

Abstract
Velo-cardio-facial/DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a congenital anomaly disorder characterized by craniofacial anomalies including velo-pharyngeal insufficiency, facial muscle hypotonia and feeding difficulties, in part due to hypoplasia of the branchiomeric muscles. Inactivation of both alleles of mouse Tbx1, encoding a T-box transcription factor, deleted on chromosome 22q11.2, results in reduction or loss of branchiomeric muscles. To identify downstream pathways, we performed gene profiling of microdissected pharyngeal arch one (PA1) from Tbx1+/+ and Tbx1−/− embryos at stages E9.5 (somites 20–25) and E10.5 (somites 30–35). Basic helix–loop–helix (bHLH) transcription factors were reduced, while secondary heart field genes were increased in expression early and were replaced by an increase in expression of cellular stress response genes later, suggesting a change in gene expression patterns or cell populations. Lineage tracing studies using Mesp1Cre and T-Cre drivers showed that core mesoderm cells within PA1 were present at E9.5 but were greatly reduced by E10.5 in Tbx1−/− embryos. Using Tbx1Cre knock-in mice, we found that cells are lost due to apoptosis, consistent with increase in expression of cellular stress response genes at E10.5. To determine whether Tbx1 is required autonomously in the core mesoderm, we used Mesp1Cre and T-Cre mesodermal drivers in combination with inactivate Tbx1 and found reduction or loss of branchiomeric muscles from PA1. These mechanistic studies inform us that Tbx1 is required upstream of key myogenic genes needed for core mesoderm cell survival and fate, between E9.5 and E10.5, resulting in formation of the branchiomeric muscles.
INTRODUCTION
Velo-cardio-facial syndrome (MIM # 192430)/DiGeorge syndrome (MIM# 188400), also known as 22q11.2 deletion syndrome (22q11DS), is caused by a 1.5–3 million base pair hemizygous 22q11.2 deletion and is characterized by cardiac, immune and craniofacial anomalies. Craniofacial malformations consist of submucous or overt cleft palate, platybasia and velo-pharyngeal insufficiency (VPI) (1–3). VPI symptoms include feeding or swallowing difficulties during infancy and hypernasal speech later. One cause of VPI is the existence of a submucous cleft palate and/or muscle hypotonia, both present in most 22q11DS patients (3–5). In addition to VPI, some 22q11DS children have asymmetric crying facies, which is characterized by drooping of one side of the mouth during crying. It is thought that this phenotype is caused in part by branchiomeric muscle hypoplasia (6).
The branchiomeric muscles are specific to the head and neck, but do not include extraocular muscles or the tongue. When compared with the somites in the body, branchiomeric muscles derive from unsegmented cranial paraxial mesoderm in the central core of the pharyngeal arches during embryogenesis (7,8). The core mesoderm cells within pharyngeal arch 1 (PA1) form the muscles of mastication required for chewing. Basic helix–loop–helix (bHLH) transcription factors are central to branchiomeric myogenesis. Four bHLH transcription factor genes, Tcf21 (Capsulin), Msc (MyoR), Myf5 and MyoD, play a critical role in craniofacial muscle formation. Tcf21 and Msc work redundantly to regulate the first step of the specification of the branchiomeric muscles; in their combined absence, the muscles do not form from the core mesoderm Tcf21 and Msc act upstream of Myf5 and MyoD needed for differentiation (9,10).
In addition to the bHLH transcription factors, additional transcription factor genes play critical roles in forming the branchiomeric muscles and they include Pitx2 and Isl1 (11–16). Pitx2 is required early for craniofacial muscle development (14,15). Lhx2 encodes another homeodomain transcription factor required for branchiomeric muscle development (17). Thus far, Tcf21, Msc, Pitx2, Isl1 and Lhx2 are the known key transcription factor genes required for early craniofacial muscle development upstream of Myf5 and MyoD; none lies in the 22q11.2 region deleted in affected individuals.
The TBX1 gene is located within the 22q11.2 region that is hemizygously deleted in patients. It encodes a transcription factor that is part of the T-box gene family. Haploinsufficiency of TBX1 is believed to be responsible for most of the physical defects in 22q11DS patients, including craniofacial anomalies (18–20). Mouse models have been useful in dissecting Tbx1 function during embryogenesis. Tbx1+/− mice have mild defects at reduced penetrance, while Tbx1−/− mice die at birth with overt cleft palate and reduced or missing branchiomeric muscles, absent thymus and parathyroid glands and a single cardiac outflow tract instead of a separate aorta and pulmonary trunk (18–20). The distal pharyngeal arches do not form in Tbx1−/− mutant embryos, and thus, their derivatives do not form. The first pharyngeal arch is the only one present in mutant embryos and it appears grossly normal. The muscles of mastication form from the mandibular portion of pharyngeal arch one (PA1) and include the masseter, pterygoid and temporalis muscles, which are intermittently absent in Tbx1−/− embryos (21). Relevant to this, mouse genetic studies suggest that Tbx1 acts downstream of Tcf21, Pitx2 and Isl1 and upstream of Lhx2, Myf5 and MyoD (14,17,21,22). Tbx1 is not required for initial core mesoderm formation or specification, because core mesodermal cells are present at E9.5, but it is required later to pattern the muscles (21). The mechanism responsible for craniofacial muscle loss in Tbx1 mutant embryos is unknown.
Major goals here are to identify new genes and pathways downstream of Tbx1 so as to determine why the muscles of mastication do not form in PA1 and finally, whether Tbx1 is required cell autonomously, as was raised in the past for chick (23), but not conclusively proved in mouse models. We found that Tbx1 is required for determining cell fate as well as survival, between E9.5 and E10.5, in the core mesoderm needed autonomously to form the muscles of mastication. This provides mechanistic evidence explaining why the craniofacial muscles do not form when Tbx1 is inactivated.
RESULTS
Expression of PA1 core mesoderm genes reduced in Tbx1−/− embryos
In situ hybridization was performed on Tcf21, Msc, Myf5 and MyoD in Tbx1+/+ and Tbx1−/− mutant embryos at E9.5 (somites, 20–25) and E10.5 (somites, 30–35) as shown in Figure 1. Whole-mount in situ hybridization at E10.5 was followed by analysis of tissue sections to visualize the pattern of expression inside the embryos (Fig. 1). Expression appeared to be in the core mesoderm. All the four genes were expressed in PA1 at both stages in Tbx1+/+ embryos, except MyoD, whose expression was not detected in PA1 at E9.5 (Fig. 1). The Tcf21 and Msc genes were expressed in PA1 in Tbx1−/− embryos at E9.5, but not E10.5 (Fig. 1). In contrast, Myf5 was absent in null mutant embryos at E9.5 and E10.5 (Fig. 1). Frontal whole-mount images are shown depicting gene expression patterns with respect to Tbx1 showing expression of the four bHLH genes in a similar region of PA1 (Fig. 1).
Figure 1.

Basic HLH transcription factor genes, Tcf21, Msc, Myf5 and MyoD, in Tbx1+/+ versus Tbx1−/− embryos at E9.5 and E10.5. Lateral views of Tbx1+/+ versus Tbx1−/− embryos at E9.5 (somite count, 20–25) and E10.5 (somite count, 30–40) are shown following whole-mount in situ hybridization with probes for Tcf21, Msc, Myf5 and MyoD. Coronal histological sections of the PA1 region from E10.5 embryos are shown next to the whole-mount images. Arrow points to core mesoderm expression domain of Tcf21 in PA1. The image below shows whole-mount coronal views of the first and second pharyngeal arch in Tbx1+/+ embryos at E9.5 and E10.5 after whole-mount in situ hybridization, using antisense probes to the four bHLH genes. Cartoons are shown below to depict expression patterns of individual probes.
To build molecular connections between Tbx1 and the four bHLH genes as well as to identify new genes for branchiomeric myogenesis, gene expression profiling was performed from microdissected PA1 tissues from Tbx1+/+ and Tbx1−/− mutant embryos at E9.5. We did not perform microarray analysis on Tbx1+/− embryos because they have no obvious craniofacial muscle defects (21,24). We first examined genes that were reduced in expression in the microarrays (Supplementary Material, Table S1). Ingenuity pathway analysis (IPA) and hand curation was done to create a gene network as shown in Figure 2A. The gene network included the genes that were reduced in expression in Tbx1−/− embryos (gray fill; Fig. 2A), the four bHLH genes discussed above (Fig. 1) and connecting genes identified by IPA irrespective of known function (Fig. 2A). We also evaluated gene expression levels in PA1 by quantitative reverse transcriptase–polymerase chain reaction (qRT–PCR; Fig. 2B). There are some differences between in situ hybridization, microarray and qRT–PCR results, but overall they showed a consistent differential expression pattern. For example, expression of Tcf21 as detected by whole-mount in situ hybridization was not visibly changed in Tbx1−/− embryos at E9.5, but was lost in mutant embryos at E10.5 (Fig. 1). In contrast, Tcf21 expression was significantly reduced in Tbx1−/− embryos by microarray (Supplementary Material, Table S1) and qRT–PCR (Fig. 2B) analyses at E9.5 and E10.5. MyoD expression was not detected by in situ hybridization analysis of Tbx1+/+ embryos, nor was expression detected in the microarrays at E9.5 (somites 20–25). MyoD probes failed in the microarray at E10.5, possibly due to technical problems with the specific probe. Expression of MyoD was detected by qRT–PCR and it was reduced in Tbx1−/− embryos at E9.5 and E10.5 (Fig. 2B).
Figure 2.

Gene network of PA1 genes reduced in expression at E9.5 in Tbx1−/− embryos. (A) The network of genes that were reduced in expression in Tbx1−/− embryos when compared with Tbx1+/+ embryos (>1.5-fold change, P < 0.05) was created using IPA (http://www.ingenuity.com/products/ipa). The top network is depicted here. The genes reduced in expression are highlighted by gray fill, and additional genes connected to them based on IPA network analysis are indicated as diamonds. The lines indicate relationships between genes that could directly or indirectly interact. Genes reduced in expression but not linked to this network can be found in the Supplementary Material, Table S1. (B) Quantitative RT-PCR of PA1 tissue from Tbx1+/+ and Tbx1−/− embryos at E9.5. The X-axis indicates genes analyzed and Y-axis represents fold change of expression in PA1 tissue from Tbx1−/− relative to Tbx1+/+controls at E9.5. Error bars, s.e.m. (n = 3). (C) Genes reduced in expression in Tbx1−/− embryos at E9.5 were analyzed at the Genemania website (http://www.genemania.org/) to identify top enriched functions. We provide the FDR (false discovery rate) score estimated in Genemania by the Benjamini–Hochberg correction.
The IPA network contains genes that have been previously found to be genetically or molecularly connected to the differentially expressed genes in this study, but they may not show expression change in our system. This is because the IPA network builds connections based upon genes and their relationships from papers reported in the literature. For example, Shh and Ash2l were part of the IPA network but were not changed in expression by microarray or qRT–PCR. We included a list of top gene ontology networks identified from IPA analysis (Fig. 2C). The most significant category identified was embryonic organ morphogenesis with 16 of 215 genes connected to this particular network (Fig. 2C). Similarly, genes important for mesenchymal and neural crest cell development were reduced in expression (Fig. 2C).
We performed a similar gene profiling experiment at E10.5 to determine whether the same or different genes were reduced in expression (Supplementary Material, Table S2). We found that 20 genes were changed similarly between Tbx1+/+ and Tbx1−/− embryos at stages E9.5 and E10.5. A subset of genes changed in the microarray as well as the four bHLH genes were tested by qRT–PCR (Supplementary Material, Fig. S1; Supplementary Material, Table S2). The qRT–PCR results (Supplementary Material, Fig. S1) were consistent with the microarray results, except for the expression of Msc and MyoD, for which were not significantly changed (or detected; MyoD) by microarray analysis, but there expression was reduced by qRT–PCR analysis. Based upon the gene lists from E9.5 and E10.5 (Supplementary Material, Tables S1 and S2) and qRT–PCR analysis (Fig. 2B; Supplementary Material, Fig. S1), we performed in situ hybridization to detect patterns of Foxc1, Foxc2, Sim2, Lhx2, Lrrn1 and Chrdl1 gene expression in Tbx1+/+ and Tbx1−/− embryos.
We evaluated the spatial and temporal pattern of expression of these six genes in PA1 at E9.5 and E10.5. We separated the analysis to those known to be expressed in the core mesoderm from those expressed in the neural crest mesoderm. Two of the genes, Lhx2 and Lrrn1, were expressed in the core mesoderm at E9.5 and three of the genes, Lhx2, Chrdl1 and Lrrn1, were expressed at E10.5 in Tbx1+/+ embryos (Fig. 3). The gene Lhx2 encodes a LIM homeobox transcription factor that is required for craniofacial muscle formation. Recent studies showed that Lhx2 acts downstream of Tbx1, Pitx2 and Tcf21 in the core mesoderm for branchiomeric muscle formation (17); however, we wanted to check expression in our Tbx1 mutant allele. Expression of the Pitx2 gene, although not changed in the PA1 microarray study, was slightly but significantly increased by qRT–PCR analysis (Fig. 2B). This data was included in the network shown in Figure 2A. Lhx2 was expressed in the core mesoderm in Tbx1+/+ embryos at E9.5 and E10.5 but absent from PA1 in Tbx1−/− embryos at both stages (Fig. 3).
Figure 3.

Tbx1, Lhx2, Lrrn1 and Chrdl1 expression in the core mesoderm in Tbx1+/+ versus Tbx1−/− embryos at E9.5 and E10.5. Lateral views of whole-mount in situ hybridization of Tbx1+/+ and Tbx1−/− embryos at E9.5 and E10.5 with antisense probes for Tbx1 (Wt only), Lhx2, Lrrn1 and Chrdl1. Coronal tissue sections from a subset of the whole-mount stained embryos are shown adjacent to the whole-mount images. Chrdl1 is not expressed in the core mesoderm in Tbx1+/+ embryos at E9.5; thus, no images are shown for Tbx1−/− embryos at this stage. Lhx2, Lrrn1 and Chrdl1 were not expressed in the core mesoderm in Tbx1−/− embryos so only whole-mount images are shown.
The Lrrn1 (neuronal leucine-rich repeat-1, NLRR-1) gene encodes a transmembrane glycoprotein localized on the cell surface. The gene is expressed in developing somites (25). The Lrrn1 gene was weakly expressed in the core mesoderm of PA1 in Tbx1+/+ embryos at E9.5 and strongly expressed at E10.5. Expression of Lrrn1 was absent in Tbx1−/− embryos at E9.5 and E10.5 (Supplementary Material, Tables S1 and 2; Fig. 3). Of interest, a previous report demonstrated that Lrrn1 expression was reduced in MyoD−/− myogenic cells in culture (26). This suggests a possible functional role of Lrrn1 in branchiomeric myogenesis.
The Chrdl1 (Chordin like 1) gene is a closely related Bone morphogenic protein (Bmp) antagonist gene family member to Chordin (27). Chrdl1 is expressed in the neural plate, forebrain and pharyngeal apparatus core mesoderm among other tissues (27). Chrdl1 was not specifically expressed in PA1 in Tbx1+/+ embryos at E9.5 as determined by in situ hybridization analysis (Fig. 3). Although we did not find specific PA1 expression by in situ hybridization in Tbx1+/+ embryos at E9.5, core PA1 expression was observed in a different report (26). This difference could be due to slight embryo stage differences, mouse strain or probe quality differences. On the other hand, specific PA1 expression by this method was observed at E10.5 (Fig. 3), similar to what was previously reported (26). PA1 expression was lost in Tbx1−/− embryos at E10.5, although forelimb bud expression remained (Fig. 3). Chrdl1 expression as determined by microarray and qRT–PCR analysis was present in PA1 of Tbx1+/+ embryos at E9.5 and E10.5 and it was reduced in Tbx1−/− embryos at both stages (Supplementary Material, Tables S1 and 2; Fig. 2B; Supplementary Material, Fig. S1).
The Fgf10 gene, important downstream of Tbx1 in multiple tissues, was previously shown to be expressed in the core mesoderm of the pharyngeal arches (28,29) and is part of the gene network shown in Figure 2A. Fgf10 shows a very small reduction of expression in PA1 in Tbx1−/− embryos by microarray analysis (Supplementary Material, Table S1) and qRT–PCR at E9.5 (Fig. 2B). Consistent with this, we previously found that Fgf10 is present in the core mesoderm in Tbx1+/+ embryos but diffusely expressed in PA1 tissue of Tbx1−/− embryos at E9.5, rather than being absent (30).
Reduced expression of genes for craniofacial development in Tbx1−/− embryos
Neural crest cell (NCC) mesoderm lies in the first pharyngeal arch between the core mesoderm and surrounding epithelia. The Foxc1 and Foxc2 genes encode proteins that belong to the forkhead family of transcription factors. Both genes act redundantly for cardiac and vascular development in neural crest mesoderm as well as somitogenesis (31). As mentioned above, we found that expression of the Foxc1 gene was reduced in Tbx1−/− when compared with Tbx1+/+ embryos at E9.5 and E10.5 by both microarray and qRT–PCR analyses (Supplementary Material, Figs S1 and 2B). We performed in situ hybridization to determine whether the pattern of expression changed in Tbx1−/− embryos at E9.5 and at E10.5. The Foxc1 gene was broadly expressed in Tbx1+/+ embryos at E9.5 and more specifically expressed in the caudal PA1 at E10.5 (Fig. 4A and B). Expression was similar between Tbx1+/+ when compared with Tbx1−/− embryos at E9.5, but localized to a smaller region in Tbx1−/− embryos at E10.5 (Fig. 4A).
Figure 4.
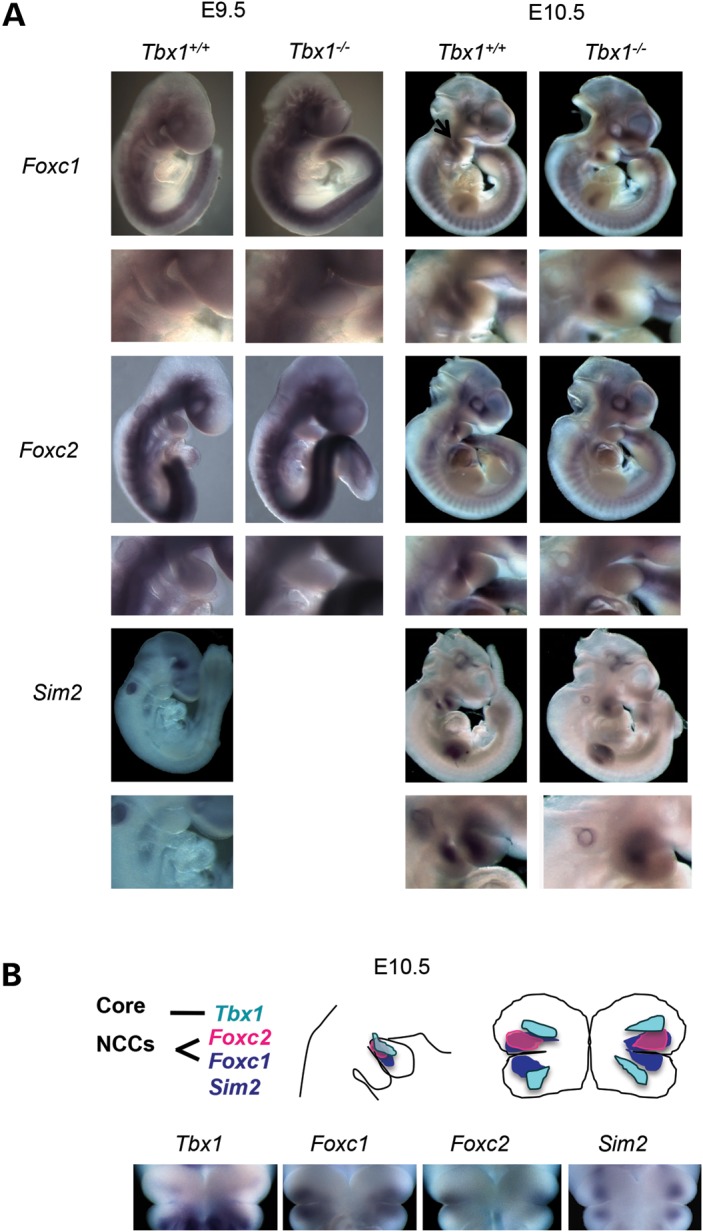
Foxc1, Foxc2 and Sim2 expression in PA1 in Tbx1+/+ versus Tbx1−/− embryos at E9.5 and E10.5. (A) Lateral views of whole-mount in situ hybridization of probes for Foxc1, Foxc2 and Sim2 in Tbx1+/+ and Tbx1−/− embryos at E9.5 and E10.5. Enlarged lateral views of the PA1 region in the whole-mount specimens is shown below each whole embryo view. (B) Cartoon of lateral and coronal views (shown below) of Tbx1, Foxc1, Foxc2 and Sim2. Expression domain of Tbx1 is in the core mesoderm.
In contrast to Foxc1, we did not detect a change in expression of Foxc2 at E9.5 in the microarray experiment (Supplementary Material, Table S1) and did not perform qRT–PCR at this stage. We observed a similar diffuse pattern of expression in PA1 for Foxc2, as for Foxc1 in Tbx1+/+ and Tbx1−/− embryos by in situ hybridization at E9.5 (Fig. 4A). Consistent with microarray and qRT–PCR findings (Supplementary Material, Table S2 and Fig. S1), there was loss of expression in PA1 at E10.5 in Tbx1−/− embryos by in situ hybridization analysis (Fig. 4A; Supplementary Material, Fig. S1).
The Sim2 gene encodes a bHLH transcription factor gene (single-minded, Drosophila) expressed in the pharyngeal arches and is required to form the secondary palate, tongue and pterygoid process (32). The muscles of mastication are still present in Sim2−/− mutant mice, and thus, it is not required for branchiomeric myogenesis (32). We did not observe expression of Sim2 by in situ hybridization or microarray analysis in Tbx1+/+ embryos at E9.5 (Fig. 4A). Sim2 is expressed in the pharyngeal arches at E10.5 in Tbx1+/+ embryos. It appears to be adjacent to the Tbx1 expression domain, although we cannot rule out that Sim2 expression may partially overlap with that of Tbx1 (Fig. 4B). The area of expression is reduced in Tbx1−/− embryos but not absent at this stage (Fig. 4). At E10.5, expression of Sim2, as determined by microarray (Supplementary Material, Table S2) and qRT–PCR (Supplementary Material, Fig. S1) analysis, was reduced in Tbx1−/− embryos. The specific patterns of expression in PA1 tissue of Foxc1, Foxc2, Sim2 and Tbx1 are shown in lateral and frontal views in the cartoon in Figure 4B. Changes in expression of all three genes implicate possible roles downstream of Tbx1 in either craniofacial muscle and/or bone development.
Increased expression of PA1 genes in Tbx1−/− embryos
We examined genes increased in expression in Tbx1+/+ versus Tbx1−/− mutant embryos at E9.5 by gene expression microarray analysis (Fig. 5). As for genes reduced in expression described above, we performed gene network analysis (Fig. 5A). Many of the genes increased in expression at E9.5 (gray fill; Fig. 5A) are known genes for cardiac development and differentiation (Fig. 5B). A subset of the genes that were increased in expression in the microarray in Tbx1−/− mutant embryos at E9.5 were tested and confirmed by qRT–PCR of dissected PA1 tissue (Fig. 2B). Based upon the results, it is possible that loss of Tbx1 results in a change in fate of the core mesoderm from the craniofacial pattern to the cardiac differentiation program. To test this, we performed in situ hybridization analysis. Ectopic expression of Gata4, Gata5, Gata6 and Tbx20 transcription factor genes extended rostrally to the caudal tip of PA1, but were not detected in the core mesoderm (Fig. 5C). This suggests that increase in expression may not be due to cell fate change in the core mesoderm from the craniofacial to the cardiac specific developmental pathways. Furthermore, additional cardiac specific genes were also ectopically expressed rostrally in Tbx1−/− embryos, such as Myl7, Actc1 and Tbx5 (33). They were expressed in the caudal part of PA1 but were not detected in the core mesoderm (33).
Figure 5.

Ectopic expression of cardiac genes in PA1 in Tbx1+/+ versus Tbx1−/− embryos at E9.5. (A) The network downstream of Tbx1 was created using genes that had a >1.5-fold increase, P < 0.05 by microarray analysis (gray fill). Genes not changed in expression significantly in the microarray or qRT–PCR assay, but connected to those based upon the software generated connections, are indicated as uncolored circles. The lines indicate relationships between genes that could be direct or indirect in nature. Genes increased in expression but not linked to the network were removed but can be found in the Supplementary Material, Table S1. (B) Genes increased in expression in Tbx1−/− embryos at E9.5 were analyzed at the Genemania website to identify top enriched functions. We provide the FDR score estimated in Genemania by the Benjamini–Hochberg correction. (C) Whole-mount in situ hybridization was performed on in Tbx1+/+ versus Tbx1−/− embryos at E9.5 with probes to the genes indicated. Tissue sections are shown adjacent to whole embryo images. The arrows in Tbx20 images point to regions of ectopic expression. Regions of ectopic expression of Gata5 and Gata6 in Tbx1+/+ versus Tbx1−/− embryos are shown as arrows in the cropped images.
At E10.5, we found a different set of genes increased in expression by gene profiling of PA1 tissue in Tbx1−/− embryos versus Tbx1+/+ embryos. We detected enrichment of stress response and glucose metabolism genes (Fig. 6). Four genes were increased in expression in the microarrays at E9.5 and E10.5 in PA1 tissue in Tbx1−/− embryos: Mapk13, Rarres2, Lipg and Vldlr (Supplementary Material, Table S2). We validated enhanced expression of Mapk13 at E9.5 and E10.5 in Tbx1−/− mutant embryos by qRT–PCR, albeit with some differences in the extent of upregulation in expression between microarray and the qRT–PCR assays (Fig. 2B; Supplementary Material, Fig. S1). Mapk13 encodes mitogen-activated protein kinase 13 (p38 delta) and it is important for promoting cell survival in response to cell stress (34). Rarres2 encodes a retinoic acid receptor responder (tazarotene induced) 2, also referred to as Chemerin, an adipokine, and is a retinoic acid responsive gene (29). Lipg encodes a lipase and Vldlr encodes very low density lipoprotein receptor and it is upregulated in response to hypoxia (35). Several genes increased in expression belong to the Vegf, hypoxia response pathway and Akt, cellular survival pathways as indicated (Fig. 6). This may implicate the presence of cellular stress or apoptosis.
Figure 6.

Ectopic expression of hypoxia and stress response pathway genes in PA1 in Tbx1+/+ versus Tbx1−/− embryos at E10.5. (A) The network of genes that were reduced in expression in Tbx1−/− embryos when compared with Tbx1+/+ embryos (>1.5-fold change, P < 0.05) was created using IPA. The top network is depicted here. The genes increased in expression are highlighted in orange. The lines indicate relationships between genes that could directly or indirectly interact. Genes increased in expression but not linked to this network can be found in the Supplementary Material, Table S2. (B) Genes increased in expression in Tbx1−/− embryos at E10.5 were uploaded to the Genemania website to identify top enriched functions.
Core mesodermal tissue is lost in Tbx1−/− embryos between E9.5 and E10.5
To determine the embryonic stage when tissue forming these muscles is reduced or lost in Tbx1−/− mutant embryos, we crossed T-Cre; Mesp1Cre/+; Tbx1+/− mice with RCEEGFP/EGFP; Tbx1+/− mice and dissected embryos to perform lineage tracing. We combined the two mesodermal Cre drivers together to ensure more complete fate mapping of the lineage (Meso-Cre). Fortunately, neither T-Cre nor Mesp1Cre/+ results in ectopic loxP site recombination in vivo (28,36). At E9.5, the mesodermal lineage was seen in the pharyngeal apparatus and heart, as expected (Fig. 7I–P). Core mesodermal tissue was apparent in PA1 in Tbx1+/+ and Tbx1−/− embryos (Fig. 7K, L, O and P). Existence of core mesodermal cells (21) is supported by the presence of Tcf21 and Msc mRNA expression at E9.5 in Tbx1−/− embryos (Fig. 1).
Figure 7.

Mesodermal fate mapping in Tbx1+/+ versus Tbx1−/− embryos at E9.5 and E10.5. Mesodermal fate mapping was performed using Mesp1Cre with T-Cre alleles and RCEEGFP/+ mice in Tbx1+/+ versus Tbx1−/− littermates at E9.5 (I–P) and E10.5 (A–H). Lateral views of Tbx1+/+ (A–D and I–L) and Tbx1−/− (E–H and M–P) mutants are shown as bright (A, E, I and M) and darkfield (B, F, J and N) views. The red arrow points to the region of the presence or absence of core mesodermal tissue in Tbx1+/+ versus Tbx1−/− at E9.5 and E10.5. An enlarged image of the PA1 region is shown to the right of the images (C, G, K and O). On the far right are coronal cryosections of the same embryos depicting the core mesodermal cells (D, H, L and P). Dapi stain was used to visualize PA1 at E9.5 (L and P).
The situation was different at E10.5, where the mesodermal lineage covered a larger area of the embryo, including the somites (Fig. 7A–D); however, the cell lineages were largely reduced or absent in the same region from Tbx1−/− embryos (Fig. 7E–H). This is consistent with loss of mRNA expression of all core mesodermal genes at E10.5 (Supplementary Material, Table S2). The results suggest that the core mesoderm cells are greatly reduced or lost between E9.5 and E10.5. We observed a small patch of unstained cells surrounded by robustly labeled NCCs in Tbx1−/− embryos using a Wnt1-Cre driver and a ROSA26-LacZ reporter to trace the NCCs in Tbx1−/− embryos at E10.5, indicating that at least some mesodermal cells are present, perhaps of a different lineage (28).
Increased apoptosis in the core mesoderm of Tbx1−/− embryos at E9.5–E10
To determine whether there is a change in cell proliferation or apoptosis accounting for loss of core mesodermal gene expression, we used Tbx1Cre/+ mice (37) to perform lineage tracing. Tbx1Cre/+ is a knock-in and there is a replacement of part of the Tbx1 coding region with Cre recombinase on one allele, thereby inactivating Tbx1 (37). Previous studies showed that proliferation of mesenchymal cells in the pharyngeal apparatus was reduced at E8.5 in Mesp1Cr-e-mediated Tbx1 conditional null mutant embryos (38), suggesting cell autonomous differences in proliferation early in development. We evaluated cell proliferation from E9.5 (S20–25) to E10.0 (S26–29) in Tbx1Cre/+; RCEEGFP/+ (heterozygous control) and Tbx1Cre/−; RCEEGFP/+ (mutant) embryos by dual immunofluorescence with antibodies against GFP and anti-phospho Histone H3 (Ser10). Tbx1+/− mice have no defects in craniofacial muscles, making it possible to use the knock-in line for comparative lineage tracing. We calculated the average number of Tbx1 lineage cells (GFP fluorescence) and proliferating cells (Rhodamine fluorescence) per tissue section for heterozygous controls (WT; Fig. 8B) and mutant embryos (KO; Fig. 8B). At E9.5, heterozygous control embryos had similar numbers of Tbx1 lineage and proliferating cells (19.3/section, 15.0/section; n = 3) with homozygous null mutant embryos (17.4/section, 14.5/section; n = 4). Closer to E10, heterozygous controls tended to have more Tbx1 lineage cells than in homozygous mutant embryos (39.9/section, n = 3 versus.16.8/section, n = 6; P = 0.12), while proliferating cells were similar between the two genotypes (31.0/section; n = 3 versus. 33.1/section; n = 6). We noted qualitatively that proliferation occurred in few cells and evenly across the entire PA1 tissue in control and mutant embryos at E9.5 (S20–21; data not shown) and E9.5–E10 (S24–26; Fig. 8). We therefore concluded that loss of Tbx1 did not significantly affect PA1 cell proliferation at E9.5 or E10.
Figure 8.

Proliferation and apoptosis of PA1 in Tbx1+/+ versus Tbx1−/− embryos. (A) Immunofluorescence images of tissue sections to visualize the Tbx1 lineage (GFP, green) and either cell proliferation (anti-phospho Histone H3 (Ser10); red fluorescence; top) or apoptosis (TUNEL); red fluorescence; bottom) in Tbx1 heterozygous (Het) and homozygous mutant (KO) embryos are shown. Dapi fluorescent stain to visualize nuclei and identify the tissue is shown in blue. The somite counts of the Tbx1+/+ and Tbx1−/− embryos are indicated (S24; S25). (B) Cell proliferation analysis. Statistical analysis was performed to determine whether cell proliferation was the same or different between Tbx1 heterozygous and homozygous null mutant embryos. The bar graph depicts average count per section with error bars from Student's t-test indicated.
We next examined apoptosis using TUNEL assays on histological sections at E9.5 (S20–21; heterozygous controls, n = 3; mutant embryos, n = 3) and E9.5–10.0 (S23–26; heterozygous controls, n = 5; homozygous mutant embryos, n = 6), using the same approach as for proliferation analysis. We noted intense TUNEL staining in histological sections from homozygous mutant embryos at E9.5–E10 near the GFP cell lineage (Fig. 8). We observed a large, intense region of TUNEL staining in the center of the PA1 in the vicinity of the Tbx1 lineage in Tbx1Cre/− mutant embryos that was not observed in heterozygous controls as shown in the representative image in Figure 8 and Supplementary Material, Figure S2. This suggests that there is loss of PA1 cells by apoptosis. When taken together with the in situ hybridization experiments, microarray, qRT–PCR analysis, lineage tracing and our previous report showing no difference in proliferation or apoptosis at E10.5 (28), current results suggest that core mesoderm cells are lost prior to E10.5 due to apoptosis.
Tbx1 is required in the core mesoderm to form muscles of mastication
It has been established that development of the branchiomeric muscles is disrupted in Tbx1−/− mutant embryos (21,24). Surprisingly, the muscles were present as normal, after inactivation of Tbx1 using Mesp1Cre or T-Cre mesodermal drivers (23,28,38). Thus, inactivation of Tbx1 was not complete using either allele alone (23,28,39). This is in contrast to the chick model where Tbx1 is required in the mesoderm to promote craniofacial muscle formation (23). It is important to understand these differences, because it would help decipher molecular mechanisms. In mouse models, significant Tbx1 expression remained in conditional mutant embryos using Mesp1Cre mice to drive mesodermal inactivation (38). It may be because of sub-optimal recombination of loxP sites or that different mesodermal lineages comprise the core mesoderm. To get around this problem, we crossed T-Cre; Mesp1Cre/+; Tbx1+/− mice with Tbx1flox/flox mice to obtain MesoTbx1flox/+ or MesoTbx1flox/− mice, which were heterozygous for both mesodermal drivers of Cre. The masseter, pterygoid and temporalis muscles were absent on both sides in one embryo (Supplementary Material, Fig. S3), absent on one side and hypoplastic on the other in another embryo (Fig. 9 and Supplementary Material, Fig. S3), and hypoplastic in both in a third and fourth embryos (Supplementary Material, Fig. S3). Variable penetrance of craniofacial muscles is similar to the situation in Tbx1−/− embryos (21). Resulting mutant embryos showed a major reduction of Tbx1 expression in the mesoderm, although its expression was still not completely lost (Fig. 9B). The second and distal pharyngeal arches were missing in most mutant embryos (MesoTbx1flox/−; Fig. 9C). Myf5 expression, normally in the core mesoderm, was lost in the MesoTbx1flox/− embryos at E10.5 (Fig. 9C), indicating that mesoderm loss of Tbx1 behaves similar to that of the global null mutant.
Figure 9.

Mesodermal Tbx1 is required to form the muscles of mastication. (A) Transverse histological sections of Mesp1Cre/+; T-Cre; Tbx1flox/+ and Mesp1Cre/+; T-Cre; Tbx1flox/− (MesoTbx1flox/+ or MesoTbx1flox/−) embryos at E17.5 stained with hematoxylin and eosin. Adipocytes have largely replaced muscles of mastication (right). The masseter muscle is present unilaterally, but hypoplastic, and a pterygoid muscle is present unilaterally (additional images are in the Supplementary Material, Fig. S3), but again, is severely hypoplastic. ma = masseter. (B) Lateral views of whole-mount in situ hybridization of antisense probe for Tbx1 in MesoTbx1flox/+ and MesoTbx1flox/− littermates at E9.5. Mesodermal expression of Tbx1 in heterozygous embryos is greatly reduced, but still present in conditional mutants. (C) Lateral views of whole-mount in situ hybridization of antisense probe for Myf5 in MesoTbx1flox/+ and MesoTbx1flox/− littermates at E10.5. Enlarged pharyngeal region from in situ hybridization images are shown below whole embryo views. Arrow points to the Myf5 core mesoderm expression domain. Expression was diminished in conditional null mutant embryos.
DISCUSSION
In this report, we attempted to understand the mechanistic basis for the loss of branchiomeric muscles in Tbx1−/− embryos. This is because once the mechanism is understood, we may be able to identify the molecular pathogenesis of muscle hypotonia in 22q11DS patients. In order to generate hypotheses for possible mechanisms, we performed gene profiling at two developmental stages, at E9.5 and at E10.5, which turned out to be critical stages for Tbx1 function, because at these stages core mesodermal gene expression declines coincident with loss of cells due to apoptosis.
Tbx1 may be required to maintain Tcf21 and Msc gene expression
We found that at stages E9.5 and/or E10.5, Tcf21, Msc, Myf5 and MyoD were decreased in expression. This is somewhat surprising for Tcf21, especially at E9.5, since it is known that when Tcf21 is inactivated Tbx1 expression is reduced, suggesting that it acts downstream of Tcf21 (17). This work suggests that Tbx1 might be required to maintain Tcf21 expression at E9.5 to E10.5. Expression of both Tcf21 and Msc were absent from PA1 at E10.5. Our observation that Tcf21 expression was lost in Tbx1−/− embryos at E10.5 is in contrast to a previous report indicating that Tcf21 was still expressed in their Tbx1−/− mutant embryos at this stage (40). This discrepancy could be due to differences in somite counts for which ranges define stage, difference in alleles or to differences in genetic backgrounds. Overall, the new findings in this experiment are that Tcf21 and Msc are reduced in expression at E10.5 in our Tbx1−/− embryos.
In previous studies, when mouse Tcf21 and Msc were both inactivated, muscles of mastication did not form due to apoptosis of core mesoderm cells by E10.5 (41). The timing of both gene expression changes and enhanced apoptosis suggests that loss of tissue in Tbx1−/− null mutant embryos might be explained, in part, by failure to maintain Tcf21 and Msc expression. This is because Tcf21 and Msc act upstream of Myf5 and MyoD and are required for survival of the core mesoderm (10). Reduction in Myf5 and MyoD expression in Tbx1−/− mutant embryos could be a direct or indirect result of loss of Tcf21 and Msc expression. This would implicate direct or indirect transcriptional regulation of Tcf21 and Msc. Relevant to this, it has remained an enigma in the field as to whether Tbx1 has cell autonomous functions in the core mesoderm. In the chick model, downregulation of Tbx1 in the core mesoderm tissue in vitro resulted in loss of branchiomeric muscles, but in the mouse model, use of single mesodermal drivers of Cre recombinase was unable to completely disrupt Tbx1 resulting in continued presence of the muscles of mastication (23). By simultaneously using two different mesodermal Cre drivers, we were able to demonstrate that Tbx1 has cell autonomous functions in promoting myogenesis in this tissue. This suggests that a main function of Tbx1 is to maintain direct or indirect regulation of Tcf21 and Msc expression, as well as other genes, in the core mesoderm of PA1.
One question is whether this is a direct effect or whether Tbx1 in the mesodermal core indirectly signals to neural crest mesenchymal cells, which then inhibit myogenesis (21). Although we cannot directly answer this question, we do know that complete inactivation of Tbx1 or partial inactivation of Tbx1 using T-Cre mice (mesodermal inactivation) alters patterns of expression pattern of many neural crest expressed genes and results in ectopic expression of Hoxa2 (28). However, the muscles of mastication still formed in T-Cre mediated Tbx1 conditional mutants (28). When taken together, this suggests that changes in neural crest mesenchymal gene patterns alone cannot account for loss of core mesoderm tissue, and likely Tbx1 has cell autonomous roles in the core mesodermal cells.
Genes acting downstream of Tbx1 in the core mesoderm
In addition to Tcf21, Msc, Myf5 and MyoD, several interesting genes were altered when Tbx1 was inactivated that might also explain resulting phenotypes. We identified two newly recognized core mesodermal genes, Chrdl1 and Lrrn1, which were reduced in expression in Tbx1−/− embryos. Both are interesting genes that have not been studied with respect to formation of the muscles of mastication. The Chrdl1 gene encodes a Bmp antagonist that has many different developmental functions. The human CHRDL1 gene maps to the X chromosome. Recently, mutations in CHRDL1 have been identified in humans causing X-linked megalocornea (MGC1; MIM#309300) (42). This is consistent with its expression pattern in the developing eye (43). There is not a mouse knockout allele available for Chrdl1 to further analyze the craniofacial muscles. However, craniofacial muscle defects have not been reported in humans with inactivating mutations. It is possible, based upon the human patient studies that Chrdl1 may not be essential because other genes serve redundant functions. Chordin (Chrd) encodes another member of the gene family, but it is not expressed in the core mesodermal tissue, implicating other Bmp antagonists as possibly serving redundant functions. The Grem1 gene encodes a Bmp antagonist and we found by microarray and qRT–PCR that it was reduced in expression in Tbx1−/− embryos. Inactivation of Grem1 causes embryonic lethality with kidney and skeletal defects (44). Grem1 is also important for regulating myogenic progenitor cell proliferation in skeletal muscle (45). The gene was weakly expressed in the mesoderm of PA1 in Tbx1+/+ as well as Tbx1−/− embryos (21), suggesting that it may not be greatly affected by the Tbx1 mutation. More work would need to be performed to understand the function of Chrdl1, Grem1 and other Bmp antagonists downstream of Tbx1.
Lrrn1, encoding a transmembrane protein, was reduced in expression at E9.5 in Tbx1−/− embryos. It has a specific expression pattern in the telencephalon, core mesoderm and somites. Despite its intriguing expression pattern, Lrrn1−/− mice survive to adulthood (46). This would indicate that it is not essential for craniofacial muscle development. As for Chrdl1, Lrrn1 might act redundantly with another gene(s). Relevant to this, there are three Lrrn family members in the mouse genome: Lrrn1, Lrrn2 and Lrrn3 (25), indicating that Lrrn2 or Lrrn3 might act redundantly. Chick Lrrn2 is expressed in the second pharyngeal arch (PA2) core mesoderm but not PA1 (47). Neither Lrrn2 nor Lrrn3 is expressed in PA1 in mouse embryos at similar stages (25). Other more distant family members exist, sharing leucine-rich repeats (LRRs) and immunoglobulin-like (Ig) domains (48), suggesting that perhaps some of these might act redundantly with Lrrn1.
The two additional genes of interest that were identified are Tcf12 and Ash2l. Both proteins TCF12 and ASH2L separately interact physically with myogenic regulatory factors, such as TCF21. The TCF12 protein, like the myogenic regulatory factors, is a bHLH E-box class of transcription factors, and as such can heterodimerize. Specifically, TCF21 can physically interact with TCF12 (49) implicating a possible molecular interaction. Ash2l (absent, small or homeotic-like, Drosophila) encodes a transcription regulator with histone methyltransferase activity. MYF5 and ASH2L are present in a shared protein–protein complex within cultured primary mouse myoblasts (50). Of interest, ASH2L physically interacts with TBX1 as identified using a yeast two hybrid screen (51). This suggests a possible molecular connection between ASH2L, TBX1 and MYF5 proteins. Another chromatin modifier, SETD7 (SET domain containing lysine methyltransferase 7), physically interacts with MYOD for myoblast differentiation (52) and separately interacts with TBX1 (53). Ash21 and Setd7 genes identified as part of the Tbx1 gene network in PA1 were not changed in mRNA expression in Tbx1−/− embryos at E9.5 (Fig. 2B), but we may speculate that there are posttranslational modifications that alter protein–protein interactions.
One reason for loss of craniofacial muscle gene expression could be due to a change in cell fate, such as the cardiac lineage. This is because we found ectopic expression of cardiac morphogenetic genes, such as GATA4, GATA5, GATA6 and Tbx5. However, these genes are not expressed in the core mesoderm, but are expressed in the caudal part of PA1 in Tbx1−/− embryos. The data are more consistent with loss of the core mesodermal lineage, as determined by fate mapping studies and apoptosis assays. Once precursors undergo apoptosis, the branchiomeric muscles can no longer form.
Genes downstream of Tbx1 for pharyngeal development
There are other functions of Tbx1 in craniofacial development besides forming the branchiomeric muscles. One important function is to control formation of the aortic arch arteries and their remodeling to form the asymmetric aortic arch. Tbx1 is required for development and remodeling of the aortic arch arteries and mutant mice have similar defects as seen in 22q11DS patients (19,36,54). Tbx1 was recently shown to act upstream of the Gbx2 homeodomain transcription factor gene in the pharyngeal ectoderm and Gbx2 acts upstream of Slit and Robo signaling in the neural crest cells for development of pharyngeal arch arteries 3, 4 and 6 (54). We found that Gbx2, Robo1 and Slit1 were reduced in expression in PA1 at E9.5 and/or E10.5, suggesting possible defects in PA1 artery formation.
There are other genes important for vascular development reduced in expression in Tbx1−/− embryos. The Foxc1 and Foxc2 as well as Cxcl12 genes were reduced in expression in PA1 at E9.5 and/or E10.5. The two Forkhead transcription factor genes act redundantly upstream of CXCL12 ligand and its receptor CXCR4, and these proteins are required for chemotaxic motility of endothelial cells to form blood vessels (55). We observed a change in pattern of Foxc1 expression and diminished Foxc2 expression in the PA1 tissue in Tbx1−/− embryos at E10.5 coincident with microarray results. Neural crest cells do migrate into PA1 as determined by lineage tracing using Wnt1-Cre and a ROSA26-LacZ reporter gene in Tbx1−/− embryos and appear to be grossly normal (28). This work suggests that there is a change in cell fate rather than loss of tissue.
These genes may have roles in addition to that of vascular development. Specifically, knockout of Foxc2 in the mouse results in cleft palate along with malformed mandible, skull bones and absent middle ear bones, similar to what is observed in Tbx1−/− embryos (31). Related to this, Sim2 was reduced in expression in Tbx1−/− embryos at E10.5. Sim2 encodes a transcription factor expressed in neural crest mesenchymal cells required for forming bones of the craniofacial region, including the palate (32). Based upon the in situ hybridization experiments presented in this report, we cannot rule out partial expression of Sim2 in non-neural crest mesoderm; however, there are no obvious craniofacial muscle defects in Sim2−/− embryos (31). It is possible that altered Foxc2 and Sim2 expression in PA1 in Tbx1−/− embryos could contribute to the presence of a cleft palate and other craniofacial bone defects in later staged Tbx1−/− embryos. Similarly, changes in expression of these genes might help understand the basis of defects in the proximal mandible in PA1 in Tbx1−/− embryos (28). We previously found that Fgf8 and Bmp4 expression in the PA1 ectoderm at E10.5, as well as some downstream effector genes, was shifted laterally. We did not detect significant changes in gene expression levels of Fgf8 and Bmp4 (28). Additional genes that were reduced in expression that are expressed in neural crest mesenchymal cells but not yet linked to Tbx1 are Sox10, Six1 or Six2 (56–58). It is possible that change in Fgf8 and Bmp4 expression patterns could be a cause or effect of changes in expression pattern of these genes.
Hypoxic stress in PA1 of Tbx1−/− embryos
The mechanism by which loss of Tbx1 results in increased apoptosis is not known. Some clues for this are derived from gene profiling at E10.5, where we found increased expression of hypoxic stress response genes. One trivial possibility for this increase is that PA1 cells undergo hypoxic stress secondary to vascular problems when Tbx1 is inactivated (36,54). Tbx1 is expressed early in development, before E8.5, but yet apoptosis does not take place until E9.5-E10.0. Similarly, many stress response genes were increased in expression by E10.5 but not at E9.5. This suggests that Tbx1 may act with other genes to promote cell survival and stress in surrounding cells at these critical stages. Further studies of novel and known genes altered in expression or function between E9.5 and E10.5 should shed light on the relative hierarchy of stress response genes downstream of Tbx1.
SUMMARY
Our work demonstrates a key function of Tbx1 in the core mesoderm itself for maintenance of cells and when inactivated, results in loss of muscles of mastication. We developed a gene network in which genes responsible for mesenchymal cell differentiation were highlighted as those reduced in expression or downregulated in the absence of Tbx1. We found that genes for cell stress are increased by E10.5 at a time period when the core mesoderm cells have undergone apoptosis. This sets the stage for future studies to identify direct transcriptional target genes and fully elucidate its role in myogenesis and or cell survival.
MATERIALS AND METHODS
Mouse mutants
The following mouse mutant alleles used in this study have been described previously: Tbx1+/− (20), RCEEGFP/+ (59), Tbx1flox/+ (39), Tbx1Cre/+ (60), (Mesp1Cre/+ (61) and T-Cre (62). Tbx1+/− mice, used to obtain Tbx1−/− embryos, have been maintained congenic in the Swiss Webster background. To generate T-Cre; Mesp1Cre/+; Tbx1flox/− embryos, T-Cre transgenic mice were crossed to Tbx1+/− mice, on a mixed Swiss Webster background, to obtain T-Cre; Tbx1+/− mice, and these were then crossed with Mesp1Cre/+mice to generate T-Cre; Mesp1Cre/+; Tbx1+/− mice. These mice were then further crossed with the Tbx1flox/flox mice in a Swiss Webster background. Wild-type and T-Cre; Mesp1Cre/+; Tbx1flox/+ littermates were used as controls in relevant experiments. All other mouse strains used were maintained in a mixed Swiss Webster background. PCR strategies for mouse genotyping have been described in the original reports.
Histology
Mouse embryos were isolated in phosphate-buffered saline (PBS) and fixed in 10% neutral buffered formalin (Sigma) overnight. Following fixation, the embryos were dehydrated through a graded ethanol series, embedded in paraffin and sectioned at 5 µm. All sections were stained with hematoxylin and eosin using standard protocols. Some of the work was performed in the Einstein Histopathology Core Facility (http://www.einstein.yu.edu/histopathology/page.aspx).
RNA in situ hybridization
Whole-mount and tissue section RNA in situ hybridization with non-radioactive probes was performed as previously described (63,64), using PCR-based probes (Supplementary Material, Table S3).
Gene profiling
To obtain enough RNA for microarray hybridization experiments, dissected mandibular arches from three Tbx1+/+ and three Tbx1−/− E9.5 and E10.5 embryos were pooled according to genotype, with three microarrays performed at each stage, in total. The tissue was homogenized in Buffer RLT (QIAGEN). Total RNA was isolated with the RNeasy Micro Kit according to the manufacturer's protocol. Quality and quantity of total RNA were determined using an Agilent 2100 Bioanalyzer (Agilent) and an ND-1000 Spectrophotometer (NanoDrop), respectively. Biotinylated single-stranded cDNA targets were amplified from 100 ng (ng) starting total RNA using the Ovation RNA Amplification System V2 and FL- Ovation cDNA Biotin Module V2 (NuGEN). A total of 3.75 μg of cDNA from the last step was hybridized to the GeneChip Test3 array (Affymetrix) to test the quality of the labeled target. Nucleic acid samples that passed quality control were then hybridized to the Affymetrix Mouse GeneST 1.0 (E9.5) or Affymetrix GeneChip Mouse Genome 430 2.0 Arrays (E10.5). Hybridization, washing, staining and scanning were performed in the Genomics Core at Einstein (http://www.einstein.yu.edu/genetics/CoreFacilities.aspx?id=23934) according to the Affymetrix manual. The microarray data have been uploaded to the GEO database (GSE30980 GSE35013).
Microarray statistical analysis for Affymetrix mouse gene ST arrays
GeneChip data were analyzed with oligo and limma (Linear Models for Microarray Analysis), which are two libraries present in the R package. Briefly, the original Affymetrix GeneChip CEL files generated by the Genomics Core were imported and summarized at the probe set level or at the transcript cluster level using the oligonucleotide library. Robust multi-array average (RMA) method was used to normalize, background correct and summarize. The data were converted to logarithmic scale and the significance analysis was performed using the two-sample t-test with a cut-off of unadjusted P-value of <0.05. The statistical analysis was performed using the Limma package.
Pathway and network analysis
Genes with a nominal P-value of <0.05 and a fold change of ≥1.3 were considered to be differently expressed and used to find associated and enriched biological processes and network pathways using IPA and Genemania (http://www.ncbi.nlm.nih.gov/pubmed/20576703) for gene pathway groups shown in Figs 2C and 5.
Quantitative reverse transcriptase–polymerase chain reaction
To obtain enough total RNA and minimize the variability of gene expression in individual embryos, each tube contained microdissected PA1s at E10.5 from four embryos. Total RNA was isolated from dissected tissues from three independent biological replicates using the RNeasy Micro Kit (Qiagen) and analyzed for sample purity and integrity using BioAnalyzer instrumentation. The mRNA levels were measured by TaqMan Gene Expression Assays (Applied Biosystems) for each gene and were carried out in triplicate using Ipo8 for the E9.5 and Gapdh for the E10.5 studies as normalization controls. TaqMan probes and primer sets were obtained from the Applied Biosystems' Gene Expression Assay database (http://allgenes.com). Samples were run in 96-well plates (10 μl final volume per reaction) on an ABI 7900HT Q-PCR apparatus. The sodium dodecyl sulfate 2.2 software platform (Applied Biosystems) was used for the computer interface with the ABI 7900HT PCR System to generate normalized data, compare samples and calculate the relative quantity. Statistical significance of the difference in gene expression was estimated using the two-tailed t-test.
Proliferation and apoptosis on tissue sections
After fixation as described for whole-mount direct fluorescence and embryo freezing, frozen sections were obtained at a thickness of 10 μm and then permeabilized in 0.5% Triton X-100 for 5 min. Blocking was performed with 5% serum (goat or donkey) in PBS/0.1% Triton X-100 (PBT) for 1 h. Primary antibody was diluted in blocking solution (1:500) and incubated for 1 h. Proliferation of cells was assessed by immunofluorescence using the primary antibody anti-phospho Histone H3 (Ser10), a mitosis marker (06–570 Millipore). Sections were washed in PBT and incubated with secondary antibody for 1 h. Secondary antibody was Alexa Fluor 568 goat α-rabbit IgG (A11011 Invitrogen) at 1:500. Slides were mounted in hard-set mounting medium with DAPI (Vector Labs H-1500). Images were captured using a Zeiss Axio Observer microscope. To perform statistical analysis of cell proliferation, we first calculated the average cell counts per tissue section for each embryo. Then, we estimated the mean and standard error of the average cell counts for control and mutant groups and compared them using the t-test. Apoptosis was assessed on frozen 10 μm thick sections by using DeadEnd Fluorometric TUNEL System (G3250 Promega) following the manufacturer's instructions. Natural GFP from the reporter or antibodies to GFP was used to distinguish the core mesodermal cells in both assays described above.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the National Institutes of Health (R01HL088698 and P01HD070454, B.E.M.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Department of Surgery at Montefiore Medical Center for providing salary support for Dr Carpenter. We thank Dr Antonio Baldini for the Tbx1Cre mice. We thank Dr Mark Lewandoski for the T-Cre mice. We thank Nousin Haque and Harpreet Singh for their technical help. We acknowledge the Einstein Histopathology Core for embedding embryos and the Genomics Core for performing the gene expression profiling.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Bryson-Richardson R.J., Currie P.D. The genetics of vertebrate myogenesis. Nat. Rev. Genet. 2008;9:632–646. doi: 10.1038/nrg2369. [DOI] [PubMed] [Google Scholar]
- 2.Shprintzen R.J. Velo-cardio-facial syndrome: 30 years of study. Dev. Dis. Res. Rev. 2008;14:3–10. doi: 10.1002/ddrr.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shprintzen R.J., Marrinan E. Velopharyngeal insufficiency: diagnosis and management. Curr. Opin. Otolaryngol. Head Neck Surg. 2009;17:302–307. doi: 10.1097/MOO.0b013e32832cbd6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zim S., Schelper R., Kellman R., Tatum S., Ploutz-Snyder R., Shprintzen R. Thickness and histologic and histochemical properties of the superior pharyngeal constrictor muscle in velocardiofacial syndrome. Arch. Facial Plast. Surg. 2003;5:503–510. doi: 10.1001/archfaci.5.6.503. [DOI] [PubMed] [Google Scholar]
- 5.Lypka M., Bidros R., Rizvi M., Gaon M., Rubenstein A., Fox D., Cronin E. Posterior pharyngeal augmentation in the treatment of velopharyngeal insufficiency: a 40-year experience. Ann. Plast. Surg. 2010;65:48–51. doi: 10.1097/SAP.0b013e3181c1fec6. [DOI] [PubMed] [Google Scholar]
- 6.Akcakus M., Ozkul Y., Gunes T., Kurtoglu S., Cetin N., Kisaarslan A.P., Dundar M. Associated anomalies in asymmetric crying facies and 22q11 deletion. Genet. Couns. 2003;14:325–330. [PubMed] [Google Scholar]
- 7.Noden D.M. The embryonic origins of avian cephalic and cervical muscles and associated connective tissues. Am. J. Anat. 1983;168:257–276. doi: 10.1002/aja.1001680302. [DOI] [PubMed] [Google Scholar]
- 8.Trainor P.A., Tan S.S., Tam P.P. Cranial paraxial mesoderm: regionalisation of cell fate and impact on craniofacial development in mouse embryos. Development. 1994;120:2397–2408. doi: 10.1242/dev.120.9.2397. [DOI] [PubMed] [Google Scholar]
- 9.Lu J., Webb R., Richardson J.A., Olson E.N. MyoR: a muscle-restricted basic helix-loop-helix transcription factor that antagonizes the actions of MyoD. Proc. Natl. Acad. Sci. USA. 1999;96:552–557. doi: 10.1073/pnas.96.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moncaut N., Cross J.W., Siligan C., Keith A., Taylor K., Rigby P.W., Carvajal J.J. Musculin and TCF21 coordinate the maintenance of myogenic regulatory factor expression levels during mouse craniofacial development. Development. 2012;139:958–967. doi: 10.1242/dev.068015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semina E.V., Reiter R., Leysens N.J., Alward W.L., Small K.W., Datson N.A., Siegel-Bartelt J., Bierke-Nelson D., Bitoun P., Zabel B.U., et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 12.Kioussi C., Briata P., Baek S.H., Wynshaw-Boris A., Rose D.W., Rosenfeld M.G. Pitx genes during cardiovascular development. Cold Spring Harb. Symp. Quant. Biol. 2002;67:81–87. doi: 10.1101/sqb.2002.67.81. [DOI] [PubMed] [Google Scholar]
- 13.Ai D., Liu W., Ma L., Dong F., Lu M.-F., Wang D., Verzi M.P., Cai C., Gage P.J., Evans S., et al. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev. Biol. 2006;296:437–449. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong F., Sun X., Liu W., Ai D., Klysik E., Lu M.F., Hadley J., Antoni L., Chen L., Baldini A., et al. Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development. 2006;133:4891–4899. doi: 10.1242/dev.02693. [DOI] [PubMed] [Google Scholar]
- 15.Shih H.P., Gross M.K., Kioussi C. Cranial muscle defects of Pitx2 mutants result from specification defects in the first branchial arch. Proc. Natl. Acad. Sci. USA. 2007;104:5907–5912. doi: 10.1073/pnas.0701122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathan E., Monovich A., Tirosh-Finkel L., Harrelson Z., Rousso T., Rinon A., Harel I., Evans S.M., Tzahor E. The contribution of Islet1-expressing splanchnic mesoderm cells to distinct branchiomeric muscles reveals significant heterogeneity in head muscle development. Development. 2008;135:647–657. doi: 10.1242/dev.007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harel I., Maezawa Y., Avraham R., Rinon A., Ma H.Y., Cross J.W., Leviatan N., Hegesh J., Roy A., Jacob-Hirsch J., et al. Pharyngeal mesoderm regulatory network controls cardiac and head muscle morphogenesis. Proc. Natl. Acad. Sci. USA. 2012;109:18839–18844. doi: 10.1073/pnas.1208690109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerome L.A., Papaioannou V.E. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 19.Lindsay E.A., Vitelli F., Su H., Morishima M., Huynh T., Pramparo T., Jurecic V., Ogunrinu G., Sutherland H.F., Scambler P.J., et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–9101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 20.Merscher S., Funke B., Epstein J.A., Heyer J., Puech A., Lu M.M., Xavier R.J., Demay M.B., Russell R.G., Factor S., et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 21.Kelly R.G., Jerome-Majewska L.A., Papaioannou V.E. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum. Mol. Genet. 2004;13:2829–2840. doi: 10.1093/hmg/ddh304. [DOI] [PubMed] [Google Scholar]
- 22.Grifone R., Jarry T., Dandonneau M., Grenier J., Duprez D., Kelly R.G. Properties of branchiomeric and somite-derived muscle development in Tbx1 mutant embryos. Dev. Dyn. 2008;237:3071–3078. doi: 10.1002/dvdy.21718. [DOI] [PubMed] [Google Scholar]
- 23.Dastjerdi A., Robson L., Walker R., Hadley J., Zhang Z., Rodriguez-Niedenfuhr M., Ataliotis P., Baldini A., Scambler P., Francis-West P. Tbx1 regulation of myogenic differentiation in the limb and cranial mesoderm. Dev. Dyn. 2007;236:353–363. doi: 10.1002/dvdy.21010. [DOI] [PubMed] [Google Scholar]
- 24.Liao J., Kochilas L., Nowotschin S., Arnold J.S., Aggarwal V.S., Epstein J.A., Brown M.C., Adams J., Morrow B.E. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum. Mol. Genet. 2004;13:1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- 25.Haines B.P., Gupta R., Jones C.M., Summerbell D., Rigby P.W. The NLRR gene family and mouse development: Modified differential display PCR identifies NLRR-1 as a gene expressed in early somitic myoblasts. Dev. Biol. 2005;281:145–159. doi: 10.1016/j.ydbio.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 26.Seale P., Ishibashi J., Holterman C., Rudnicki M.A. Muscle satellite cell-specific genes identified by genetic profiling of MyoD-deficient myogenic cell. Dev. Biol. 2004;275:287–300. doi: 10.1016/j.ydbio.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 27.Coffinier C., Tran U., Larrain J., De Robertis E.M. Neuralin-1 is a novel Chordin-related molecule expressed in the mouse neural plate. Mech. Dev. 2001;100:119–122. doi: 10.1016/s0925-4773(00)00507-4. [DOI] [PubMed] [Google Scholar]
- 28.Aggarwal V.S., Carpenter C., Freyer L., Liao J., Petti M., Morrow B.E. Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev. Biol. 2010;344:669–681. doi: 10.1016/j.ydbio.2010.05.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mussig K., Staiger H., Machicao F., Thamer C., Machann J., Schick F., Claussen C.D., Stefan N., Fritsche A., Haring H.U. RARRES2, encoding the novel adipokine chemerin, is a genetic determinant of disproportionate regional body fat distribution: a comparative magnetic resonance imaging study. Metabolism, 2009;58:519–524. doi: 10.1016/j.metabol.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Aggarwal V.S., Liao J., Bondarev A., Schimmang T., Lewandoski M., Locker J., Shanske A., Campione M., Morrow B.E. Dissection of Tbx1 and Fgf interactions in mouse models of 22q11DS suggests functional redundancy. Hum. Mol. Genet. 2006;15:3219–3228. doi: 10.1093/hmg/ddl399. [DOI] [PubMed] [Google Scholar]
- 31.Kume T., Jiang H., Topczewska J.M., Hogan B.L. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001;15:2470–2482. doi: 10.1101/gad.907301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shamblott M.J., Bugg E.M., Lawler A.M., Gearhart J.D. Craniofacial abnormalities resulting from targeted disruption of the murine Sim2 gene. Dev. Dyn. 2002;224:373–380. doi: 10.1002/dvdy.10116. [DOI] [PubMed] [Google Scholar]
- 33.Liao J., Aggarwal V.S., Nowotschin S., Bondarev A., Lipner S., Morrow B.E. Identification of downstream genetic pathways of Tbx1 in the second heart field. Dev. Biol. 2008;316:524–537. doi: 10.1016/j.ydbio.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozawa S., Ito S., Kato Y., Kubota E., Hata R. Human p38 delta MAP kinase mediates UV irradiation induced up-regulation of the gene expression of chemokine BRAK/CXCL14. Biochem. Biophys. Res. Commun. 2010;396:1060–1064. doi: 10.1016/j.bbrc.2010.05.072. [DOI] [PubMed] [Google Scholar]
- 35.Shakib K., Norman J.T., Fine L.G., Brown L.R., Godovac-Zimmermann J. Proteomics profiling of nuclear proteins for kidney fibroblasts suggests hypoxia, meiosis, and cancer may meet in the nucleus. Proteomics. 2005;5:2819–2838. doi: 10.1002/pmic.200401108. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z., Cerrato F., Xu H., Vitelli F., Morishima M., Vincentz J., Furuta Y., Ma L., Martin J.F., Baldini A., et al. Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development. 2005;132:5307–5315. doi: 10.1242/dev.02086. [DOI] [PubMed] [Google Scholar]
- 37.Balmer J.E., Blomhoff R. Gene expression regulation by retinoic acid. J. Lip. Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z., Huynh T., Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development. 2006;133:3587–3595. doi: 10.1242/dev.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold J.S., Werling U., Braunstein E.M., Liao J., Nowotschin S., Edelmann W., Hebert J.M., Morrow B.E. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development. 2006;133:977–987. doi: 10.1242/dev.02264. [DOI] [PubMed] [Google Scholar]
- 40.Sambasivan R., Gayraud-Morel B., Dumas G., Cimper C., Paisant S., Kelly R.G., Tajbakhsh S. Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev. Cell. 2009;16:810–821. doi: 10.1016/j.devcel.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 41.Lu J.R., Bassel-Duby R., Hawkins A., Chang P., Valdez R., Wu H., Gan L., Shelton J.M., Richardson J.A., Olson E.N. Control of facial muscle development by MyoR and capsulin. Science. 2002;298:2378–2381. doi: 10.1126/science.1078273. [DOI] [PubMed] [Google Scholar]
- 42.Webb T.R., Matarin M., Gardner J.C., Kelberman D., Hassan H., Ang W., Michaelides M., Ruddle J.B., Pennell C.E., Yazar S., et al. X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am. J. Hum. Genet. 2012;90:247–259. doi: 10.1016/j.ajhg.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakuta H., Suzuki R., Takahashi H., Kato A., Shintani T., Iemura S., Yamamoto T.S., Ueno N., Noda M. Ventroptin: a BMP-4 antagonist expressed in a double-gradient pattern in the retina. Science. 2001;293:111–115. doi: 10.1126/science.1058379. [DOI] [PubMed] [Google Scholar]
- 44.Michos O., Panman L., Vintersten K., Beier K., Zeller R., Zuniga A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development. 2004;131:3401–3410. doi: 10.1242/dev.01251. [DOI] [PubMed] [Google Scholar]
- 45.Frank N.Y., Kho A.T., Schatton T., Murphy G.F., Molloy M.J., Zhan Q., Ramoni M.F., Frank M.H., Kohane I.S., Gussoni E. Regulation of myogenic progenitor proliferation in human fetal skeletal muscle by BMP4 and its antagonist Gremlin. J. Cell Biol. 2006;175:99–110. doi: 10.1083/jcb.200511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang T., Li L., Tang J., Li Y., Lin W.Y., Martin F., Grant D., Solloway M., Parker L., Ye W., et al. A mouse knockout library for secreted and transmembrane proteins. Nat. Biotech. 2010;28:749–755. doi: 10.1038/nbt.1644. [DOI] [PubMed] [Google Scholar]
- 47.Andreae L.C., Lumsden A., Gilthorpe J.D. Chick Lrrn2, a novel downstream effector of Hoxb1 and Shh, functions in the selective targeting of rhombomere 4 motor neurons. Neural Dev. 2009;4:27. doi: 10.1186/1749-8104-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Homma S., Shimada T., Hikake T., Yaginuma H. Expression pattern of LRR and Ig domain-containing protein (LRRIG protein) in the early mouse embryo. Gene Exp. Pattn. 2009;9:1–26. doi: 10.1016/j.gep.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Tandon P., Miteva Y.V., Kuchenbrod L.M., Cristea I.M., Conlon F.L. Tcf21 regulates the specification and maturation of proepicardial cells. Development. 2013;140:2409–2421. doi: 10.1242/dev.093385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKinnell I.W., Ishibashi J., Le Grand F., Punch V.G., Addicks G.C., Greenblatt J.F., Dilworth F.J., Rudnicki M.A. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat. Cell Biol. 2008;10:77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stoller J.Z., Huang L., Tan C.C., Huang F., Zhou D.D., Yang J., Gelb B.D., Epstein J.A. Ash2l interacts with Tbx1 and is required during early embryogenesis. Exp. Biol. Med. (Maywood) 2010;235:569–576. doi: 10.1258/ebm.2010.009318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tao Y., Neppl R.L., Huang Z.P., Chen J., Tang R.H., Cao R., Zhang Y., Jin S.W., Wang D.Z. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J. Cell Biol. 2011;194:551–565. doi: 10.1083/jcb.201010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L., Fulcoli F.G., Ferrentino R., Martucciello S., Illingworth E.A., Baldini A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012;8:e1002571. doi: 10.1371/journal.pgen.1002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calmont A., Ivins S., Van Bueren K.L., Papangeli I., Kyriakopoulou V., Andrews W.D., Martin J.F., Moon A.M., Illingworth E.A., Basson M.A., et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development. 2009;136:3173–3183. doi: 10.1242/dev.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayashi H., Kume T. Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration. Biochem. Biophys. Res. Commun. 2008;367:584–589. doi: 10.1016/j.bbrc.2007.12.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kutejova E., Engist B., Mallo M., Kanzler B., Bobola N. Hoxa2 downregulates Six2 in the neural crest-derived mesenchyme. Development. 2005;132:469–478. doi: 10.1242/dev.01536. [DOI] [PubMed] [Google Scholar]
- 57.Gavalas A., Trainor P., Ariza-McNaughton L., Krumlauf R. Synergy between Hoxa1 and Hoxb1: the relationship between arch patterning and the generation of cranial neural crest. Development. 2001;128:3017–3027. doi: 10.1242/dev.128.15.3017. [DOI] [PubMed] [Google Scholar]
- 58.Oliver G., Wehr R., Jenkins N.A., Copeland N.G., Cheyette B.N., Hartenstein V., Zipursky S.L., Gruss P. Homeobox genes and connective tissue patterning. Development. 1995;121:693–705. doi: 10.1242/dev.121.3.693. [DOI] [PubMed] [Google Scholar]
- 59.Sousa V.H., Miyoshi G., Hjerling-Leffler J., Karayannis T., Fishell G. Characterization of Nkx6–2-derived neocortical interneuron lineages. Cere. Cortex. 2009;19(Suppl 1):1–10. doi: 10.1093/cercor/bhp038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vitelli F., Huynh T., Baldini A. Gain of function of Tbx1 affects pharyngeal and heart development in the mouse. Genesis. 2009;47:188–195. doi: 10.1002/dvg.20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saga Y., Miyagawa-Tomita S., Takagi A., Kitajima S., Miyazaki J., Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- 62.Perantoni A.O., Timofeeva O., Naillat F., Richman C., Pajni-Underwood S., Wilson C., Vainio S., Dove L.F., Lewandoski M. Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development. 2005;132:3859–3871. doi: 10.1242/dev.01945. [DOI] [PubMed] [Google Scholar]
- 63.Hidai H., Bardales R., Goodwin R., Quertermous T., Quertermous E.E. Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech. Dev. 1998;73:33–43. doi: 10.1016/s0925-4773(98)00031-8. [DOI] [PubMed] [Google Scholar]
- 64.Alappat S.R., Zhang Z., Suzuki K., Zhang X., Liu H., Jiang R., Yamada G., Chen Y. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev. Biol. 2005;277:102–113. doi: 10.1016/j.ydbio.2004.09.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.