

Abstract
Creating covalent protein conjugates is an active area of research due to the wide range of uses for protein conjugates spanning everything from biological studies to protein therapeutics. Protein Farnesyltransferase (PFTase) has been used for the creation of site-specific protein conjugates, and a number of PFTase substrates have been developed to facilitate that work. PFTase is an effective catalyst for protein modification because it transfers Farnesyl diphosphate (FPP) analogues to protein substrates on a cysteine four residues from the C-terminus. While much work has been done to synthesize various FPP analogues, there are few reports investigating how mutations in PFTase alter the kinetics with these unnatural analogues. Herein we examined how different mutations within the PFTase active site alter the kinetics of the PFTase reaction with a series of large FPP analogues. We found that mutating either a single tryptophan or tyrosine residue to alanine results in greatly improved catalytic parameters, particularly in kcat. Mutation of tryptophan 102β to alanine caused a 4-fold increase in kcat and a 10-fold decrease in KM for a benzaldehyde-containing FPP analogue resulting in an overall 40-fold increase in catalytic efficiency. Similarly, mutation of tyrosine 205β to alanine caused a 25-fold increase in kcat and a 10-fold decrease in KM for a coumarin-containing analogue leading to a 300-fold increase in catalytic efficiency. Smaller but significant changes in catalytic parameters were also obtained for cyclo-octene- and NBD-containing FPP analogues. The latter compound was used to create a fluorescently labeled form of Ciliary Neurotrophic Factor (CNTF), a protein of therapeutic importance. Additionally, computational modeling was performed to study how the large non-natural isoprenoid analogues can fit into the active sites enlarged via mutagenesis. Overall, these results demonstrate that PFTase can be improved via mutagenesis in ways that will be useful for protein engineering and the creation of site-specific protein conjugates.
Introduction
The creation of protein conjugates as tools for biological studies and as effective therapeutics is currently an area of intense interest.1−3 Targets include protein–protein conjugates,4,5 protein–DNA conjugates,6,7 and protein–small molecule conjugates.8−10 There are a number of different techniques already available to create these types of protein conjugates including native protein ligation,11 non-natural amino acid expression,12 chemical synthesis,13 and enzymatic labeling.14 Most of these methods require the initial attachment of a small molecule handle that can then undergo further reaction to create the final desired conjugate.
Post-translational enzymatic labeling offers several advantages over other types of protein labeling techniques. Reaction conditions are typically mild and help to preserve the biological functionality of the protein target;15 it can occur rapidly with high specificity;16,17 and it generally gives a high yield.18 Many investigators have used enzymatic labeling to introduce non-natural functionalities onto proteins for the purpose of creating protein conjugates. This strategy works via the use of alternative substrates bearing bioorthogonal functional groups that then become enzymatically incorporated onto the protein of interest. Enzymes, which have been used for this type of protein labeling,19 include biotin ligase,20 sortase,21 lipoic acid ligase,22 and protein farnesyltransferase.23
Protein farnesyltransferase (PFTase) is an α/β heterodimer enzyme which catalyzes the attachment of a farnesyl group, from farnesyl diphosphate (FPP), through a thioether bond to a cysteine near the C-terminus of a number of proteins (Figure 1).24
Figure 1.
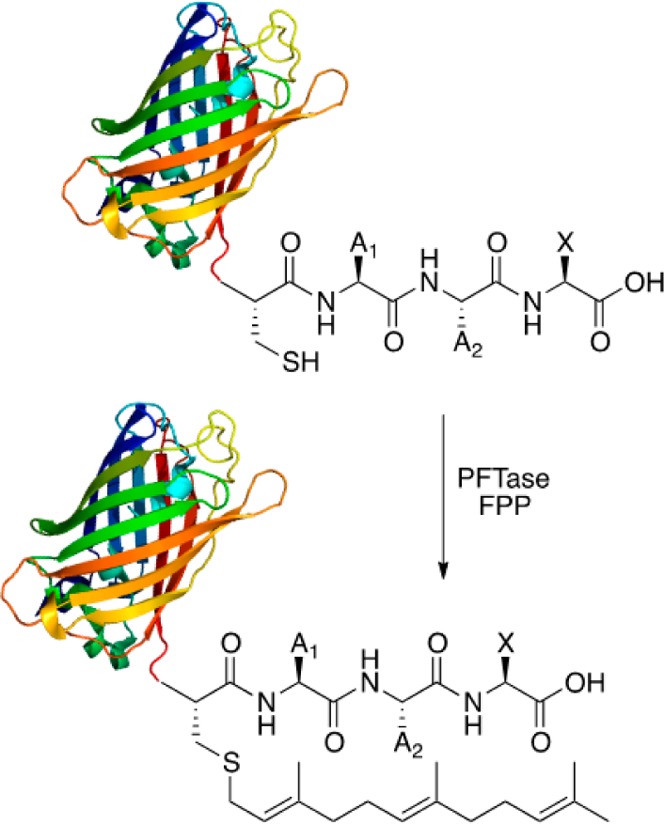
Scheme of protein prenylation. The terminal four amino acid sequence (CaaX) serves as the recognition site for the enzyme which then catalyzes the attachment of the farnesyl moiety onto the cysteine residue.
This cysteine is located four residues upstream from the C-terminus, and the sequence of the next three amino acids determines whether the protein becomes farnesylated or prenylated with a longer geranylgeranyl isoprenoid by protein geranylgeranyltranserfase I (PGGTase I).25 This post-translation modification plays a role in anchoring proteins to the cellular membrane and also modulates protein–protein interactions. The process of protein prenylation has attracted significant attention since many diseases including cancer require prenylated proteins for their effects.26,27
Recently, PFTase has been used by a number of groups for selective labeling of a variety of different proteins including several with potential therapeutic applications.28,29 Interestingly, PFTase is promiscuous with regard to its isoprenoid specificity and can tolerate a variety of modifications, predominantly in the third isoprenoid unit of the FPP.30−35 A number of different groups have synthesized a wide variety of probes for use in protein labeling.36 These include FPP analogues that contain azides and alkynes,23,37 for the click reaction; aldehydes and ketones for aminooxy conjugation;28,38 NBD39 and anthranilate40 for fluorescent studies; and benzophenone41,42 and diazotrifluoropropanoyl43 for photolabeling. Highly unusual non-isoprenoidal analogues can also be accommodated in some cases.44
One of the limitations to using non-natural FPP analogues with bioorthogonal functionality is that they have low activity when compared to natural FPP. This is especially true with the FPP analogues that contain bulkier moieties. In order for these types of analogues to be more readily used in protein labeling it would be helpful to have PFTase variants that could catalyze the incorporation of different FPP analogues into proteins more efficiently compared to the wild-type enzyme. Additionally, having different PFTases that can catalyze the reaction preferentially for different FPP analogues would be advantageous for multiple substrate labeling experiments.
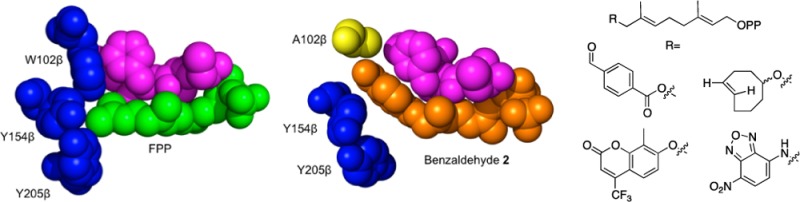
In order to allow for bulkier isoprenoid analogues to be efficiently processed by PFTase, the first step is to understand the binding of FPP in the PFTase binding pocket, specifically looking at the interactions between the third isoprenoid unit and PFTase. The PFTase binding pocket consists mainly of large aromatic amino acids that act as the backstop for FPP (Figure 2). There are three specific residues in the binding pocket that come into close contact with the third isoprenoid unit of FPP. All those residues lie within the β subunit of the enzyme and include a tryptophan at position 102, a tyrosine at position 154, and a second tyrosine at position 205. The other molecular structures that come in close contact with the third isoprenoid unit are amino acids in the CaaX box substrate itself.
Figure 2.
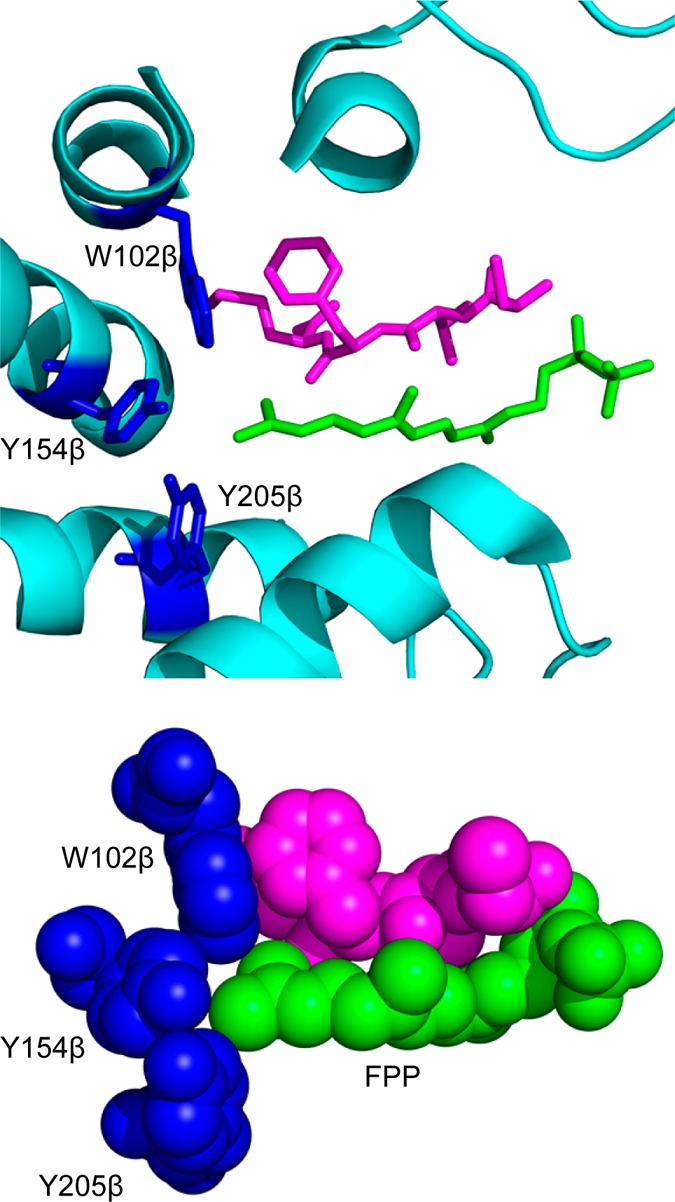
FPP binding pocket of PFTase (1JCR). (Above) The FPP binding pocket of PFTase. The isoprenoid and key residues are shown in stick representations with the protein secondary structure given in a cartoon form. Color scheme: the isoprenoid portion of FPP is shown in green. A peptide inhibitor CVFM that is bound in the CaaX substrate binding pocket is shown in purple. Key amino acid residues in the binding pocket are shown in blue. (Below) Space filling representation of the PFTase binding pocket. The color scheme is the same as that used in the top panel.
Previously, mutants of the PFTase binding pocket have primarily been used to study how prenyltransferases distinguish between FPP and GGPP45,46 and how they alter CaaX-box peptide specificity.47 While generally similar, comparison of the structures of PGGTase I and PFTase revealed that PFTase contains a tryptophan residue at position 102 (W102β) whereas PGGTase I has a threonine residue at the analogous location. Terry et al. have shown that by mutating the tryptophan at 102β to a threonine and the tyrosine at 361β to a phenylalanine, which was also observed from an overlay of the binding pockets, that PFTase could employ GGPP as a substrate.48 Additionally, it has been shown that by mutating W102β, Y154β, and Y205β amino acids all to threonine, there is greater incorporation of both biotin- and nitrobenzofurazan-containing FPP analogues by the resulting mutant PFTases, although the kinetic behavior of these mutants was not investigated in detail.49 Overall, these studies suggest that it is possible to engineer PFTase to accept FPP analogues.
In the present study, we extend this work to isoprenoid analogues that contain bioorthogonal functionality that would be useful for site-specific protein labeling applications. Hence, here we report on the preparation of several mutant forms of PFTase including W102Aβ, Y154Aβ, and Y205Aβ and their reactivity with aldehyde-, cyclo-octene-, NBD- and coumarin-containing FPP analogues. Significant increases in catalytic efficiency with the aldehyde- and coumarin-containing substrates were obtained suggesting that protein engineering is a fruitful approach for improving PFTase for biocatalysis.
Results and Discussion
Mutant Creation
In order to explore the different binding capabilities of PFTase, we designed and synthesized three different mutants of rat PFTase to enlarge the isoprenoid binding pocket. Our studies focused on three amino acids in the substrate binding pocket including W102β, Y154β, and Y205β. As can be seen in Figure 2, these three residues (shown in blue) form the bottom of the binding pocket and limit the length of the isoprenoid (shown in green) that can fit into the PFTase active site along with the protein substrate (shown in pink). Since our group has developed a number of different isoprenoid substrates that are longer than FPP (Figure 3), we hypothesized that we could improve the activity of PFTase with these analogues by creating mutants that contained smaller residues at these three positions that would enlarge the active site. As shown in Table 1, the volume occupied by these different analogues is 5–25% greater than the volume of FPP. By mutating the large residues found in the PFTase binding pocket, we reasoned that it should be possible to accommodate these larger analogues. Additionally, we wanted to explore which of these mutations were the most important for improving the catalytic properties of PFTase toward these analogues. We reasoned that it might be possible to create “tunable” PFTases where the different mutations would have different analogue specificities.
Figure 3.

Structures of farensyl diphosphate and analogues. (Left) Lewis structure representation. (Right) Extended conformation space fill model. Structures include 1 (FPP); 2 (an aryl aldehyde-containing analogue; 3 (a cyclooctene-containing analogue); 4 (a coumarin-containing analogue); and 5 (an NBD-containing analogue). Carbon is shown in green, oxygen in red, nitrogen in blue, phosphorus in orange, and fluorine in light blue.
Table 1. Molecular Volume of FPP and Four Synthetic Analoguesa.
| functionality | compound | molecular volume (Å3) |
|---|---|---|
| None (FPP) | 1 | 306.0 |
| Benzaldehyde | 2 | 333.0 |
| Cyclooctene | 3 | 322.0 |
| Coumarin | 4 | 382.0 |
| NBD | 5 | 338.0 |
Volumes were calculated using the molecular modeling program Schrodinger and the values are given in units of cubic angstroms (Å3).
Accordingly, three mutants W102Aβ, Y154Aβ, and Y205Aβ were created using site directed mutagenesis and expressed in E. coli as His-tagged polypeptides. The resulting mutant proteins were purified via immobilized metal affinity chromatography to yield the desired proteins in good yield (20 mg/L of culture broth).
Kinetic Analysis with FPP
The three mutant proteins were initially assayed for enzymatic activity using a continuous fluorescence assay using N-dansyl-GCVLS and FPP as substrates. This assay monitors the increase in dansyl group fluorescence as the peptide substrate becomes modified by the hydrophobic isoprenoid. These experiments were performed at a constant concentration of peptide substrate while the concentration of FPP was varied to obtain the kcat and KM parameters listed in Table 2. Inspection of those values shows a 2- to 3-fold reduction in both kcat and KM suggesting that mutation of those residues has minimal effect on the catalytic efficiency of PFTase when FPP is the substrate.
Table 2. Kinetic Values for PFTase and Three Mutants Using FPP as the Isoprenoid Substratea.
| protein | kcat (s–1) | KM (μM) | kcat/KM (μM s–1) | rel. kcat/KM |
|---|---|---|---|---|
| WT | 0.27 ± 0.02 | 0.5 ± 0.2 | 0.5 ± 0.2 | 1 |
| W102A | 0.083 ± 0.005 | 0.26 ± 0.09 | 0.3 ± 0.1 | 0.59 |
| Y154A | 0.11 ± 0.007 | 0.27 ± 0.09 | 0.4 ± 0.1 | 0.76 |
| Y205A | 0.18 ± 0.01 | 0.5 ± 0.2 | 0.4 ± 0.1 | 0.67 |
All values are apparent kinetic parameters since the peptide concentration was held constant at 2 μM. Values are reported here as the averages and standard deviations from three separate experiments.
Kinetic Analysis with Benzaldehyde Analogue (2)
Next, the three mutants were assayed using an aryl aldehyde-containing FPP analogue (2). In previous work, we have shown that 2 can be used to incorporate an aldehyde functional group into a number of different proteins. In contrast to azide- and alkyne-containing analogues that require Cu(I) or synthetically complex strained-ring reagents for their subsequent derivatization, aldehydye-based compounds can be easily functionalized with commercially available hydrazine- or alkoxyamine-containing moieties. In addition, using recently developed aryl amine catalysts, they react rapidly with half-lives of less than 2 min.28 Given their larger size relative to FPP (see Figure 3), we hypothesized that they should fit better into the enlarged active sites generated by mutagenesis described here.
The results of kinetic studies with benzaldehyde 2 using the continuous fluorescence described above are given in Table 3. As was previously reported, wild-type PFTase catalyzes the transfer of the benzaldehyde-containing isoprenoid 2 several hundred-fold (kcat/KM) less efficiently than FPP. However, importantly, the W102Aβ mutant manifests a (43-fold) increase in catalytic efficiency in processing 2 compared with FPP. That increase is derived from a 4-fold increase in kcat and a 12-fold decrease in KM. In contrast, the Y154Aβ and Y205Aβ mutants showed only small increases in catalytic efficiency. In those cases, decreases in KM values were offset by decreases in kcat. These observations suggest that increasing the active site dimensions allows more favorable interactions between the substrate and enzyme to occur resulting in lower KM values. However, modulation of kcat is clearly dependent on other factors including the precise conformation of the substrate which is harder to predict. Nevertheless, these findings are quite exciting given the relatively large increase in catalytic efficiency obtained with the W102Aβ mutation.
Table 3. Kinetic Values for PFTase and Three Mutants Using the Benzaldehyde Containing Analogue (2) as the Substratea.
| protein | kcat (s–1) | KM (μM) | kcat/KM (μM s–1) | rel. kcat/KM |
|---|---|---|---|---|
| WT | 0.0021 ± 0.0002 | 2.4 ± 0.6 | 0.0008 ± 0.0002 | 1 |
| W102A | 0.0076 ± 0.0005 | 0.2 ± 0.1 | 0.04 ± 0.02 | 43 |
| Y154A | 0.0014 ± 0.0009 | 0.7 ± 0.2 | 0.002 ± 0.001 | 2.3 |
| Y205A | 0.0012 ± 0.0006 | 0.5 ± 0.1 | 0.002 ± 0.001 | 2.7 |
All values are presented as apparent kinetic parameters with the peptide concentration held constant at 2 μM. Values are reported here as the averages and standard deviations from three separate experiments.
Kinetic Analysis with Cyclooctene Analogue (3)
Next, we choose to examine the ability of the mutants to process an FPP analogue appended with a moiety different in shape from the planar benzaldehyde group present in 2. Compound 3 incorporates a trans cyclo-octene group for subsequent reaction via an inverse electron demand Diels–Alder reaction. That type of conjugation reaction has the advantage of having a rate of ligation of 2000 M–1 s–1,50 substantially faster than other traditionally used ligation reactions including the “click” reaction. However, compared with azides and alkynes, the cyclo-octene moiety is larger and must be in the trans configuration. Hence, we reasoned that it would be a good candidate to evaluate with the mutant PFTases described here that have enlarged active sites.
Our initial screen of the mutant PFTases using the aforementioned continuous fluorescence assay showed that, at high concentrations of the cyclo-octene analogue, only the Y205Aβ mutant catalyzed the incorporation of 3 at a faster rate compared to the wild-type enzyme; the W102Aβ and Y154Aβ enzymes did catalyze the transfer of 3 but at a slower rate (data not shown). Hence a more detailed kinetic analysis was performed only with the wild-type enzyme and the Y205Aβ mutant; kinetic constants from those experiments are summarized in Table 4.
Table 4. Kinetic Values for PFTase and the Y205A Mutant Using the Cyclo-Octene Containing Analogue (3) as the Substratea.
| protein | kcat (s–1) | KM (μM) | kcat/KM (μM s–1) | rel. kcat/KM |
|---|---|---|---|---|
| WT | 0.017 ± 0.002 | 0.2 ± 0.1 | 0.08 ± 0.04 | 1 |
| Y205A | 0.082 ± 0.007 | 0.5 ± 0.2 | 0.16 ± 0.07 | 2 |
All values are presented as kinetic parameters with the peptide concentration held constant at 2 μM. Values are reported here as the averages and standard deviations from three separate experiments.
Interestingly, for the wild-type enzyme, the KM value for 3 (0.2 μM) is 2.5-fold less than that observed for FPP (0.5 μM, see Table 1). For the Y205Aβ mutant, the trend is reversed with the KM value for 3 being higher than that for FPP. However, perhaps more importantly, the kcat value for 3 is almost 5-fold higher for the Y205Aβ mutant compared with the wild-type enzyme. Since the Y205Aβ mutant manifests a KM value that is submicromolar, it is relatively easy to saturate the enzyme with 3 and hence fully capitalize on the ca. 5-fold improvement in kcat for in vitro protein labeling applications.
PFTase FRET Assay Using a Protein Substrate and a Fluorescent Isoprenoid
In addition to analogues 2 and 3 which contain bioorthogonal functionality, we also wanted to evaluate some alternative substrates that contain intrinsically fluorescent moieties. These latter compounds are particularly interesting since their incorporation could, in principle, allow direct fluorescent labeling of proteins without the need for a secondary modification reaction to install the fluorophore. Several types of fluorescent FPP analogues have been previously used to label proteins. However, those molecules interfere with the continuous fluorescence assay used for the experiments described above rendering kinetic analyses of their activity difficult.
To solve this problem, we developed a continuous fluorescence assay that can monitor the attachment of a coumarin-containing FPP analogue (4) to a GFP variant equipped with a C-terminal CVIA sequence that makes it a substrate for PFTase. Because the emission spectrum of the coumarin fluorophore overlaps with the excitation spectrum of GFP, PFTase mediated prenylation with the coumarin analogue results in fluorescence resonance energy transfer (FRET). Figure 4a shows that when GFP-CVIA and the coumarin analogue are mixed together in the absence of PFTase and the reaction is excited at 330 nm (a wavelength suitable to excite the coumarin) emission occurs at 460 nm due to the coumarin emission as well as a smaller amount at 510 nm due to GFP fluorescence; this latter emission occurs because there is a low level of absorption at 330 nm by GFP. However, upon addition of PFTase, a significant increase in the 510 nm emission occurs along with a concomitant decrease in 460 nm fluorescence signaling the covalent attachment of the coumarin moiety to GFP. Figure 4b shows that the enzymatic reaction can be monitored by exciting the coumarin fluorophore and monitoring the increase in GFP fluorescence intensity using this FRET-based process. This assay has the benefit of not only allowing continuous monitoring of coumarin incorporation, but also does so using a whole protein in lieu of a small peptide. The presence of the peptide sequence at the C-terminus of a protein more accurately represents the true context in which prenylation occurs compared with short peptide sequences since upstream sequences are known to modulate substrate affinity such as in the case of K-Ras.51
Figure 4.
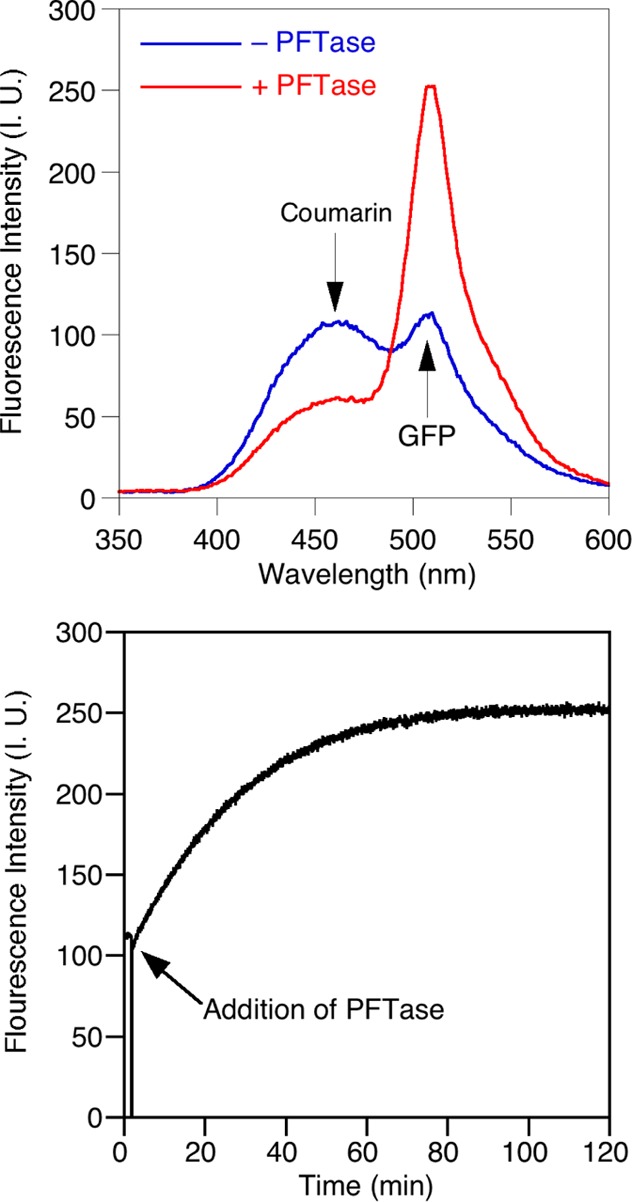
Development of a continuous FRET assay for PFTase: (Above) Emission spectra of a reaction mixture containing GFP-CVIA and coumarin FPP analogue before and after the addition of PFTase showing overall decrease in the fluorescence intensity of the coumarin at 460 nm with simultaneous increase in the fluorescence intensity of GFP at 510 nm (λex = 330 nm). (Below) Continuous FRET assay based on labeling of GFP with the coumarin analogue showing an increase in the fluorescence intensity at 510 nm after the addition of PFTase (λex = 330 nm).
In order to confirm that the observed fluorescence increase was due to covalent attachment of the coumarin moiety to the GFP, a variant of GFP that contained a CVLL sequence instead of a CVIA tag was employed. The CVLL CaaX-box is preferentially recognized by GGTase-I and is not a good substrate for PFTase. If the observed increase in fluorescence was due to association of GFP with either PFTase or the courmarin analogue, then changing the CaaX-box should not affect the rate of fluorescence increase. However, when the assay was performed using GFP-CVLL (see SI Figure S9), a much slower increase in fluorescence was observed compared with that seen with GFP-CVIA. This is consistent with the fact that GFP-CVLL is a poor substrate for PFTase. Finally, to definitively determine whether the fluorescence increase was coming from the covalent modification of the GFP, mass spectrometry was performed to confirm that GFP-CVIA was being modified with the coumarin analogue. The ESI-MS results (see SI Figure S10) showed that unmodified GFP had a mass of 27 334 Da, whereas after reaction with the coumarin analogue and PFTase, the GFP had been transformed to a species with mass of 27 713 Da. That difference, 379 Da, is consistent with covalent attachment of the courmain-isoprenoid to the GFP. Based on these results it can be concluded that PFTase is catalyzing the attachment of coumarin resulting in the FRET interaction between the GFP-fluorophore and the coumarin moiety on the isoprenoid.
Kinetic Analysis with the Coumarin Analogue
This FRET assay described above was used to examine the rate of reaction for all three mutant enzymes at high coumarin analogue (4) concentrations (data not shown). Similar to results obtained with the cyclo-octene-containing compound (3), only the Y205Aβ mutant showed an increased rate and was further investigated to determine its kinetic parameters. As seen in Table 5, the Y205Aβ mutant enzyme manifests both a significant (ca. 25-fold) increase in kcat and a decrease (10-fold) in KM resulting in an overall impressive 300-fold increase in catalytic efficiency.
Table 5. Kinetic Values for PFTase and Three Mutants Using Coumarin Containing Analogue (4) as the Substratea.
| protein | kcat (min–1) | Km (μM) | kcat/KM (μM min–1) | rel. kcat/KM |
|---|---|---|---|---|
| WT | 0.08 ± 0.01 | 1.4 ± 0.3 | 0.06 ± 0.01 | 1 |
| Y205A | 2.1 ± 0.3 | 0.12 ± 0.08 | 20 ± 10 | 300 |
These values were generated using the PFTase FRET assay described herein. All values are reported as apparent kinetic parameters with the GFP-CVIA concentration held constant at 2.5 μM. Values are reported here as the averages and standard deviations from three separate experiments.
CNTF NBD Labeling
Next, we examined the activity of a different fluorescent FPP analogue that incorporates an NBD group developed by Waldmann and co-workers.39 Unfortunately, that compound cannot be assayed in the continuous fluorescence assay or the FRET assay described above due to incompatible spectral properties. Hence, a discontinuous assay based on SDS-PAGE and fluorescence scanning of the resulting gel was employed. Given the effort involved in performing this protocol, we elected to use a form of cilliary neurotrophic factor (CNTF) tagged with a C-terminal CVIA PFTase recognition sequence as a substrate. CNTF is a neurotrophic factor that is currently being investigated for a number of therapeutic applications,52−54 and hence fluorescently labeled forms of CNTF would be useful for a variety of studies.
CNTF was prenylated with the NBD analogue (5) using both the wild-type enzyme and the Y205Aβ mutant. The reaction was monitored as described above using a gel-based assay coupled with fluorescence scanning to detect the desired product. A plot illustrating the production of NBD-labeled CNTF as a function of time for the two different enzymes is shown in Figure 5. Inspection of that data reveals that the Y205Aβ mutant enzyme catalyzes the reaction approximately 1.5-fold faster than the wild-type. Since the reactions were performed at high (10 μM) concentrations of 5, it is likely that this effect is due to an increase in kcat. While the effect of mutation is not as substantial for this FPP analogue, these results are still significant from a practical perspective since use of the Y205Aβ mutant allows for a reduction in reaction time. Such a decrease would be helpful in decreasing potential proteolytic degradation that is sometimes observed in enzymatic labeling procedures.
Figure 5.
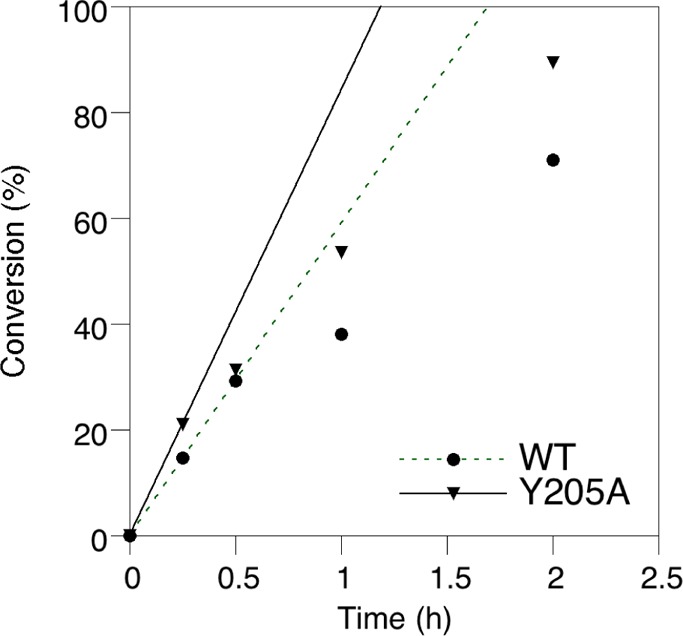
Labeling of CNTF with NBD analogue 5 by wild-type and Y205Aβ mutant PFTase. Fluorescence intensity of the fluorescence scan shown in the SI was calculated using ImageJ software. Conversion is based on the ratio of fluorescence at the given time point to the fluorescence at its maximum. The lines on the graph indicate the initial rate of enzyme-catalyzed NBD incorporation determined in the first 15 min of reaction.
Computational Modeling of Analogue Binding
To better understand the interactions between the different FPP analogues studied here and their recognition by mutant PFTases, a series of computational modeling studies were performed. Models for the different PFTase mutant proteins were generated using the crystal structure of wild-type PFTase, and then the various FPP analogues were modeled in the isoprenoid binding site using a molecular docking program. Of the four FPP analogues studied, aldehyde 2 and coumarin 4 manifested decreased KM values for different mutant enzymes relative to their activity with the wild type, suggesting that mutation improves their binding affinity. Consequently, modeling focused on those analogues.
When analogue 2 is docked into the wild-type enzyme active site (Figure 6a, red), the benzaldehyde group becomes occluded by the indole side chain of W102β amino acid which prevents the analogue from adopting an extended conformation of the type observed for FPP (green). Instead, the structure is “kinked” in the region of the second isoprenoid unit relative to the normal FPP-bound structure. It is quite likely that this perturbation is deleterious for binding and for efficient catalysis. In contrast, when 2 is docked into the W102β mutant structure (orange), sufficient space is available for the benzaldehyde moiety to fit into the area previously occupied by the indole, resulting in an overall conformation similar to that for FPP with minimal changes in the isoprenoid structure in the region where catalysis occurs.
Figure 6.
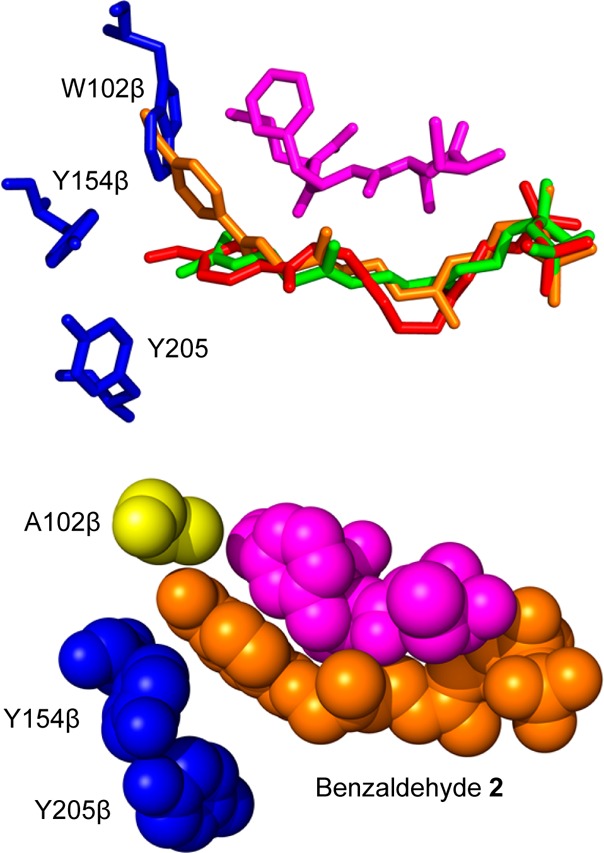
Computational modeling structures of benzaldehyde analogues in PFTase. (Above) Overlays of benzaldehyde analogue docked in WT structure (red), benzaldehyde analogue docked in W102A mutant (orange), and FPP bound in the WT structure (green). The peptide substrate (purple) and the three relevant amino acids (blue) are also displayed. Note the benzaldehyde analogue docked into mutant structure comes into contact with the tryptophan residue in the overlay. (Below) Benzaldehyde analogue docked into W102Aβ mutant binding pocket. The structure is shown as spheres to show the benzaldehyde moiety binding into the place occupied by the tryptophan residue in the WT structure. Benzaldehyde analogue showed in orange, peptides substrate shown in purple, residues Y154β and Y205β shown in blue, and the mutated W102β shown in yellow.
The docking results for the coumarin analogue 4 show a more drastic change in binding conformation (Figure 7). When 4 is docked into the wild-type structure, the highest scoring pose is one in which the coumarin ring is positioned outside the active site (red). Interestingly, that putative binding site partially overlaps with a second isoprenoid binding site present on PFTase previously identified in X-ray crystallographic experiments; in the PFTase catalytic cycle, it has been hypothesized that the isoprenoid group from a nascent prenylated protein is translocated to this second site prior to product release. Thus, it is reasonable that the coumarin analogue could bind in this site although not in a catalytically productive fashion. Lower scoring poses do show binding of 4 within the active site, but in those cases, the structure of the isoprenoid is distorted in a way that is likely to be less productive for enzymatic reaction as was observed with 2, described above. A more likely explanation for how 4 binds in the active site is that side chain rearrangements occur that allow the analogue to be better accommodated; this has been previously observed in the binding of other probes to PFTase.43 In contrast, when the coumarin analogue is docked into the Y205A mutant structure (orange), the aromatic moiety fits neatly into the space previously occupied by the tyrosyl phenolic side chain. This in turn allows for the rest of the isoprenoid chain to adopt a conformation similar to that manifested by FPP (green). Taken together, these computational results provide a compelling rationale for understanding the behavior of these mutant PFTase variants and suggest that docking experiments should be useful in predicting the effects of mutations as this enzyme is further engineered.
Figure 7.
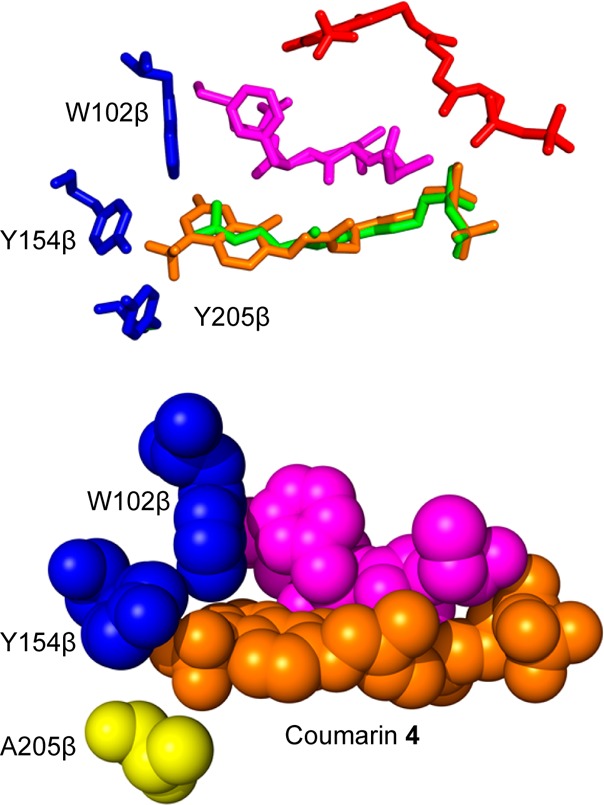
Computational modeling structures of coumarin analogues in PFTase. (Above) Overlays of coumarin analogue docked in WT structure (red), courmarin analogue docked in Y205A mutant (orange), and FPP bound in the WT structure (green). The peptide substrate (purple) and the three relevant amino acids (blue) are also displayed with the surface of the WT enzyme. (Below) Coumarin analogue docked into Y205A mutant binding pocket. The structure is shown as spheres to show the coumarin moiety binding into the place occupied by the tyrosine residue in the WT structure. Coumarin analogue showed in orange, peptides substrate shown in purple, residues W102β and Y154β shown in blue, and the mutated Y205β shown in yellow.
Conclusion
In conclusion, site directed mutagenesis has been used to enlarge the isoprenoid binding site of PFTase. Those mutant enzymes are able to process several FPP analogues that are significantly larger than FPP more efficiently than the wild-type enzyme. In some cases the catalytic efficiency has been increased ca. 300-fold. Much of this improved catalytic activity has come from an improvement in kcat, which is important if PFTase is used for a labeling reagent in vitro. For use in in vivo studies where the substrate concentration of the FPP analogue is at lower concentrations, decreases in KM will also be important for improving the enzyme labeling. One of these mutants has been used to prepare a fluorescently labeled form of CNTF, a therapeutically important protein. Computational docking experiments have been used to provide a rationale for understanding the interactions between the larger FPP analogues and the mutant enzymes. As PFTase labeling becomes more widely used and the number of FPP analogues grows, these enzymes should contribute significantly to the development of more advanced protein therapeutics.
Experimental Section
General Synthesis
FPP analogues were synthesized as previously reported.31,39,55,56 Reagents were purchased from Sigma-Aldrich unless otherwise noted.
Mutant Creation
PFTase mutants were created using the Quikchange II Site Directed Mutagenesis Kit from Agilent (Catalogue # 200523). The plasmid used as the template for mutants has been previously described.57 In brief, primers were designed containing an alanine mutation for each of the three amino acid positions for W102β, Y154β, and Y205β (see SI). The manufacturer’s protocol was followed for PCR conditions, DpnI DNA digestion, and E. coli transformation. Plasmids were purified using Wizard Plus SV Minipreps DNA Purification System (Catalogue # A1460). Mutations were confirmed via Sanger type DNA sequencing from the University of Minnesota Genomics center. Plasmids containing the proper mutation were transformed in electrocompetent JM109(DE3) cells and stored at −80 °C for long-term storage.
Protein Purification
JM109(DE3) cells were plated on LB-agar plates containing 50 μg/mL streptomycin and grown overnight at 37 °C. Single colonies were used to inoculate 50 mL of LB media containing 50 μg/mL streptomycin and grown overnight at 37 °C shaking at 250 rpm. 10 mL of that overnight culture was added to 1 L LB media containing 50 μg/mL streptomycin and grown to an OD600 of 0.8, by shaking at 250 rpm at 37 °C, which took approximately 3 h. Cells were induced to express the PFTase by addition of IPTG and ZnCl2 to final concentrations of 1 mM, and 500 μM, respectively. Cells were grown for 4 h at 37 °C, shaken at 250 rpm. Cells were harvested by centrifugation at 5400g for 10 min. Pelleted cells were stored at −80 °C prior to use.
The enzyme purification procedure for the various PFTases was performed as previously described.58 Briefly, the cell pellet from 1 L of liquid broth was suspended in 50 mL of lysis buffer containing 50 mM Tris·HCl (pH 7.0), 200 mM NaCl, 5 μM ZnCl2, 5 mM MgCl2, 20 mM imidazole, and 1 mM β-mercaptoethanol. To this, 1 mL of protease inhibitor cocktail was added (Sigma-Aldrich), and the cells were sonicated at 50 W for 5 min (10 s on/10 s off). The resulting solution was centrifuged at 13 000g for 30 min and the soluble fraction was then loaded onto a 30 mL Ni-NTA column bed equilibrated with lysis buffer at a rate of approximately 2 mL/min. The column was washed with lysis buffer until the A280 reached a minimum level where no further was observed (approximately 200 mL) and the PFTase enzyme was eluted using the above lysis buffer supplemented with 250 mM imidazole. Fractions containing PFTase were pooled together and concentrated using an Amicon Ultra-15 centrifugal filter from Millipore, and concentrated to 4 mL. This was diluted 10-fold with buffer containing 50 mM Tris·HCl, 200 mM NaCl, 5 μM ZnCl2, 5 mM MgCl2, and 1 mM β-mercaptoethanol and concentrated again to a volume of 4 mL; this dilution/concentration process was repeated a total of three times. The enzyme was stored in the latter buffer containing 50% glycerol at −80 °C. This purification typically yielded approximately 20 mg/L of liquid culture of PFTase.
Continuous fluorescence Assay for PFTase Activity Measurement
The assay for PFTase activity employed here using FPP and analogues 2 and 3 as substrates is based on the previously published assay by Pompliano et al.59 and later refined by the Poulter group.60N-Dansyl-GCVLS was preincubated with DTT for 30 min to ensure that no disulfide was present. After incubation, the peptide/DTT solution was used to make an assay solution that had a final concentration of 2 μM dansyl-CVLS, 5 mM DTT, 50 mM Tris·HCl (pH 7.5), 10 mM MgCl2, 10 μM ZnCl2, 0.2% n-octyl-β-d-glucoside, and varying concentrations of the FPP analogue being investigated, ranging from 0 to 25 μM. The enzyme concentration varied between 5 nM to 50 nM depending on the FPP analogue being studied.
The reactions were performed using a DTX 880 Multimode Detector plate reader (Beckman Coulter) in a black pinch bar 96-well plate (Nunc 237105) with a reaction volume of 250 μL. The reactions were performed by addition of 100 μL of a concentrated reaction solution (5 μM dansyl-GCVLS, 12.5 mM DTT, 125 mM Tris·HCl (pH 7.5), 25 mM MgCl2, 25 μM ZnCl2, 0.5% n-octyl-β-d-glucoside). The diphosphate analogue was added at varying concentrations along with H2O to give a volume in the well of 230 μL. An initial fluorescent measurement was taken using the excitation filter A340/10 to excite at 340 nm and emission was monitored at 505 nm using the F535/25 filter. Then, 20 μL of PFTase in enzyme buffer (50 mM Tris·HCl (pH 7.5), 50 μM ZnCl2, 5 mM MgCl2, 20 mM KCl, 1 mM DTT, 1 mg/mL BSA) was used to initiate the enzymatic reaction. The fluorescence was monitored until it reached a plateau signifying that the reaction was complete.
The data was exported from the Multimode Analysis Software, and the initial rate values were calculated using Microsoft Excel. This rate was calculated using the linear region of the progress curve. This was usually in the first 20 min of the assay, but for assays containing low substrate concentrations this time period was shorter. The enzymatic rate of each reaction was determined by converting the rate obtained in fluorescence intensity units (FIU/s) to μM/s with the equation vi = RP/ΔF, where vi is the enzymatic rate in μM/s, R is the measured rate in FIU/s, and P is the amount of product formed in the assay. ΔF is the change in fluorescence intensity between the start of the assay and when the reaction is complete. This rate data was exported to KaliedaGraph 3.6 and the kcat and Km were determined using a nonlinear least-squares analysis. All kinetic values reported here are apparent values because they were measured at single fixed concentration of peptide substrate.
FRET Assay for PFTase Activity Using Coumarin-Containing FPP Analogue 4
PFTase activity with coumarin-containing analogue 4 was detected using a FRET based assay. A 1 mL mixture containing 2.4 μM GFP-CVIA, 5 mM DTT, 10 mM MgCl2, 10 μM ZnCl2 and 50 mM Tris·HCl, pH 7.5, was transferred to a cuvette and the emission at 510 nm (λex = 330 nm) was monitored until a stable baseline was obtained. The reaction was initiated with 50 nM PFTase of the wild-type enzyme and 10 nM for the mutant enzyme and the increase in the fluorescence was monitored until a plateau was reached. To determine the kinetic parameters, the concentration of 4 was varied from 0.1 to 20 μM.
Gel-Based Assay for PFTase Activity Using NBD-Containing FPP Analogue 5
PFTase activity with the NBD-containing analogue 5 was measured using a gel-based assay. A reaction mixture containing 2 μM CNTF-CVIA, 50 mM Tris·HCl (pH 7.5), 10 mM MgCl2, 50 μM ZnCl2, 15 mM DTT, 20 mM KCl, 10 μM 5, and 5 nM PFTase was incubated at 30 °C and 500 μL aliquots were removed at various times, flash frozen, and lyophilized to dryness. The resulting residue was resuspended in 20 μL SDS-PAGE loading buffer and fractionated on a 12% SDS-PAGE gel. The labeled protein was visualized by scanning the gel for fluorescence using a Storm 840 fluorescence scanner with 450 nm excitation wavelength and 520 nm long pass emission filter. Quantitation of fluorescent images were performed using ImageJ software.
Computational Modeling
For modeling of FPP analogues into the active site of PFTase (pdb file 1JCR), docking was performed using MacroModel v 9.9 and its program Glide. The PFTase crystal structure was prepared using the default settings in the protein preparation wizard as part of the Maestro (Schrodinger, version 9.3) package. To generate models of the mutant enzymes, the PFTase structure generated from the protein wizard was used and only the mutant modification was made in Maestro. Once that was accomplished, the mutant amino acid was subjected to a local minimization search using the MacroModel feature to find the most likely conformation of the amino acid within the binding pocket. A receptor grid large enough to encompass the entire binding site for the FPP analogues was generated from the prepared PFTase enzyme or the prepared mutant enzymes. A standard precision docking parameter was set and 10 000 ligand poses per docking were run per analogue per enzyme model. The conformations with the overall highest binding score were chosen for display here.
Molecular volumes calculations of FPP and FPP analogues were performed using the volume calculation script from Schrodinger, version 9.3.
Acknowledgments
The authors thank Dr. Mohammad Rashidian for the preparation of compound 2 and Dr. Jake Vick for help in mutant construction. This work was funded in part by the National Institutes of Health (GM058442, GM084152, and T32 GM008700), an award from the Research Corporation for Science Advancement (#20888) and support from the Minnesota Supercomputer Institute.
Supporting Information Available
Description of primers for PFTase mutants, MS results for NBD labeled CNTF, gel results for NBD labeling of CNTF, and stereoscopic images for all structural figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hackenberger C. P. R.; Schwarzer D. (2008) Chemoselective ligation and modification strategies for peptides and proteins. Angew. Chem., Int. Ed. 47, 10030–10074. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendoerfer G. G. (2005) Site-specific polymer modification of therapeutic proteins. Curr. Opin. Chem. Biol. 9, 555–560. [DOI] [PubMed] [Google Scholar]
- Witte M. D.; Cragnolini J. J.; Dougan S. K.; Yoder N. C.; Popp M. W.; Ploegh H. L. (2012) Preparation of unnatural N-to-N and C-to-C protein fusions. Proc. Natl. Acad. Sci. U.S.A. 109, 11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudak J. E.; Barfield R. M.; de Hart G. W.; Grob P.; Nogales E.; Bertozzi C. R.; Rabuka D. (2012) Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew. Chem., Int. Ed. 51, 4161–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudiuk S.; Venancio-Marques A.; Baigl D. (2012) Enhancement and modulation of enzymatic activity through higher-order structural changes of giant DNA–protein multibranch conjugates. Angew. Chem., Int. Ed. 51, 12694–12698. [DOI] [PubMed] [Google Scholar]
- Sacca B.; Niemeyer C. M. (2011) Functionalization of DNA nanostructures with proteins. Chem. Soc. Rev. 40, 5910–5921. [DOI] [PubMed] [Google Scholar]
- Ban H.; Nagano M.; Gavrilyuk J.; Hakamata W.; Inokuma T.; Barbas C. F. (2013) Facile and stabile linkages through tyrosine: bioconjugation strategies with the tyrosine-click reaction. Bioconjugate Chem. 24, 520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagath R. J.; Helga R.; Suzanna C.; Sunil B.; Douglas D. L.; Sylvia W.; Yvonne C.; Michelle S.; Siao Ping T.; Mark S. D.; Yanmei L.; Meng Y. G.; Carl N.; Jihong Y.; Chien C. L.; Eileen D.; Jeffrey G.; Viswanatham K.; Amy K.; Kevin M.; Kelly F.; Rayna V.; Sarajane R.; Susan D. S.; Wai Lee W.; Henry B. L.; Richard V.; Mark X. S.; Richard H. S.; Paul P.; William M. (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 26, 925–932. [DOI] [PubMed] [Google Scholar]
- Marta F.-S.; Hemanta B.; Laura M.-H.; Kathleen T. X.; Jeremy M. B.; Carolyn R. B.; Alice Y. T. (2007) Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nat. Biotechnol. 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolin L.; Schroeder H.; Itzen A.; Goody R. S.; Niemeyer C. M.; Becker C. F. W. (2013) Protein–DNA arrays as tools for detection of protein–protein interactions by mass spectrometry. ChemBioChem 14, 92–99. [DOI] [PubMed] [Google Scholar]
- de Graaf A. J.; Kooijman M.; Hennink W. E.; Mastrobattista E. (2009) Nonnatural amino acids for site-specific protein conjugation. Bioconjugate Chem. 20, 1281–1295. [DOI] [PubMed] [Google Scholar]
- Khalili H.; Godwin A.; Choi J.; Lever R.; Brocchini S. (2012) Comparative binding of disulfide-bridged PEG-Fabs. Bioconjugate Chem. 23, 2262–2277. [DOI] [PubMed] [Google Scholar]
- Rabuka D. (2010) Chemoenzymatic methods for site-specific protein modification. Curr. Opin. Chem. Biol. 14, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavoff S. A.; Liu D. S.; Cohen J. D.; Ting A. Y. (2011) Imaging protein-protein interactions inside living cells via interaction-dependent fluorophore ligation. J. Am. Chem. Soc. 133, 19769–19776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R. G.; Dostal P.; McCafferty D. G. (2004) Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 326, 42–48. [DOI] [PubMed] [Google Scholar]
- Gillet S. M. F. G.; Pelletier J. N.; Keillor J. W. (2005) A direct fluorometric assay for tissue transglutaminase. Anal. Biochem. 347, 221–226. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Guo X.; Guo Z. (2011) Sortase A-catalyzed peptide cyclization for the synthesis of macrocyclic peptides and glycopeptides. Chem. Commun. 47, 9218–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M.; Dozier J. K.; Distefano M. D. (2013) Enzymatic labeling of proteins: techniques and approaches. Bioconjugate Chem. 24, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth M.; Takao K.; Hayashi Y.; Ting A. Y. (2005) Targeting quantum dots to surface proteins in living cells with biotin ligase. Proc. Natl. Acad. Sci. U. S. A. 102, 7583–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiji S.; Nagamune T. (2009) Sortase-mediated ligation: a gift from gram-positive bacteria to protein engineering. ChemBioChem 10, 787–798. [DOI] [PubMed] [Google Scholar]
- Fernández-Suárez M.; Baruah H.; Martínez-Hernández L.; Xie K. T.; Baskin J. M.; Bertozzi C. R.; Ting A. Y. (2007) Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nat. Biotechnol. 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth B. P.; Zhang Z.; Hosokawa A.; Distefano M. D. (2007) Selective labeling of proteins by using protein farnesyltransferase. ChemBioChem 8, 98–105. [DOI] [PubMed] [Google Scholar]
- Hightower K. E.; Casey P. J. (2011) The enzymology of CAAX protein prenylation. Enzymes 30, 1–11. [Google Scholar]
- Park H.-W.; Beese L. S. (1997) Protein farnesyltransferase. Curr. Opin. Struct. Biol. 7, 873–880. [DOI] [PubMed] [Google Scholar]
- Gelb M. H.; Brunsveld L.; Hrycyna C. A.; Michaelis S.; Tamanoi F.; Van Voorhis W. C.; Waldmann H. (2006) Therapeutic intervention based on protein prenylation and associated modifications. Nat. Chem. Biol. 2, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt N.; Hamilton A. D.; Sebti S. M. (2011) Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M.; Mahmoodi M. M.; Shah R.; Dozier J. K.; Wagner C. R.; Distefano M. D. (2013) A highly efficient catalyst for oxime ligation and hydrazone-oxime exchange suitable for bioconjugation. Bioconjugate Chem. 24, 333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan R.; Labadie G. R.; Poulter C. D. (2013) Regioselective covalent immobilization of catalytically active glutathione S-transferase on glass slides. Bioconjugate Chem. 24, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadie G. R.; Viswanathan R.; Poulter C. D. (2007) Farnesyl diphosphate analogues with ω-bioorthogonal azide and alkyne functional groups for protein farnesyl transferase-catalyzed ligation reactions. J. Org. Chem. 72, 9291–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashidian M.; Song J. M.; Pricer R. E.; Distefano M. D. (2012) Chemoenzymatic reversible immobilization and labeling of proteins without prior purification. J. Am. Chem. Soc. 134, 8455–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen U. T. T.; Cramer J.; Gomis J.; Reents R.; Gutierrez-Rodriguez M.; Goody R. S.; Alexandrov K.; Waldmann H. (2007) Exploiting the substrate tolerance of farnesyltransferase for site-selective protein derivatization. ChemBioChem 8, 408–423. [DOI] [PubMed] [Google Scholar]
- Krzysiak A. J.; Rawat D. S.; Scott S. A.; Pais J. E.; Handley M.; Harrison M. L.; Fierke C. A.; Gibbs R. A. (2007) Combinatorial modulation of protein prenylation. ACS Chem. Biol. 2, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehade K. A. H.; Andres D. A.; Morimoto H.; Spielmann H. P. (2000) Design and synthesis of a transferable farnesyl pyrophosphate analogue to Ras by protein farnesyltransferase. J. Org. Chem. 65, 3027–3033. [DOI] [PubMed] [Google Scholar]
- Chehade K. A. H.; Kiegiel K.; Isaacs R. J.; Pickett J. S.; Bowers K. E.; Fierke C. A.; Andres D. A.; Spielmann H. P. (2002) Photoaffinity analogues of farnesyl pyrophosphate transferable by protein farnesyl transferase. J. Am. Chem. Soc. 124, 8206–8219. [DOI] [PubMed] [Google Scholar]
- Placzek A. T.; Krzysiak A. J.; Gibbs R. A. (2011) Chemical probes of protein prenylation. Enzymes 30, 91–127. [Google Scholar]
- Duckworth B. P.; Xu J.; Taton T. A.; Guo A.; Distefano M. D. (2006) Site-specific, covalent attachment of proteins to a solid surface. Bioconjugate Chem. 17, 967–974. [DOI] [PubMed] [Google Scholar]
- Rashidian M.; Dozier J. K.; Lenevich S.; Distefano M. D. (2010) Selective labeling of polypeptides using protein farnesyltransferase via rapid oxime ligation. Chem. Commun. 46, 8998–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dursina B.; Reents R.; Delon C.; Wu Y.; Kulharia M.; Thutewohl M.; Veligodsky A.; Kalinin A.; Evstifeev V.; Ciobanu D.; Szedlacsek S. E.; Waldmann H.; Goody R. S.; Alexandrov K. (2006) Identification and specificity profiling of protein prenyltransferase inhibitors using new fluorescent phosphoisoprenoids. J. Am. Chem. Soc. 128, 2822–2835. [DOI] [PubMed] [Google Scholar]
- Maalouf M. A.; Wiemer A. J.; Kuder C. H.; Hohl R. J.; Wiemer D. F. (2007) Synthesis of fluorescently tagged isoprenoid bisphosphonates that inhibit protein geranylgeranylation. Bioorg. Med. Chem. 15, 1959–1966. [DOI] [PubMed] [Google Scholar]
- Turek T. C.; Gaon I.; Distefano M. D.; Strickland C. L. (2001) Synthesis of farnesyl diphosphate analogues containing ether-linked photoactive benzophenones and their application in studies of protein prenyltransferases. J. Org. Chem. 66, 3253–3264. [DOI] [PubMed] [Google Scholar]
- Völkert M.; Uwai K.; Tebbe A.; Popkirova B.; Wagner M.; Kuhlmann J.; Waldmann H. (2003) Synthesis and biological activity of photoactivatable N-Ras peptides and proteins. J. Am. Chem. Soc. 125, 12749–12758. [DOI] [PubMed] [Google Scholar]
- Hovlid M. L.; Edelstein R. L.; Henry O.; Ochocki J.; DeGraw A.; Lenevich S.; Talbot T.; Young V. G.; Hruza A. W.; Lopez-Gallego F.; Labello N. P.; Strickland C. L.; Schmidt-Dannert C.; Distefano M. D. (2010) Synthesis, properties, and applications of diazotrifluropropanoyl-containing photoactive analogs of farnesyl diphosphate containing modified linkages for enhanced stability. Chem. Biol. Drug Des. 75, 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian T.; Liu S.; Troutman J. M.; Andres D. A.; Spielmann H. P. (2008) Protein farnesyltransferase-catalyzed isoprenoid transfer to peptide depends on lipid size and shape, not hydrophobicity. ChemBioChem 9, 2872–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett J. S.; Bowers K. E.; Hartman H. L.; Fu H.-W.; Embry A. C.; Casey P. J.; Fierke C. A. (2003) Kinetic studies of protein farnesyltransferase mutants establish active substrate conformation. Biochemistry 42, 9741–9748. [DOI] [PubMed] [Google Scholar]
- Fu H. W.; Beese L. S.; Casey P. J. (1998) Kinetic analysis of zinc ligand mutants of mammalian protein farnesyltransferase. Biochemistry 37, 4465–4472. [DOI] [PubMed] [Google Scholar]
- Hougland J. L.; Gangopadhyay S. A.; Fierke C. A. (2012) Expansion of protein farnesyltransferase specificity using ″tunable″ active site interactions: development of bioengineered prenylation pathways. J. Biol. Chem. 287, 38090–38100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry K. L.; Casey P. J.; Beese L. S. (2006) Conversion of protein farnesyltransferase to a geranylgeranyltransferase. Biochemistry 45, 9746–9755. [DOI] [PubMed] [Google Scholar]
- Nguyen U. T. T.; Zhong G.; Christine D.; Yaowen W.; Celine D.; Benjamin F.; Robin S. B.; Wulf B.; Roger S. G.; Herbert W.; Dirk W.; Kirill A. (2009) Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat. Chem. Biol. 5, 227–235. [DOI] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. (2008) Tetrazine ligation: fast bioconjugation based on inverse-electron-demand diels-alder reactivity. J. Am. Chem. Soc. 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S. B.; Casey P. J.; Beese L. S. (2000) The basis for K-Ras4B binding specificity to protein farnesyl-transferase revealed by 2 Å resolution ternary complex structures. Structure 8, 209–222. [DOI] [PubMed] [Google Scholar]
- Sleeman M. W.; Garcia K.; Liu R.; Murray J. D.; Malinova L.; Moncrieffe M.; Yancopoulos G. D.; Wiegand S. J. (2003) Ciliary neurotrophic factor improves diabetic parameters and hepatic steatosis and increases basal metabolic rate in db/db mice. Proc. Natl. Acad. Sci. U. S. A. 100, 14297–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W.; Wen R.; Goddard M. B.; Sherman S. D.; O’Rourke P. J.; Stabila P. F.; Bell W. J.; Dean B. J.; Kauper K. A.; Budz V. A.; Tsiaras W. G.; Acland G. M.; Pearce-Kelling S.; Laties A. M.; Aguirre G. D. (2002) Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest. Ophthalmol. Visual Sci. 43, 3292–3298. [PubMed] [Google Scholar]
- Alcalá-Barraza S. R.; Lee M. S.; Hanson L. R.; McDonald A. A.; Frey W. H.; McLoon L. K. (2010) Intranasal delivery of neurotrophic factors BDNF, CNTF, EPO, and NT-4 to the CNS. J. Drug Targeting 18, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollack J. W., Monson B., Dozier J. K., Dalluge J. J., Poss K., Distefano M. D. (2014) Site-specific labeling of proteins and peptides with trans-cyclooctene containing handles capable of tetrazine ligation. Chem. Biol. Drug Des. DOI: 10.1111/cbdd.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. P. C.; Chen Y.-H.; Liu H.-P.; Li Y.-C.; Chen C.-T.; Liang P.-H. (2002) Synthesis and application of a fluorescent substrate analogue to study ligand interactions for undecaprenyl pyrophosphate synthase. J. Am. Chem. Soc. 124, 15217–15224. [DOI] [PubMed] [Google Scholar]
- DeGraw A. J.; Hast M. A.; Xu J.; Mullen D.; Beese L. S.; Barany G.; Distefano M. D. (2008) Caged protein prenyltransferase substrates: tools for understanding protein prenylation. Chem. Biol. Drug Des. 72, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozier J. K.; Distefano M. D. (2012) An enzyme-coupled continuous fluorescence assay for farnesyl diphosphate synthases. Anal. Biochem. 421, 158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompliano D. L.; Gomez R. P.; Anthony N. J. (1992) Intramolecular fluorescence enhancement: a continuous assay of Ras farnesyl:protein transferase. J. Am. Chem. Soc. 114, 7945–7946. [Google Scholar]
- Cassidy P. B.; Dolence J. M.; Dale Poulter C. (1995) Continuous fluorescence assay for protein prenyltransferases. Methods Enzymol. 250, 30–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.