

Abstract
Peptide nucleic acids (PNA) are a unique class of synthetic molecules that have a peptide backbone and can hybridize with nucleic acids. Here, a versatile method has been developed for the automated, in situ synthesis of PNA from a porous silicon (PSi) substrate for applications in gene therapy and biosensing. Nondestructive optical measurements were performed to monitor single base additions of PNA initiated from (3-aminopropyl)triethoxysilane attached to the surface of PSi films, and mass spectrometry was conducted to verify synthesis of the desired sequence. Comparison of in situ synthesis to postsynthesis surface conjugation of the full PNA molecules showed that surface mediated, in situ PNA synthesis increased loading 8-fold. For therapeutic proof-of-concept, controlled PNA release from PSi films was characterized in phosphate buffered saline, and PSi nanoparticles fabricated from PSi films containing in situ grown PNA complementary to micro-RNA (miR) 122 generated significant anti-miR activity in a Huh7 psiCHECK-miR122 cell line. The applicability of this platform for biosensing was also demonstrated using optical measurements that indicated selective hybridization of complementary DNA target molecules to PNA synthesized in situ on PSi films. These collective data confirm that we have established a novel PNA–PSi platform with broad utility in drug delivery and biosensing.
Introduction
Peptide nucleic acids (PNA) are synthetic nucleic acid analogues wherein the negatively charged sugar-phosphate backbone is replaced with charge-neutral amide linkages.1 Nucleobases (A, C, T, and G) are spaced along the peptide backbone such that PNA hybridization with DNA and RNA obeys the rules of Watson–Crick base pairing.2,3 PNA offer several advantages over DNA and RNA, including greater binding affinity for complementary oligos,4 innate resistance to both nuclease and protease degradation,5,6 and the ability to form hybrids with less sensitivity to changes in temperature, pH, and ionic strength.3,7 These attributes make PNA oligomers ideal candidates for application as antisense therapeutics that block expression of complementary mRNA,8,9 therapeutic inhibitors of post-transcriptional gene regulatory micro-RNA (miRNA),9 and biosensor probes for detection of target nucleic acid hybridization.10−12
A critical requirement for therapeutic or biosensing applications of PNA is stable conjugation to either an intracellular therapeutic delivery platform or a biosensing substrate. In the absence of a delivery vector, PNA therapeutic efficacy is hindered by poor intracellular bioavailability and lack of activity.13,14 It is therefore necessary to chemically modify PNA by fusion with cell penetrating peptides or formulation into delivery systems that can mediate cellular internalization and cytoplasmic release.9,11,15,16 Similarly, biosensing applications require stable integration of PNA at high surface densities onto analytical devices capable of reproducible and sensitive detection of hybridization events.10,12
This communication describes a versatile, automated method of synthesizing PNA from a nanostructured material, porous silicon (PSi). The large internal surface area (>100m2/cm3), biocompatibility, tunable pore geometry, and biodegradability of PSi have motivated a large body of research into PSi technologies for drug delivery and label-free biosensing.17−21 However, the only reported method of PNA attachment to PSi thus far has been nonspecific adsorption,22 and there are no published studies for delivery of PNA-based therapeutics using PSi delivery vehicles. Methods have been established for peptide synthesis directly from PSi,23−25 and our group has shown that base-by-base synthesis of DNA directly within PSi films (referred to as in situ synthesis) significantly increases DNA loading relative to attachment of presynthesized oligos.26 Herein, the first use of PSi as a platform for the automated synthesis and label-free characterization of PNA is reported. It is shown that in situ PNA synthesis increases PNA loading relative to conjugation of the presynthesized molecule. The advantage of this approach is demonstrated for intracellular delivery of a well-characterized anti-miR-122 PNA,27 which targets a liver-specific miRNA whose suppression has been linked to decreased hepatitis C viremia.28 Application of this conjugation strategy in selective, label-free nucleic-acid biosensing is also successfully accomplished using a model 16mer PNA probe.
Results and Discussion
In Situ PNA Synthesis from PSi
PNA synthesized in situ from PSi was compared to PNA loading into PSi using conventional physical adsorption and direct nucleic acid conjugation strategies (detailed methods available in Supporting Information). PSi films were etched from p-type Si (0.01 Ω-cm) using 15% hydrofluoric acid in ethanol to form 10-μm-thick single layers (70% porosity, 30 nm average pore diameter) that were then thermally oxidized at 800 °C for 30 min.26 Anti-miR122 PNA (NH2-ACA AAC ACC ATT GTC ACA CTC CA-COOH) was synthesized from the PSi substrate following functionalization with 3-aminopropyltriethoxysilane (APTES). For postsynthesis conjugation and physical adsorption to PSi, the same PNA was synthesized from Rink Amide LL resin (EMD Millipore). For postsynthesis conjugation, cysteine was added to the C-terminus (referred to as “cys-PNA”) to provide a reactive thiol used for conjugating to APTES-functionalized PSi via an amine-to-sulfhydryl heterobifunctional cross-linker, N-succinimidyl 3-(2-pyridyldithio) propionate (SPDP). All PNA syntheses were performed using fluorenylmethyloxycarbonyl chloride (Fmoc) solid phase chemistry within a PS3 automated peptide synthesizer (Protein Technologies).2
In situ PNA synthesis was characterized by reflectometry and matrix assisted laser desorption-ionization mass spectrometry (MALDI-MS) (Figure 1; experimental methods available in Supporting Information). Reflectometry was used to monitor PNA synthesis from the PSi film by measuring PSi film optical thickness (Figure 1a). Increases in optical thickness are directly proportional to the amount of monomer coupled to growing PNA oligos within the PSi film.19 Addition of the first 9 bases resulted in a stepwise increase in optical thickness, while the change in optical thickness was increasingly nonlinear for the addition of bases 10–23.
Figure 1.
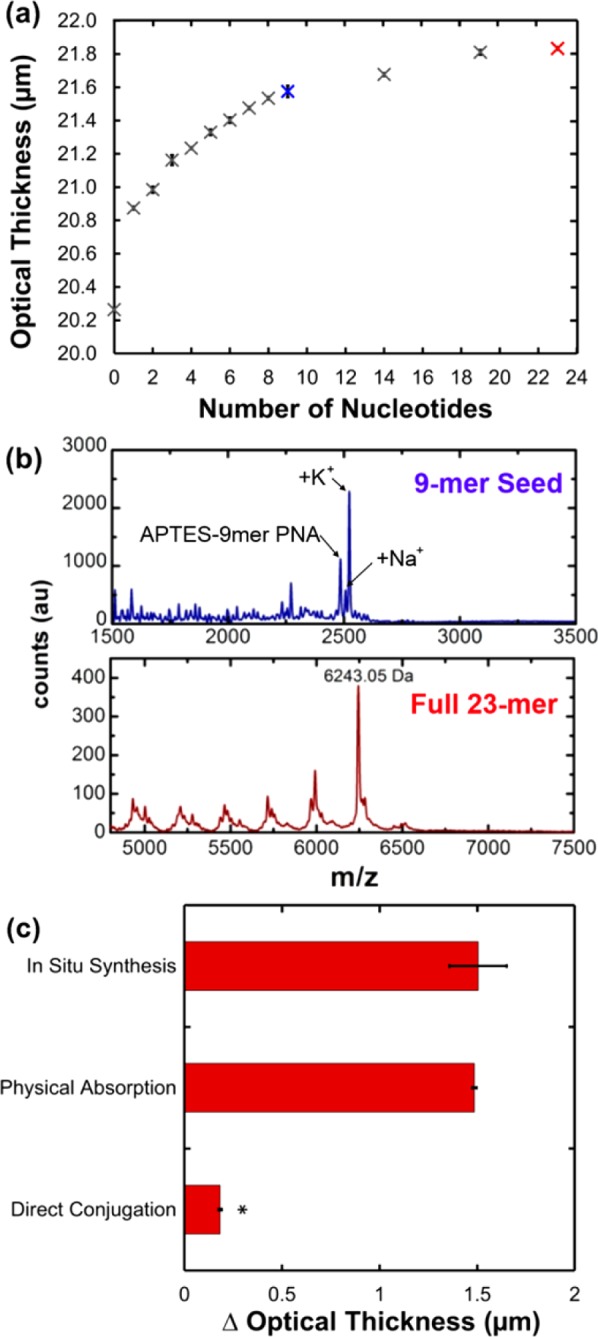
(a) Nondestructive optical characterization of in situ synthesis of 23-mer anti-miR122 PNA using reflectometry and Fourier transform analysis to determine the optical thickness of the loaded PSi film. (b) MALDI-TOF mass spectrometry of in situ synthesized 9-mer miRNA seed targeting sequence (top) and the full 23-mer PNA (bottom). (c) Comparison of the PNA loading capacity achieved using in situ synthesis, physical adsorption, and direct conjugation of a 23-mer PNA to PSi. Loading was calculated based on increase in optical thickness due to the addition of PNA to the PSi surface (* P < 0.0001).
To verify synthesis of the desired PNA sequence, in situ synthesized PNA was analyzed by mass spectrometry at two critical points during synthesis: after completion of the 9-mer miRNA seed-targeting sequence and after the full 23-mer anti-miR (Figure 1b). The seed-targeting sequence of therapeutic PNA is the most important miRNA-binding motif,27 whereas the full 23-mer sequence improves PNA target specificity and anti-miR activity.27 MALDI spectra of the 9-mer seed-targeting sequence contained a single peak corresponding to the expected molecular weight of the 3-APTES-conjugated PNA, 2485 Da, and two additional, closely spaced peaks corresponding to Na+ and K+ adducts were resolved at 2507 and 2523 Da, respectively. MALDI spectra of the full 23-mer PNA revealed an intense peak at the expected m/z for the full PNA oligo, 6243 Da, as well as a series of less intense peaks grouped at molecular weights corresponding to PNA sequences that were truncated during synthesis.
It is postulated that steric crowding of the growing PNA oligos within nanosized pores causes the partial truncation seen during in situ synthesis.29 The molecular length of 3-APTES is approximately 8 Å,30 and each PNA base extends a growing oligo by approximately 3.5 Å.31 Thus, the full, 23-mer PNA is estimated to be 8.9 nm in length. Electrochemical porosification of Si produces a Gaussian-like distribution of pore sizes, so while the average pore diameter of PSi films in this study is 30 nm, it is estimated that approximately 40% of the pores are between 10 and 25 nm in diameter.32 To investigate whether the yield of longer oligos can be improved by increasing the minimum pore size, anti-miR122 PNA was synthesized from a PSi film that was first exposed to a pore-widening solution of potassium hydroxide (KOH) in ethanol at room temperature for 20 min (Figure S4a in the Supporting Information). Pore widening increased the average pore diameter by 5 nm and decreased the number of pores with diameter less than 25 nm by ∼20%, resulting in increased shifts in optical thickness during in situ synthesis (Figure S4b in the Supporting Information). These data suggest that longer oligos can be more efficiently synthesized within a porous silicon matrix by increasing the average pore size, and although it was outside the scope of our testing, it may also be beneficial to utilize a fabrication method that yields more homogeneous pores. Since pore widening improved synthesis of the 23-mer anti-miR122, it was implemented for in vitro functional studies, i.e., cytotoxicity and bioactivity assays (all other studies were done with non-pore-widened substrates). Overall, these results, combined with subsequent experimental readouts, are a promising demonstration of successful synthesis of functional 23-mer PNA from PSi, with highly efficient growth of the vital anti-miR122 PNA seed sequence.
Optical thickness measurements were also used to determine the amount of anti-miR122 PNA loaded within PSi through in situ synthesis compared to standard methods of physical adsorption and conjugation of presynthesized PNA (Figure 1e). Consistent with our previous studies of DNA conjugation to PSi,26 PNA loading from in situ synthesis was 8-fold greater than the amount of PNA loading achieved from conjugation to a PSi surface. This corresponds to a yield of 8.6 × 10–4 mol PNA/g PSi, or 5.3 g PNA/g PSi for anti-miR122 synthesized in situ in non-pore-widened PSi, quantified by measuring the absorbance at 260 nm of PNA released after dissolution of the PSi matrix. It is postulated that during PNA loading by direct conjugation, PNA preferentially attaches nearer to the pore openings at the film surface, thereby restricting access of PNA deeper into the pores. Interestingly, there was no significant difference in the loading efficiency of in situ synthesis and physical adsorption within the pores. It is hypothesized that, in the case of nonspecific PNA loading by adsorption, lack of PNA covalent binding allows PNA to freely diffuse further into the pores and achieve a level of loading similar to in situ synthesis. Although nonspecific adsorption yielded a similar level of loading, we show in the following sections that the covalent attachment achieved from in situ synthesis provides more sustained release in the context of therapeutic PNA delivery and a more stable and reproducible substrate for biosensing.
Intracellular Delivery of In Situ Synthesized Therapeutic PNA
To evaluate the potential of therapeutic delivery of in situ synthesized PNA from PSi, the in vitro PNA temporal release profiles were compared for physically adsorbed and in situ synthesized PNA from PSi films (Figure 2a). Release of PNA synthesized in situ was significantly (p < 0.005) less than adsorbed PNA at every time point within the first 24 h, and overall, the PNA synthesized in situ showed reduced initial burst and more sustained release profiles. Because in situ synthesized PNA are covalently attached to pore walls, it is anticipated that hydrolytic degradation of the PSi matrix is the primary mechanism of PNA release, providing a means for more sustained PNA delivery when compared to physical adsorption.18,25 Mass spectra in Figure 1b provide support for a surface-degradation PNA release mechanism, as the molecular weight of the PNA released from in situ films corresponds to 3-APTES-conjugated PNA. For eventual in vivo applications, it is anticipated that reducing the burst release upon initial contact with physiological (aqueous) environments will ultimately improve drug bioavailability at the site of action, especially for intravenous delivery applications.
Figure 2.

(a) Cumulative release of physically adsorbed PNA and in situ synthesized PNA in PBS at 37 °C. Release of in situ synthesized PNA was significantly lower (p < 0.005) for time points within the boxed region. (b) PNA–PSNP size distribution characterized by NTA. Inset: Representative SEM image demonstrating PSNP size and morphology (scale bar = 400 nm). (c–f) Confocal micrographs of Huh7 cells incubated for either 1 or 24 h with (c and d) free PNA, or (e and f) PNA–PSNPs at a 2 μM dose of anti-miR-122 PNA (3.7 μg/mL PSNPs for PNA–PSNP treatments). Labels: Hoechst nuclear dye (blue), Alexa-488 labeled PNA (green). (g) PSNPs had no significant effect on Huh7 cell viability relative to untreated negative control samples, in contrast to a 2′OMe-modified AMO delivered with Fugene 6 (p < 0.05). (h) Anti-miR122 activity indicated by luciferase activity in Huh7 psiCHECK-miR122 cells 44 h after treatment with free PNA, empty PSNPs, or PNA–PSNPs (2 μM dose of anti-miR-122 PNA, 3.7 μg/mL PSNPs). Results are normalized to cell number and expressed relative to luciferase activity in Huh7 psiCHECK-miR122 control cells without treatment. Dashed green line indicates anti-miR122 activity 44 h after treatment with 2 μM of an optimized anti-miR-122 AMO34 delivered via the Fugene 6 commercial transfection reagent.
PNA anti-miR activity is dependent upon delivery into the cytoplasm where active micro-RNA is located.27 To demonstrate the application of in situ PNA synthesis for therapeutic purposes, anti-miR122-loaded multilayer PSi films (Supporting Information Figure S5) were ultrasonically fractured to produce PNA-functionalized PSi nanoparticles (PSNPs). The resultant PSNP mean size (236 nm) and size distribution were determined by Nanoparticle Tracking Analysis33 (NTA) and confirmed by scanning electron microscopy (SEM) (Figure 2b).
Huh 7 human hepatic carcinoma cells were selected for evaluation of PNA–PSNP cellular uptake, cytotoxicity, and anti-miR activities because they express elevated levels of miR122 and thus provide a good platform for testing anti-miR122 therapies (detailed cell culture and biological assay methods can be accessed in the Supporting Information).27,35 Intracellular delivery of a 2 μM dose of fluorescently labeled free or in situ synthesized PSNP anti-miR122 PNA was evaluated by confocal microscopy (Figure 2c–f). At each treatment time, cells treated with PNA–PSNPs showed greater intracellular delivery of PNA relative to those treated with free PNA. Additionally, a lactate dehydrogenase enzymatic assay was used to compare the cytotoxicity of PNA–PNSPs to that of a 2′OMe-modified anti-miR oligonucleotide (AMO) delivered by the commercial transfection reagent, Fugene 6 (Promega) (Figure 2g). Consistent with previous studies on the cytotoxicity of thermally oxidized PSi nanoparticles,36 no reduction in cell viability was observed for the PSNP concentration investigated in this study (2 μM PNA, 3.7 μg/mL), whereas the AMO treatment led to a 25% decrease in cell viability at an equivalent AMO dose of 2 μM.
Finally, Huh7 cells stably transfected with a psiCHECK-miR122 Renilla luciferase reporter for endogenous miR122 activity35 were used to determine the anti-miR activity of PNA in situ synthesized on pore-widened PSi prior to ultrasonic particle formation (Figure 2h). With this reporter, luciferase is translated upon inhibition of miR122. PSNP delivery of a 2 μM dose of in situ synthesized anti-miR122 PNA triggered a significant, 44% increase in luciferase activity at 44 h after treatment, while treatment with an equivalent dose of free PNA or empty PSNPs had no significant effect relative to untreated controls. These results provide evidence that PSNPs improve the intracellular delivery and bioactivity of therapeutic PNA without the need for fusion with cell-penetrating peptides, or use of cytotoxic transfection reagents, such as that used to deliver the AMO control in this study.
DNA Biosensing using in Situ Synthesized PNA Probes
To demonstrate the utility of in situ PNA synthesis in biosensing, a model 16-mer probe PNA was synthesized from a 10 μm thick single-layer PSi film. For biosensing applications, PNA probes must be stably bound to the PSi substrate to achieve accurate and reproducible detection;37 hence, the PNA adsorption approach is not an effective approach for fabricating PSi biosensors (Figure S6 in the Supporting Information). Biosensor selectivity was evaluated by incubating with 10 μM solutions containing either a 100% complementary DNA target (NH2-A CGA GGA CCA TAG CTA-COOH) or a 100% mismatch DNA sequence (NH2-G GTT TCT GAT GCT GAC-COOH). As shown in Figure 3, negligible change in optical thickness was observed following incubation with either the mismatch DNA sequence or buffer alone, suggesting that there are no significant nonspecific binding events. In contrast, incubation with the complementary DNA target significantly increased the film optical thickness, confirming that DNA hybridized to the complementary PNA probe molecules. This is the first demonstration of selective nucleic acid biosensing with a covalently functionalized PNA–PSi biosensor. Further studies will be conducted to investigate whether PSi nucleic acid biosensors based on in situ synthesized PNA have superior selectivity and sensitivity relative to traditional DNA probe molecules, as has been suggested for other biosensing platforms.10,12
Figure 3.

Optical thickness-based hybridization assay using in situ synthesized 16-mer PNA on a single-layer PSi biosensor. Sequence-specific hybridization (indicated by a positive shift in optical thickness) was detected with a complementary target sequence but not with a mismatched control sequence. (*p < 0.0005).
Conclusions
A new method for automated PNA synthesis from PSi has been developed and applied for drug delivery and biosensing. The optical properties of nanostructured PSi were exploited to nondestructively monitor synthesis progression with single-base resolution. In situ synthesis addresses the need for efficient, covalent conjugation of PNA to PSi, as demonstrated by 8-fold greater PNA loading when compared to covalent attachment of presynthesized PNA to the surface via a cross-linker. The versatility of this in situ synthesized PNA platform was demonstrated by employing it for proof-of-concept drug delivery and nucleic acid biosensing applications. In situ synthesized anti-miR122-loaded PSi nanoparticles were biocompatible and increased the intracellular delivery and bioactivity of therapeutic PNA, while PNA probes synthesized from a PSi biosensor exhibited selective hybridization to DNA target molecules. Taken together, these studies demonstrate the utility of this conjugation strategy for developing improved systems for controlled intracellular delivery of PNA therapeutics, as well as more selective and sensitive nucleic acid biosensors.
Acknowledgments
The authors gratefully acknowledge Brian Evans for helpful discussions and assistance with peptide synthesis and HPLC, and Dr. Christopher Nelson for help with confocal microscopy and cell culture. The authors would also like to thank Dr. Alexander Deiters’ group at North Carolina State University for kindly providing the cell lines used in this study, Dr. Robert Coffey’s group at Vanderbilt University for access and assistance to the Nanosight used for NTA analysis, and Dr. John McLean’s group at Vanderbilt University for providing access and technical expertise with MADLI-MS. This work was supported in part by the National Science Foundation (DMR-120701 and ECCS-0746296), the Army Research Office (W911NF-09-1-0101), and a National Science Foundation Graduate Research Fellowship to KRB. Confocal Imaging was performed in part through the use of the VUMC Cell Imaging Shared Resource, (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and Ey008126).
Glossary
Abbreviations
- PNA
peptide nucleic acid
- PSi
porous silicon
- miR
microRNA
Supporting Information Available
Detailed experimental methods and schematics for in situ synthesis, physical adsorption, and direct conjugation of PNA. Detailed mass spectrometry experimental methods. In situ PNA synthesis in pore-widened PSi. Experimental methods for in vitro PNA release study. Experimental methods for generating porous silicon nanoparticles, including characterization of multilayer PSi film by scanning electron microscopy. Cell culture protocols and experimental methods for biological assays. Adsorbed PNA surface stability study. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nielsen P. E.; Egholm M.; Berg R. H.; Buchardt O. (1991) Sequence-selective recognition of dna by strand displacement with a thymine-substituted polyamide. Science 254(5037), 1497–1500. [DOI] [PubMed] [Google Scholar]
- Braasch D. A.; Corey D. R. (2001) Synthesis, analysis, purification, and intracellular delivery of peptide nucleic acids. Methods 23(2), 97–107. [DOI] [PubMed] [Google Scholar]
- Egholm M.; Buchardt O.; Christensen L.; Behrens C.; Freier S. M.; Driver D. A.; Berg R. H.; Kim S. K.; Norden B.; Nielsen P. E. (1993) PNA hybridizes to complementary oligonucleotides obeying the watson-crick hydrogen-bonding rules. Nature 365(6446), 566–568. [DOI] [PubMed] [Google Scholar]
- Ratilainen T.; Holmen A.; Tuite E.; Nielsen P. E.; Norden B. (2000) Thermodynamics of sequence-specific binding of PNA to DNA. Biochemistry 39(26), 7781–7791. [DOI] [PubMed] [Google Scholar]
- Demidov V.; Frankkamenetskii M. D.; Egholm M.; Buchardt O.; Nielsen P. E. (1993) Sequence selective double-strand DNA cleavage by peptide nucleic-acid (PNA) targeting using nuclease S1. Nucleic Acids Res. 21(9), 2103–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidov V. V.; Potaman V. N.; Frank-Kamenetskil M.; Egholm M.; Buchard O.; Sönnichsen S. H.; Nlelsen P. E. (1994) Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 48(6), 1310–1313. [DOI] [PubMed] [Google Scholar]
- Nielsen P. E. (1999) Applications of peptide nucleic acids. Curr. Opin. Biotechnol. 10(1), 71–75. [DOI] [PubMed] [Google Scholar]
- Larsen H. J.; Bentin T.; Nielsen P. E. (1999) Antisense properties of peptide nucleic acid. BBA Gene Struct. Expr. 1489(1), 159–166. [DOI] [PubMed] [Google Scholar]
- Oh S. Y.; Ju Y.; Park H. (2009) A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol. Cells 28(4), 341–345. [DOI] [PubMed] [Google Scholar]
- Briones C.; Moreno M. (2012) Applications of peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) in biosensor development. Anal. Bioanal. Chem. 402(10), 3071–3089. [DOI] [PubMed] [Google Scholar]
- Fabbri E.; Brognara E.; Borgatti M.; Lampronti I.; Finotti A.; Bianchi N.; Sforza S.; Tedeschi T.; Manicardi A.; Marchelli R.; Corradini R.; Gambari R. (2011) miRNA therapeutics: delivery and biological activity of peptide nucleic acids targeting miRNAs. Epigenomics 3(6), 733–745. [DOI] [PubMed] [Google Scholar]
- Wang J.; Palecek E.; Nielsen P. E.; Rivas G.; Cai X.; Shiraishi H.; Dontha N.; Luo D.; Farias P. A. M. (1996) Peptide nucleic acid probes for sequence-specific DNA biosensors. J. Am. Chem. Soc. 118(33), 7667–7670. [Google Scholar]
- Wittung P.; Kajanus J.; Edwards K.; Nielsen P.; Nordén B.; Malmström B. G. (1995) Phospholipid membrane permeability of peptide nucleic acid. FEBS Lett. 365(1), 27–29. [DOI] [PubMed] [Google Scholar]
- Lennox K. A.; Behlke M. A. (2011) Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 18(12), 1111–1120. [DOI] [PubMed] [Google Scholar]
- Fang H.; Zhang K.; Shen G.; Wooley K. L.; Taylor J.-S. A. (2009) Cationic shell-cross-linked knedel-like (cSCK) nanoparticles for highly efficient PNA delivery. Mol. Pharmaceutics 6(2), 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehiri M.; Upert G.; Tripathi S.; Di Giorgio A.; Condom R.; Pandey V. N.; Patino N. (2008) An efficient biodelivery system for antisense polyamide nucleic acid (PNA). Oligonucleotides 18(3), 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T.; Godin B.; Bhavane R.; Nieves-Alicea R.; Gu J.; Liu X.; Chiappini C.; Fakhoury J. R.; Amra S.; Ewing A. (2010) In vivo evaluation of safety of nanoporous silicon carriers following single and multiple dose intravenous administrations in mice. Int. J. Pharm. 402(1–2), 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin E.; Cheng L.; Freeman W.; Sailor M. (2008) Porous silicon in drug delivery devices and materials. Adv. Drug Delivery Rev. 60(11), 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholski C. (2013) Photonic crystal sensors based on porous silicon. Sensors 13(4), 4694–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane A.; Dronov R.; Hodges A.; Voelcker N. H. (2009) Porous silicon biosensors on the advance. Trends Biotechnol. 27(4), 230–239. [DOI] [PubMed] [Google Scholar]
- Kashanian S.; Harding F.; Irani Y.; Klebe S.; Marshall K.; Loni A.; Canham L.; Fan D.; Williams K. A.; Voelcker N. H.; Coffer J. L. (2010) Evaluation of mesoporous silicon/polycaprolactone composites as ophthalmic implants. Acta Biomater. 6(9), 3566–3572. [DOI] [PubMed] [Google Scholar]
- Archer M.; Christophersen M.; Fauchet P. (2004) Macroporous silicon electrical sensor for DNA hybridization detection. Biomed. Microdevices 6(3), 203–211. [DOI] [PubMed] [Google Scholar]
- Furbert P.; Lu C.; Winograd N.; DeLouise L. (2008) Label-free optical detection of peptide synthesis on a porous silicon scaffold/sensor. Langmuir 24(6), 2908–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T. P.; van Poll M. L.; Dankers P. Y. W.; Huck W. T. S. (2004) Forced peptide synthesis in nanoscale confinement under elastomeric stamps. Angew. Chem. 116(32), 4286–4289. [DOI] [PubMed] [Google Scholar]
- McInnes S. J.; Voelcker N. (2012) Porous silicon-based nanostructured microparticles as degradable supports for solid-phase synthesis and release of oligonucleotides. Nanoscale Res. Lett. 7(1), 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrie J. L.; Xu Z.; Rong G.; Laibinis P. E.; Weiss S. M. (2009) Synthesis of DNA oligonucleotides in mesoporous silicon. Phys. Status Solidi A 206(6), 1339–1342. [Google Scholar]
- Torres A. G.; Fabani M. M.; Vigorito E.; Williams D.; Al-Obaidi N.; Wojciechowski F.; Hudson R. H. E.; Seitz O.; Gait M. J. (2012) Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs. Nucleic Acids Res. 40(5), 2152–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling C. L.; Yi M.; Lancaster A. M.; Lemon S. M.; Sarnow P. (2005) Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309(5740), 1577–1581. [DOI] [PubMed] [Google Scholar]
- Padmanabhan S.; Coughlin J.; Iyer R. (2005) Microwave-assisted functionalisation of solid supports: application in the rapid loading of nucleosides on controlled-pore-glass (CPG). Tetrahedron Lett. 46, 343–347. [Google Scholar]
- Lawrie J. L.; Jiao Y.; Weiss S. M. (2010) Size-dependent infiltration and optical detection of nucleic acids in nanoscale pores. IEEE Trans. Nanotechnol. 9(5), 596–602. [Google Scholar]
- Rasmussen H.; Liljefors T.; Petersson B.; Nielsen P. E.; Kastrup J. S. (2004) The influence of a chiral amino acid on the helical handedness of PNA in solution and in crystals. J. Biomol. Struct. Dyn. 21(4), 495–502. [DOI] [PubMed] [Google Scholar]
- Gaur G.; Koktysh D. S.; Weiss S. M. (2013) Immobilization of quantum dots in nanostructured porous silicon films: characterizations and signal amplification for dual-mode optical biosensing. Adv. Funct. Mater. 23(29), 3604–3614. [Google Scholar]
- Filipe V.; Hawe A.; Jiskoot W. (2010) Critical evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm. Res. 27(5), 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G.; Simard M. J.; Mello C. C.; Zamore P. D. (2004) Sequence-specific inhibition of small RNA function. PLoS Biol. 2(4), e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly C. M.; Thomas M.; Deiters A. (2012) High-throughput luciferase reporter assay for small-molecule inhibitors of microRNA function. J. Biomol. Screen. 17(6), 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos H. A.; Riikonen J.; Salonen J.; Mäkilä E.; Heikkilä T.; Laaksonen T.; Peltonen L.; Lehto V.-P.; Hirvonen J. (2010) In vitro cytotoxicity of porous silicon microparticles: Effect of the particle concentration, surface chemistry and size. Acta Biomater. 6(7), 2721–2731. [DOI] [PubMed] [Google Scholar]
- Zourob M. (2010) Recognition Receptors in Biosensors, Springer. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.