1RR025011) to the University of Wisconsin, Madison. Portions of this research were supported by the Wisconsin Alumni Research Foundation, the Helen Bader Foundation, Northwestern Mutual Foundation, Extendicare Foundation, and from the Veterans Administration including facilities and resources at the Geriatric Research Education and Clinical Center of the William S. Middleton Memorial Veterans Hospital, Madison, WI.
We gratefully acknowledge the assistance of Dustin Wooten, PhD, Ansel Hillmer, MSc, and Andrew Higgins with PET data production and processing; and Sandra Harding, MS, and Jennifer Bond, BA, with study data collection. In addition, we would like to acknowledge the support of researchers and staff at the Waisman Center, University of Wisconsin–Madison, where the brain scans took place. Finally, we thank participants in the Wisconsin Registry for Alzheimer’s Prevention for their continued dedication.
Footnotes
Okonkwo, Oh, Koscik, Jonaitis, Cleary, Dowling, Bendlin, LaRue, Hermann, Barnhart, Murali, Carlsson, Gallagher, Asthana, Sager, Christian, and Johnson report no conflicts of interest.
Rowley has provided consultation to and/or received honoraria from GE Healthcare, Bracco, Lundbeck, HL Gore, and Eli Lilly.
References
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia. 2012;8(2):131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Benedict RHB. Brief Visuospatial Memory Test-Revised. Odessa, FL: Psychological Assessment Resources, Inc; 1997. [Google Scholar]
- Benton AL. Neuropsychological assessment. Annual Review of Psychology. 1994;45:1–23. doi: 10.1146/annurev.ps.45.020194.000245. [DOI] [PubMed] [Google Scholar]
- Benton AL, Hamsher K, Sivan AB. Multilingual Aphasia Examination. Iowa City, IA: AJA Associates; 1976. [Google Scholar]
- Blacker D, Lee H, Muzikansky A, Martin EC, Tanzi R, McArdle JJ, Albert M. Neuropsychological measures in normal individuals that predict subsequent cognitive decline. Archives of Neurology. 2007;64(6):862–871. doi: 10.1001/archneur.64.6.862. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Mintun MA. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. Journal of Neuroscience. 2005;25(34):7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, Morris JC. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Archives of Neurology. 2009;66(12):1557–1562. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G. Alzheimer disease: Abeta-independent processes-rethinking preclinical AD. Nature Reviews Neurology. 2013;9(3):123–124. doi: 10.1038/nrneurol.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G, La Joie R, Villain N, Perrotin A, de La Sayette V, Eustache F, Vandenberghe R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. NeuroImage: Clinical. 2013;2(0):356–365. doi: 10.1016/j.nicl.2013.02.006. http://dx.doi.org/10.1016/j.nicl.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian BT, Vandehey NT, Floberg JM, Mistretta CA. Dynamic PET denoising with HYPR processing. Journal of Nuclear Medicine. 2010;51(7):1147–1154. doi: 10.2967/jnumed.109.073999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Annals of the New York Academy of Sciences. 2004;1019:24–28. doi: 10.1196/annals.1297.005. [DOI] [PubMed] [Google Scholar]
- Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, Dale AM. Amyloid-beta--associated clinical decline occurs only in the presence of elevated P-tau. Archives of Neurology. 2012;69(6):709–713. doi: 10.1001/archneurol.2011.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DA. Aging of the brain, entropy, and Alzheimer disease. Neurology. 2006;67(8):1340–1352. doi: 10.1212/01.wnl.0000240127.89601.83. [DOI] [PubMed] [Google Scholar]
- Elias MF, Beiser A, Wolf PA, Au R, White RF, D’Agostino RB. The preclinical phase of alzheimer disease: A 22-year prospective study of the Framingham Cohort. Archives of Neurology. 2000;57(6):808–813. doi: 10.1001/archneur.57.6.808. [DOI] [PubMed] [Google Scholar]
- Ewers M, Insel P, Jagust WJ, Shaw L, Trojanowski JQ, Aisen P, Weiner MW. CSF biomarker and PIB-PET-derived beta-amyloid signature predicts metabolic, gray matter, and cognitive changes in nondemented subjects. Cerebral Cortex. 2012;22(9):1993–2004. doi: 10.1093/cercor/bhr271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floberg JM, Mistretta CA, Weichert JP, Hall LT, Holden JE, Christian BT. Improved kinetic analysis of dynamic PET data with optimized HYPR-LR. Medical Physics. 2012;39(6):3319–3331. doi: 10.1118/1.4718669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. Mini-mental state: A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Chelune GJ, Talley JL, Kay GG, Curtiss G. Wisconsin Card Sorting Test Manual. Odessa, FL: Psychological Assessment Resources, Inc; 1993. [Google Scholar]
- Hedden T, Oh H, Younger AP, Patel TA. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology. 2013;80(14):1341–1348. doi: 10.1212/WNL.0b013e31828ab35d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers W, Vannini P, Sperling RA, C MP, Cabeza R, Daselaar SM. Explaining the encoding/retrieval flip: memory-related deactivations and activations in the posteromedial cortex. Neuropsychologia. 2012;50(14):3764–3774. doi: 10.1016/j.neuropsychologia.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurology. 2013;12(2):207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurology. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, Koeppe RA. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimer’s & Dementia. 2010;6(3):221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Schmitz TW, Moritz CH, Meyerand ME, Rowley HA, Alexander AL, Alexander GE. Activation of brain regions vulnerable to Alzheimer’s disease: the effect of mild cognitive impairment. Neurobiology of Aging. 2006;27(11):1604–1612. doi: 10.1016/j.neurobiolaging.2005.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- Kawas CH, Corrada MM, Brookmeyer R, Morrison A, Resnick SM, Zonderman AB, Arenberg D. Visual memory predicts Alzheimer’s disease more than a decade before diagnosis. Neurology. 2003;60(7):1089–1093. doi: 10.1212/01.wnl.0000055813.36504.bf. [DOI] [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Jagust WJ. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiology of Aging. 2011;32(7):1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, Jagust WJ. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75(3):230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, Jagust WJ. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Annals of Neurology. 2012;72(4):578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech R, Sharp DJ. The role of the posterior cingulate cortex in cognition and disease. Brain. 2013 doi: 10.1093/brain/awt162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YY, Ellis KA, Pietrzak RH, Ames D, Darby D, Harrington K, Maruff P. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology. 2012;79(16):1645–1652. doi: 10.1212/WNL.0b013e31826e9ae6. [DOI] [PubMed] [Google Scholar]
- Lo RY, Hubbard AE, Shaw LM, Trojanowski JQ, Petersen RC, Aisen PS, Jagust WJ. Longitudinal change of biomarkers in cognitive decline. Archives of Neurology. 2011;68(10):1257–1266. doi: 10.1001/archneurol.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–1239. doi: 10.1016/s1053-8119(03)00169-1. S1053811903001691 [pii] [DOI] [PubMed] [Google Scholar]
- Manly JJ, Schupf N, Tang MX, Stern Y. Cognitive decline and literacy among ethnically diverse elders. Journal of Geriatric Psychiatry and Neurology. 2005;18(4):213–217. doi: 10.1177/0891988705281868. [DOI] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Mintun MA. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Archives of Neurology. 2009;66(12):1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, de Leon MJ. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72(6):513–520. doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JL, O’Keefe KM, LaViolette PS, DeLuca AN, Blacker D, Dickerson BC, Sperling RA. Longitudinal fMRI in elderly reveals loss of hippocampal activation with clinical decline. Neurology. 2010;74(24):1969–1976. doi: 10.1212/WNL.0b013e3181e3966e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo OC, Xu G, Dowling NM, Bendlin BB, Larue A, Hermann BP, Johnson SC. Family history of Alzheimer disease predicts hippocampal atrophy in healthy middle-aged adults. Neurology. 2012;78(22):1769–1776. doi: 10.1212/WNL.0b013e3182583047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo OC, Xu G, Oh JM, Dowling NM, Carlsson CM, Gallagher CL, Johnson SC. Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer’s disease. Cerebral Cortex. 2012 doi: 10.1093/cercor/bhs381. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Mathis CA. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. Journal of Cerebral Blood Flow and Metabolism. 2005;25(11):1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- Rami L, Sala-Llonch R, Sole-Padulles C, Fortea J, Olives J, Llado A, Molinuevo JL. Distinct functional activity of the precuneus and posterior cingulate cortex during encoding in the preclinical stage of Alzheimer’s disease. Journal of Alzheimer’s Disease. 2012;31(3):517–526. doi: 10.3233/JAD-2012-120223. [DOI] [PubMed] [Google Scholar]
- Reitan R, Wolfson D. The Halstead-Reitan Neuropsychological Test Battery: Theory and clinical interpretation. Tucson: Neuropsychology Press; 1993. [Google Scholar]
- Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, Johnson KA. Cognition, reserve, and amyloid deposition in normal aging. Annals of Neurology. 2010;67(3):353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, Wong DF. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74(10):807–815. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer’s disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer’s Prevention. Journal of Geriatric Psychiatry and Neurology. 2005;18(4):245–249. doi: 10.1177/0891988705281882. [DOI] [PubMed] [Google Scholar]
- Schmidt M. Rey Auditory Verbal Learning Test: A Handbook. Torrance, CA: Western Psychological Services; 1996. [Google Scholar]
- Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D’Angelo G, Mintun MA. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. Journal of Neuroscience. 2010;30(50):17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitz BE, Weissfeld LA, Lopez OL, Kuller LH, Saxton J, Singhabahu DM, Dekosky ST. Cognitive trajectories associated with beta-amyloid deposition in the oldest-old without dementia. Neurology. 2013 doi: 10.1212/WNL.0b013e31828c2fc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurology. 2012;11(11):1006–1012. doi: 10.1016/S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E, Sherman EMS, Spreen O. A compendium of neuropsychological tests. 3. Oxford: Oxford University Press; 2006. [Google Scholar]
- Trenerry M, Crosson B, DeBoe J, Leber L. Stroop Neuropsychological Screening Test. Odessa, FL: Psychological Assessment Resources, Inc; 1989. [Google Scholar]
- Vannini P, O’Brien J, O’Keefe K, Pihlajamaki M, Laviolette P, Sperling RA. What goes down must come up: role of the posteromedial cortices in encoding and retrieval. Cerebral Cortex. 2011;21(1):22–34. doi: 10.1093/cercor/bhq051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Masters CL. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurology. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Vina J, Lloret A. Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. Journal of Alzheimer’s Disease. 2010;20(Suppl 2):S527–533. doi: 10.3233/JAD-2010-100501. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Memory Scale - Revised edition. San Antonio, TX: The Psychological Corporation; 1987. [Google Scholar]
- Wechsler D. WAIS-III: Wechsler Adult Intelligence Scale. 3. San Antonio, TX: The Psychological Corporation; 1997. [Google Scholar]
- Wechsler D. Wechsler Abbreviated Scale of Intelligence. San Antonio, TX: The Psychological Corporation; 1999. [Google Scholar]
- Wilkinson G. Wide Range Achievement Test Administration Manual. 3. Wilmington, Delaware: Wide Range Incorporated; 1993. [Google Scholar]
- Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer’s disease neurodegenerative biomarkers are associated with decreased cognitive function but not beta-amyloid in cognitively normal older individuals. Journal of Neuroscience. 2013;33(13):5553–5563. doi: 10.1523/JNEUROSCI.4409-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Oh H, Mormino EC, Markley C, Landau SM, Jagust WJ. The effect of amyloid beta on cognitive decline is modulated by neural integrity in cognitively normal elderly. Alzheimer’s & Dementia. 2013 doi: 10.1016/j.jalz.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, McLaren DG, Ries ML, Fitzgerald ME, Bendlin BB, Rowley HA, Johnson SC. The influence of parental history of Alzheimer’s disease and apolipoprotein E epsilon4 on the BOLD signal during recognition memory. Brain. 2009;132(Pt 2):383–391. doi: 10.1093/brain/awn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Obuchowski N, McClish D. Statistical Methods in Diagnostic Medicine. 2. Hoboken, NJ: John Wiley & Sons, Inc; 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.