

SUMMARY
Chronic intake of Western diet has driven an epidemic of obesity and metabolic syndrome, but how it induces mortality remains unclear. Here we show that chronic intake of a high-fat diet (HFD), not a low-fat diet (LFD), leads to severe pulmonary damage and mortality in mice deficient in Toll-like receptors (TLR) 2 and 4 (DKO hereafter). Diet-induced pulmonary lesions are blocked by antibiotics treatment and transmissible to wildtype mice upon either cohousing or fecal transplantation, pointing to the existence of bacterial pathogens. Indeed, diet and innate deficiency exert significant impact on gut microbiota composition. Thus, chronic intake of HFD promotes severe pulmonary damage and mortality in DKO mice in part via gut dysbiosis, a finding that may be important for immunodeficient patients, particularly those on chemotherapy or radiotherapy, where gut microbiota-caused conditions are often life-threatening.
Keywords: Western diet, pulmonary hemorrhage and edema, mortality, transmissibility, antibiotics, cohousing, gut microbiota, cell death
INTRODUCTION
Interactions between the diet, gut microbiota and host genetics are increasingly recognized as critical determinants of nutrient metabolism, health and well-being (Clemente et al., 2012). Gut microbes have co-evolved with their hosts and developed a symbiotic relationship. Under healthy conditions, intestinal microflora plays an essential role in the digestion and metabolism of nutrients, colonization resistance of pathogens and the development and regulation of innate and adaptive immunity of the host (Hooper et al., 2012; Taur and Pamer, 2013). Changes in diets, alterations in host immunity and microbial ecology may alter host-microbial homeostasis, leading to gut dysbiosis. These pathological events have been reported in the pathogenesis of immunological diseases (Taur and Pamer, 2013), obesity and its associated complications, including insulin resistance (Vijay-Kumar et al., 2010), cardiovascular disease (Wang et al., 2011) and fatty liver disease (Henao-Mejia et al., 2012). However, the link between diet and mortality remains unclear.
Acute HFD intake causes glucose intolerance, alters adipose inflammation (Ji et al., 2012a; Sun et al., 2012a) and microbial composition (Clemente et al., 2012; Turnbaugh et al., 2009a; Turnbaugh et al., 2009b). Chronic HFD intake shifts microbial community composition in obese animals and humans, which may help increase energy absorption (Clarke et al., 2012; Turnbaugh et al., 2006). In addition, chronic HFD intake may promote the expansion of pathobionts leading to the development of colitis or hepatocellular carcinoma in various mouse models (Devkota et al., 2012; Turnbaugh et al., 2006; Yoshimoto et al., 2013) and increase gut permeability and bacterial translocation, a condition termed “metabolic endotoxemia” (Cani et al., 2007). In addition to diet, innate immunity is known to shape commensal microbiota (Jin and Flavell, 2013). Defects in the innate immunity favors the overgrowth of pathogenic bacteria that promote intestinal inflammation and induce colitis in immunocompetent wildtype (WT) mice upon cohousing with the mutants (Elinav et al., 2011; Garrett et al., 2007). Moreover, dysfunction of Paneth cells, a specialized intestinal cell type secreting antimicrobial peptides, compromises innate immunity and alters gut microbiota composition (Bevins and Salzman, 2011; Salzman et al., 2010). In mice lacking MyD88, an adaptor protein that mediates TLR signaling, expression of antimicrobial peptides is reduced (Vaishnava et al., 2008). Interestingly, a recent study showed that alterations in the microbiota of TLR-deficient mice are mediated by familial transmission rather than innate deficiency (Ubeda et al., 2012). Thus, it is likely that both innate immunity and familial transmission may play a role in shaping microbiota composition.
TLRs are important for the induction of innate and adaptive immune responses against pathogens such as viruses, fungi, bacteria and protozoa (Kumar et al., 2009). Among dozen family members, TLR2 and TLR4 are the best characterized and exhibit distinct and overlapping ligand specificities (Takeda et al., 2003). It has been proposed that obesity may be associated with increased systemic levels of ligands for TLR2 and TLR4 such as high-mobility group B1 protein, heat shock proteins, endotoxins and etc (Cani et al., 2007; Dasu et al., 2010). Loss of TLR2 or TLR4 attenuates inflammation in various tissues and improves insulin sensitivity, however the results are mixed (Jin and Flavell, 2013; Sun et al., 2012a). A recent study showed that loss of TLR2 alters gut microbiota composition, increases circulating toxin levels and promotes insulin resistance and obesity (Caricilli et al., 2011). Thus, TLR signaling may play an important role in linking gut bacteria to metabolic regulation in the context of obesity. Serendipitously, while characterizing the metabolic effect of both TLR2 and TLR4 signaling pathways in vivo, we found that chronic intake of HFD, not LFD, promotes severe pulmonary damage and mortality in mice deficient in both TLR2 and TLR4 (DKO hereafter). The lethal pulmonary damage phenotype is transmissible to WT mice following co-housing or fecal transplantation with DKO mice and is secondary to the pulmonary cell damage likely caused by toxins derived from pathogenic gut microbes.
RESULTS
Chronic intake of HFD causes severe acute pulmonary damage in DKO mice
WT and DKO mice were fed with either a 13% LFD or 60% HFD starting at the age of 6 weeks, where 13% and 60% calories were derived from fat, respectively. DKO mice fed the HFD over 38 weeks exhibited a significantly higher mortality rate with 6x higher risk of death than age- and gender-matched WT cohort: 10 out of 17 (60%) DKO mice died, while only 2 out of 22 (9%) WT mice died within 38 weeks when fed the HFD (Figure 1A). Importantly, none of the DKO and WT mice on LFD died over the course of a year (Figure 1B), suggesting that mortality is HFD-dependent. DKO mice did not die from malnourishment as they grew well on either diet and, if anything, were a bit heavier than the WT cohort (Figure S1A–B). DKO mice exhibited improved metabolic parameters compared to WT cohort in terms of fasting glucose and hepatic steatosis (Figure S1C–D) as well as other parameters (not shown).
Figure 1. Chronic intake of HFD causes severe acute pulmonary damage and mortality in DKO mice.

(A–B) Survival curves of WT and DKO mice on HFD (A) or LFD (B) over a 40–50-week period. Statistical analysis was performed using the Log-rank (Mantel-cox) test.
(C) Representative image of the thoracic cavity and lung of moribund DKO mice showing severe acute pulmonary damage. h, heart; +, diaphragm; arrow, pleural effusion.
(D) Representative images of lungs taken from WT and moribund DKO mice. Hemorrhagic pleural effusion was collected and shown in a tube. N=7.
(E) Representative flow cytometric analysis of T cells (CD4+ or CD8+), B cells (B220+) and neutrophils (Gr1+ F4/80−) in pleural effusion from moribung DKO mice. Representative data of 3 independent experiments.
(F–G) Representative H&E images of lungs from WT and moribund DKO mice on HFD at low (F) and high (G) magnifications. RBC, red blood cells; *, edema fluid in the lumen of alveoli. Representative data of 5 independent cases.
Also see Figure S1.
Affected DKO mice deteriorated quickly, exhibited respiratory distress and shortness of breath and died within days. Postmortem examination invariably revealed severe pleural effusion and diffuse pulmonary edema and hemorrhage (Figure 1C–D). The cellularity of the pleural effusion consisted of blood leukocytes including CD4+/CD8+ T, B220+ B cells and F4/80− Gr1+ neutrophils (Figure 1E). There was no notable hemorrhage or effusions in other organs and body cavities nor gross abnormalities in other organs and tissues such as the liver and spleen (Figure S1D and not shown), excluding systemic illness or sepsis as a cause of death and lung injury. Histological examination of the lungs of WT mice fed the HFD or DKO mice on the LFD revealed normal pulmonary architecture with well-aerated airways and alveolar parenchyma, and intact cellular structure (Figure 1F and S1E). By contrast, the lungs of DKO mice fed the HFD that died or were euthanized displayed severe, diffuse, acute pulmonary edema and hemorrhage (Figure 1G). Of note, two WT mice on HFD died of other causes with no signs of pulmonary damages (not shown).
Non-hematopoietic expression of TLR2 and TLR4 is critical for the protection from diet-induced pulmonary damage and premature lethality
We next performed bone marrow transplantation (BMT) to determine whether the expression of TLRs in the hematopoietic and/or nonhematopoietic compartment is responsible for diet-induced mortality. Four chimeric cohorts were generated using either WT (WT→WT and DKO→WT) or DKO (WT→DKO and DKO→DKO) mice as recipients. To prevent infection, recipients were treated with a combination of antibiotics, ciprofloxacin and metronidazole, in drinking water two weeks before and one week after the irradiation for a total of three weeks. Chimeric mice were allowed to recover for 6 weeks and then placed on HFD (Figure 2A). Within 30 weeks, DKO recipient chimeras, either WT→DKO or DKO→DKO mice, exhibited substantially higher mortality rate of 36 and 64%, respectively, while all WT recipient chimeras (DKO→WT and WT→WT) survived (Figure 2B), suggesting that non-hematopoietic TLR2/TLR4 is indispensable for diet-induced mortality. Interestingly, WT→DKO mice have reduced mortality when compared to DKO→DKO mice (Figure 2B), pointing to a partial protective role of hematopoietic TLR2/TLR4 in disease pathogenesis. While all the WT recipients exhibited normal lung architecture, the dying DKO recipients exhibited acute pulmonary damage similar to those in the DKO mice (Figure 2C–D). All chimeras survived on LFD (Figure 2E), suggesting that the mortality is diet-dependent and not caused by irradiation. Thus, we conclude that TLR2/TLR4 actions primarily in non-hematopoietic compartments protect against HFD-induced lethal pulmonary damage and mortality.
Figure 2. Non-hematopoietic expression of TLR2/TLR4 is required for the protection from diet-induced pulmonary damage and premature lethality.

(A) Schematic diagram showing the experimental procedure of bone marrow transplantation (BMT). Recipients were treated with broad-spectrum antibiotics, ciprofloxacin and metronidazole, in drinking water two weeks before and one week after the BMT. Mice were put on HFD after 6-week post-BMT recovery.
(B) Survival curves of chimeric mice. * and **, comparing WT→DKO and DKO→DKO, respectively, to the two WT recipient cohorts by the Log-rank (Mantel-cox) test.
(C) Representative H&E images of the lungs in WT recipient mice showing normal pulmonary architecture.
(D) Representative H&E images of the lungs in DKO recipient mice showing diffuse pulmonary edema and hemorrhage in airways and alveolar parenchyma. Representative data of 3 independent cases.
(E) Survival curves of chimeric mice as (B) on LFD. n.s., no significance by the Log-rank (Mantel-cox) test.
Diagnosis of the cause of death: non-cardiogenic pulmonary edema with hematogenous injury to the lungs
Pulmonary edema can be either cardiogenic as a result of heart failure and increased capillary hydrostatic pressure or noncardiogenic as a result of increased vascular permeability of the lungs (Murray, 2011; Perina, 2003). As no morphological evidence of myocardial disease and no edema in peripheral tissues were observed in moribund DKO mice (Figure S1F–G), the cause of death was likely noncardiogenic. The causes for noncardiogenic pulmonary edema are diverse, either direct injury to the lung including alveolar epithelium by drugs and toxins or indirect injury as a result of systemic inflammatory responses against pathogen infection (Perina, 2003). As tissue damages in the DKO mice fed HFD were limited to the lungs, we excluded the possibility of severe sepsis and multi-organ failure. Moreover, as the lesions were diffuse and present throughout the lungs of DKO mice (Figures 1F and 2D), we concluded that a hematogenous injury rather than an aerogenous one to the lungs (e.g. inhaled toxins or bacteria) is the most likely etiology. Indeed, recent studies showed that TLR2/TLR4 are not required for pathogen resistance following intranasal infection (Jamieson et al., 2013; Lammers et al., 2012; Skerrett et al., 2007). Thus, we conclude that DKO mice fed HFD may die from hematogenous pathogenic insults.
Antibiotic treatment rescues lethal pulmonary damage and mortality
As the mortality of DKO mice was diet-dependent and required non-hematopoietic expression of TLR2/TLR4, we next addressed the role of gut bacteria in disease pathogenesis. We administrated DKO mice orally with a broad-spectrum antibiotics consisting of ampicillin, neomycin, vancomycin and metronidazole, concurrently with HFD feeding. Administration of antibiotics expectedly reduced the levels of total fecal bacteria with no significant impact on body weight, daily food intake and body temperature (Figure S2). Dramatically, antibiotic treatment prevented the death of DKO mice following HFD intake for 38 weeks (Figure 3A) with a total prevention of pulmonary damage (Figures 3B). Antibiotic treatment had no effect on the survival of the WT cohort (Figure 3C).
Figure 3. HFD-induced lethal pulmonary damage is prevented by antibiotic treatment.
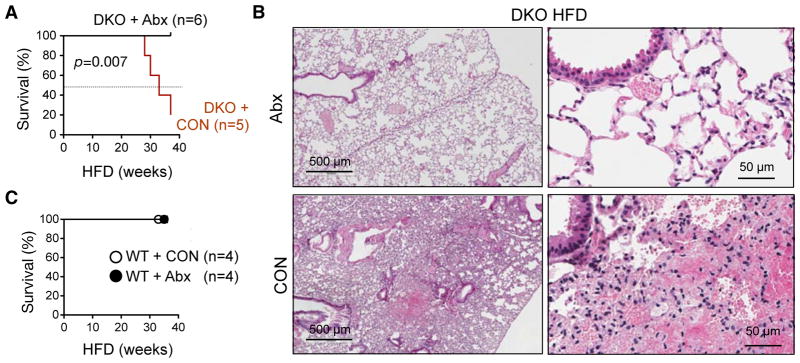
WT or DKO mice on HFD were given drinking water with or without (CON) a combination of antibiotics (Abx) including ampicillin, neomycin, vancomycin and metronidazole.
(A) Survival curves of DKO mice. P value is determined by the Log-rank (Mantel-cox) test.
(B) Representative H&E images of lungs from DKO mice on HFD with or without Abx. Representative data of 3 independent cases.
(C) Survival curves of WT mice.
Also see Figure S2.
Transmissible pulmonary damage and lethality
To further address the contribution of gut microbiota, we co-housed WT mice with DKO mice at the time of weaning, i.e. 3 weeks of age, and then placed them on HFD at the age of 6 weeks. Intriguingly, co-housed WT mice exhibited higher mortality following chronic HFD feeding with 22% mortality rate vs. 9% in single-housed (Figure 4A). The cohousing effect was more pronounced in BMT chimeric mice where WT and DKO recipients were co-housed since weaning: 7 out of 19 (37%) WT/DKO→WT chimeras co-housed with DKO chimeras died within 30-week HFD while all survived when singly housed (Figure 4B). Cohousing did not affect the mortality of WT/DKO→DKO chimeras (Figure 4B). In both studies, cohousing eliminated the statistical significance observed between single-housed cohorts (Figures 4A–B). Strikingly, histological examination revealed that these moribund WT mice exhibited similar pulmonary pathologies as those of the DKO mice (Figures 4C). Next we performed oral fecal transplantation where DKO→WT mice were orally gavaged twice a week for three weeks with feces from either WT or DKO mice on 17-week HFD, concurrently with HFD feeding. Strikingly, mice received DKO feces died within 17–19 weeks post gavage with pulmonary hemorrhage while those received WT feces survived (Figures 4D). Taken together, these data provide strong evidence for transmissible gut microbiota in DKO mice following chronic HFD feeding that are dominant in disease pathogenesis.
Figure 4. HFD-induced pulmonary damage and mortality are transmissible to wildtype mice by co-housing and fecal transplantation.

(A–B) Survival curves of single- and co-housed WT and DKO mice (A) and BMT chimeric mice (B) on HFD. Co-housed groups are shown with solid lines and their labels are boxed. Recipients with the same genotype are grouped. The Log-rank (Mantel-cox) test was used for statistical analysis.
(C) Representative H&E images of lungs of DKO→WT mice on HFD, either single-housed or co-housed with DKO→DKO mice (n=5 each).
(D) Representative H&E images of the lungs of DKO→WT chimeric mice (n=2 each) that were gavaged twice a week for a total of three weeks with feces of WT or DKO mice on HFD (for 17 weeks), showing pulmonary hemorrhage in mice received fecal transplantation from DKO mice, but not WT mice. Recipients were put on HFD concurrently with the first gavage. Recipients gavaged with DKO feces died at 17 and 19 week HFD while those with WT feces survived.
Diet and innate deficiency modify microbiota composition in DKO mice
We next determined how both HFD and innate deficiency affect fecal microbiota composition in our WT and DKO mice on either HFD or LFD for 17 weeks. We performed culture-independent PCR amplification of variable region 4 (V4) of bacterial 16S rRNA genes followed by Illumina sequencing. The quality-filtered sequences were analyzed using UniFrac. Principal coordinates analysis (PCoA) of unweighted UniFrac distance between samples revealed that HFD feeding had a profound impact on the microbiota composition in both WT and DKO cohorts when compared to age- and gender-matched mice on LFD (Figure 5A and S3). Moreover, WT and DKO mice had distinct microbial profiles regardless of diet as revealed in PCoA analysis of the unweighted UniFrac distances (Figures 5A) and relative abundance of the bacteria (Figure S3A), pointing to a likely effect of familial transmission on gut microbiota (Ubeda et al., 2012). Profiles of gut microbiota between WT and DKO cohorts were further separated from 10- to 17-week HFD (Figure 5B). While Bacteroidales (phylum: Bacteroidetes) were the major representative signature operational taxonomic unit (OTUs) accounting for the differences in gut microbiota composition between WT and DKO mice on LFD (Figure S3B), Clostridiales (phylum: Firmicutes) became the predominant signature OTUs when the mice were on HFD (highlight in red in Figure 5C). Complete list of changes at the levels of order, genus and OTUs are shown in Table S1. Among 141 OTUs that were significantly different between WT and DKO on HFD, 87 were higher in DKO mice (Table S1). Among these 87 OTUs, 56 (64%) were highly enriched in DKO mice only after chronic HFD while remained undetectable or low in other cohorts (Figure S3C), pointing to a combinatorial effect of diet and innate deficiency on gut microbiota composition. Some examples are shown in Figure 5D–F. Of note, most of these signature sequences present only in DKO mice on HFD poorly matched with any known bacterial species (Figure 5D–E).
Figure 5. Chronic HFD intake and innate deficiency affect microbiota composition.

(A) Principal coordinates analysis (PCoA) of fecal microbiota from single-housed WT and DKO mice on LFD or HFD for 17 weeks.
(B) PCoA analysis of fecal microbiota from single-housed WT and DKO mice on HFD for 10 or 17 weeks of the same set of mice.
(C) Heat map showing the relative abundance of the top bacterial OTUs representing the difference between WT and DKO mice after 17-week HFD feeding. OTUs belonging to the order of Clostridiales are highlighted in red.
(D–F) Examples of the OTUs significantly enriched only in HFD-fed DKO mice, showing the synergistic effect of HFD and TLR deficiency on the abundance of gut microbiota. D–F, OTUs belong to the order Clostridiales (D), Bacteroidales (E), and Lactobacillales (F). a.u, arbitrary units. The phylum (P), class (C), order (O) and family (F) of each OTU are indicated. A complete list of changed OTUs is shown in Table S1. N=5–6 mice each. **, p<0.01; ***, p<0.001 by Student’s t-test with Bonferroni correction.
Also see Figure S3–4 and Table S1.
We next examined gut microbiota composition in BMT cohorts to further assess the effect of innate immunity on gut bacteria. As recipients orally received ciprofloxacin and metronidazole for three weeks (Figure 2A), microbiota was expectedly to be re-shaped (Dethlefsen et al., 2008; Willing et al., 2011). Intriguingly, following 6-week HFD feeding, gut microbiota in WT→DKO chimeras were distinct from those in DKO→DKO chimeras, but not between the two WT recipient chimeras (Figure S4A–B). This finding was unexpected because the recipients – the DKO mice –were littermates from the same parents, pointing to the effect of innate deficiency on microbiota. The difference in gut microbiota composition between chimeric WT and DKO recipients on HFD was still largely driven by changes in the bacterial order Clostridiales (Figure S4C–D). Thus, host innate immunity plays an important role in influencing microbiota composition.
Cohousing modifies microbiota composition of WT mice
Cohousing at weaning for over 20 weeks altered microbiota compositions of WT mice on HFD, which became similar to the communities of DKO mice (Figure 6A). Figure 6B shows the top OTUs that changed in relative abundance in WT fecal microbiota due to cohousing. Interestingly, several OTUs that were signatures of single-housed DKO mice fed the HFD were now found in the cohoused WT (Figures 6C). Thus, cohousing modifies microbiota composition and presumably leads to the expansion of pathogenic bacteria in WT mice.
Figure 6. Cohousing modifies microbiota composition of wildtype mice.

(A) PCoA analysis of fecal microbiota from WT and DKO mice on HFD either single-housed for 17 weeks or co-housed for 24 weeks.
(B) Heat map showing the relative abundance of the top bacteria OTUs representing the difference between single- and co-housed (with DKO mice) WT mice (as A). OTUs belonging to the order of Clostridiales are highlighted in red.
(C) Examples of the OTUs (from the order Clostridiales) that were transmitted from DKO to co-housed (co-) WT mice. Dashed lines indicate that mice were single housed. a.u, arbitrary units. N=3–6 mice each. *, p<0.05; **, p<0.01; ***, p<0.001, n.s, not significant by Student’s t-test with Bonferroni correction.
Mucosal innate immunity is impaired in DKO mice fed HFD
We next further explored possible causes of gut dysbiosis. Previous studies have shown the importance of the TLR-MyD88 signaling axis in Paneth cells in the expression of antimicrobial peptides (Vaishnava et al., 2008). Indeed, examination of ileum revealed significant reduction of Paneth cell granules and numbers per crypt in the ileum of DKO mice compared to those of WT mice on both LFD and HFD (Figure S5A–B). mRNA levels of various antimicrobial peptides including Lyz1 (encoding Lysozyme), defa (Defensin α) and Cr2 (Cryptdin 2) in the small intestine of DKO mice were lower than those in WT mice (Figure S5C). More profound changes were seen in cohorts with HFD feeding (Figure S5B–C). This was further confirmed by immunofluorescent staining of Lysozyme C, a marker of Paneth cells (Figure S5D). Expression of various antimicrobial peptides in WT mice was not affected by cohousing with DKO mice (Figure S5E), suggesting that innate deficiency, rather than microbiota, may primarily account for Paneth cell defect. Although TLR signaling has been implicated in epithelial proliferation and repair in response to injuries (Abreu et al., 2005), we observed no obvious abnormalities in overall intestinal morphology (e.g. enteritis and etc) and PCNA-positive proliferating cells in the small intestine of WT and DKO cohorts (Figure S6A–B). Together, TLR2/TLR4 deficiency impairs Paneth cell function while having no effect on epithelial proliferation.
Increased bacterial translocation in DKO mice on HFD
Gut permeability of DKO mice as measured by the absorption of dextran-FITC was comparable between the two HFD cohorts (Figure 7A and S5F), so was the expression of tight junction genes in the small intestine (Figure S5G). However, serum endotoxin level was significantly higher in DKO mice on HFD (Figure 7B). These data are in line with a previous report showing increased bacterial load in the spleen of mice deficient in TLR signaling in the absence of any measureable changes in intestinal barrier function (Slack et al., 2009). Indeed, levels of TNFα-expressing activated CD8+ cytotoxic and CD4+ T cells were elevated in the spleen of DKO mice on HFD when compared to those of HFD-fed WT cohort (Figure 7C). This response was bacteria elicited as it was attenuated by antibiotics treatment (Figure 7C). Similarly, a two-fold increase of activated IFNγ+ CD4+ TH1 and CD8+ cytotoxic T cells and a four-fold increase of Gr-1+ CD11b+ neutrophils were observed in the spleen of the WT→DKO chimeras relative to those in the WT→WT chimeras (Figure 7D–E), pointing to an active immune response. As we were not able to detect bacteria in the liver of DKO mice fed the HFD using Gram staining, Q-PCR and in vitro bacterial culture (Figure S6C–D and not shown), we speculate bacteria-derived toxin(s) as the possible culprit.
Figure 7. Hyperendotoxemia and increased pulmonary cell death in DKO mice on HFD.

(A) Serum dextran-FITC levels 4 hr post-gavage in mice on LFD or 27-week HFD. N=4–12 mice each.
(B) Serum endotoxin levels in mice on LFD or 27-week HFD. N=8–10 mice each.
(C) Flow cytometric analysis of TNFα+ CD4+ and TNFα+ CD8+ T cells in the spleens of mice fed on HFD with or without Abx for 5 weeks. N=3–4 mice each.
(D–E) Flow cytometric analysis of activated IFNγ+ CD4+ and IFNγ+ CD8+ T cells and Gr1+ CD11b+ neutrophils in the spleens of BMT chimeric mice on HFD for 24 weeks. Quantitation shown in (E). N = 3–4 mice each.
(F) Representative confocal images of TUNEL staining (green) of the lungs from mice on HFD over 24 weeks. Right, q-PCR analysis of the anti-apoptotic gene Bcl-2. Bcl2, B-cell lymphoma 2. n=3 each.
(G) Q-PCR analysis of tight junction genes in the lungs of WT or moribund DKO mice on HFD for 29–36 weeks. a.u., arbitrary units. n=3 mice each. Cldn1, claudin 1; Ocln, occludin; Cdh1, cadherin 1; ZO1, tight junction protein 1; Jam1, F11 receptor.
(H) Q-PCR analysis of inflammatory genes in the lungs of WT or moribund DKO mice on HFD. Tnfa, tumor necrosis factor alpha; Il6, interleukin 6; Mip1a, chemokine (C-C motif) ligand 3; Ifng, interferon gamma; Il1b, interleukin (IL) 1 beta; IL18, IL18; Nlrp3, NLR family, pyrin domain containing 3; Asc, PYD and CARD domain containing; Nfkbia, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha; Emr1, EGF-like module containing, mucin-like, hormone receptor-like sequence 1; Il12a, IL12a; Nos2, nitric oxide synthase 2, inducible; Rantes, chemokine (C-C motif) ligand 5. n=3 mice each.
(I) Lung histology of the WT mice on LFD i.p. challenged with filter-sterilized water-soluble gut luminal contents from WT and moribund DKO mice on HFD for 35 weeks. Note pulmonary hemorrhage in lungs of WT mice injected with luminal contents from DKO mice, but not from WT mice. N=2–4.
Values, mean ± s.e.m. (A–C) statistical analyses were performed with one-way ANOVA with the Tukey Post-tests (different letters mark p < 0.05). (E–H), *, p<0.05 by Student’s t-test. Also see Figure S5–6.
Increased pulmonary cell death and reduced tight junction in the lung of DKO mice on HFD
Noncardiogenic pulmonary edema is often associated with increased cell death (Perina, 2003). To directly assess cell death, we performed TUNEL staining on the lung sections. Strikingly, massive amount of apoptotic cells were detected in the lungs of DKO mice on chronic HFD, while no apoptotic cells were present in the WT cohort (Figure 7F). Consistently, expression of a key antiapoptotic gene Bcl-2 was downregulated in DKO mice (Figure 7F). Moreover, as tight junction proteins are essential for epithelial barrier function and defects in tight junctions are known to prime the lung for pulmonary edema and hemorrhage (Koval, 2013), we next examined the expression of tight junction genes. Indeed, several genes involved in tight junctions were downregulated in the lung of DKO mice on HFD (Figure 7G). Surprisingly, transcript levels of various inflammatory genes including Tnfa, Il6, Il1b and il18 in the lungs were comparable between WT and DKO mice (Figure 7H). This finding is reminiscent of an early study showing that lethal toxin derived from C. sordellii increases lung permeability and causes lung damage in the absence of inflammation (Geny et al., 2007). Finally, pointing to a possible role of gut-derived bacterial toxin(s) in disease pathogenesis, i.p. challenge of 8-week-old WT mice with filter-sterilized water-soluble gut luminal contents from DKO mice on HFD, but not WT mice on HFD, caused pulmonary hemorrhage after 16 h (Figure 7I). Thus, toxin-mediated pulmonary cell death is likely a possible cause of lethal pulmonary damage in DKO mice on HFD.
DISCUSSION
Consumption of a Western diet, one of the driving forces for the obesity epidemic and type-2 diabetes, may promote the expansion of pathobionts which are associated with increased incidence of colitis or hepatocellular carcinoma in genetically susceptible mice (Devkota et al., 2012; Henao-Mejia et al., 2012; Ley et al., 2006; Turnbaugh et al., 2006; Vijay-Kumar et al., 2010; Wang et al., 2011; Yoshimoto et al., 2013). How obesity and Western diet are linked to lethality remains vague. Here, our data demonstrate that chronic intake of HFD promotes pulmonary damage and mortality in genetically susceptible mice. Strikingly, similar to recent reports of transmissible colitogenic and metabolic phenotypes (Couturier-Maillard et al., 2013; Elinav et al., 2011; Henao-Mejia et al., 2012; Zenewicz et al., 2013), our data show transmissibility of pulmonary damage and mortality through cohousing and fecal transplantation, pointing to the presence of a dominant bacterial factor(s) in the intestines of DKO mice fed the HFD. Our data show Paneth cell defects, alterations in microbiota composition and elevated circulating bacterial toxins in DKO mice on HFD, which are associated with reduced tight junction gene expression and elevated cell death in the lung.
Our study is in line with previous studies showing that specific changes of the TLR/MyD88 innate signaling as well as inflammasome signaling pathways may impair Paneth cell function and thereby are associated with altered gut microbiota composition (Caricilli et al., 2011; Elinav et al., 2011; Henao-Mejia et al., 2012; Vijay-Kumar et al., 2010). Our extensive microbiota sequencing data indicates that the majority of bacterial species enriched in DKO mice on HFD (vs. WT mice on HFD) are expanded during HFD feeding, thereby arguing for the role of innate immunity in shaping gut microbiota and disease pathogenesis. Moreover, sequencing analyses of gut microbiota from BMT chimeric mice following 6-week HFD reveal that gut microbiota in DKO→DKO chimeras are distinct from those in WT→DKO chimeras. This data provides strong support for the role of innate deficiency in shaping microbiota composition. Thus, our data demonstrate that host innate immunity plays an important role in influencing microbiota composition. By contrast, a recent study showed that alterations in the microbiota of TLR-deficient mice may be mediated by familial transmission rather than innate deficiency (Ubeda et al., 2012). We speculate that the discrepancy between the studies may be due to the differences in sampling points for microbiota composition and diet in these studies. In the Ubeda et al. (Ubeda et al., 2012), samples were collected 5–7 weeks after the weaning on LFD where similar microbiota compositions were observed between different genotypes in the same litter. In this study, samples were analyzed at least over 13 weeks, 20 or 24 weeks in some cases, post weaning or cohousing. Moreover, our studies were performed over the course of 3 years with several breeding pairs, during which diet-induced lethality was consistently observed. Therefore, we conclude that microbiota differences between WT and DKO cohorts in our study cannot be explained exclusively by familial transmission and that rather, HFD and TLR2/TLR4 deficiency synergistically alter the structure of bacterial community, which may favor the outgrowth of potentially virulent species or pathobionts in the gut.
Similar to any other diseases, disease etiology and pathogenesis of pulmonary damage in our study are likely to be complex. Our data indicate a connection between the acquisition of gut dysbiosis and lethal pulmonary damage in DKO mice where the lethality is associated with chronic intake of HFD, preventable by antibiotics treatment and transmissible through cohousing or fecal transplantation. In addition, we show that i.p. challenge of WT mice with water-soluble gut luminal contents from DKO mice on HFD causes damage to the lung while those from WT mice on HFD do not. Thus, these data suggest that the susceptibility to pulmonary damages observed in DKO mice may be a result of complex interactions between the gut microbiota and innate immunity at the level of specific cell types such as the intestinal epithelial cells (e.g., Paneth cells). However, the exact origin of the phenotype cannot be firmly identified with the use of whole body DKO mice. Genetic defect of TLR2/TLR4 in tissues such as the liver, lung or intestines may contribute to disease pathologies, which requires the generation and characterization of tissue- and cell-type-specific DKO mice. Moreover, we can’t conclusively exclude other possibilities such as the role of lung microflora. The functional role and implications of the lung microbiota are unknown at this point, but this is a promising avenue for future investigations, particularly given the recent literature that has linked microbiota in the lung with healthy or diseased lung (Blainey et al., 2012; Erb-Downward et al., 2011; Hilty et al., 2010).
Although pulmonary damage in our study is acute, an extended feeding period or “incubation period” (time from initial HFD feeding to death) is required before the development of severe pulmonary damage and peak mortality. This is likely attributable to a gradual change in gut microbiota composition, the overgrowth of a specific gut pathogen(s), and/or production of a threshold level of deleterious bacterial product(s) leading to systemic translocation and pulmonary alveolar endothelial damage. In addition, a recent study showed that HFD and HFD-induced obesity in WT mice do not dramatically affect inflammatory response in the lung, but do modulate sensitivity to LPS challenge (Tilton et al., 2013). Moreover, our data show decreased tight junction gene expression with elevated cell death in the lung of DKO mice on HFD. As tight junction proteins are essential for epithelial barrier function and the junctional deterioration often leads to pulmonary edema and hemorrhage (Koval, 2013), this finding provides possible molecular mechanism underlying disease pathogenesis. Future studies are required to further elucidate how tight junction genes are regulated.
Defining the role of a specific bacterial specie(s) or toxin(s) remains extremely challenging given diverse gut bacteria and complex disease pathogenesis (Kamada et al., 2013). Our bacterial sequencing reveals increased abundance of several bacteria belonging to the order Clostridiales (Phylum: Firmicutes) in DKO mice on HFD, which are found in WT mice upon cohousing. Clostridia are one of the most prominent Gram-positive and spore-forming bacteria indigenous to the murine gastrointestinal tract (Momose et al., 2009). Certain species of Clostridia secrete metabolites that are required for the optimal induction of intestinal regulatory T cells independently of TLR signaling (Atarashi et al., 2011). Clostridia species have also been implicated in the maintenance of mucosal homeostasis and the pathogenesis of IBD (Frank et al., 2007; Sokol et al., 2009). Certain members of the Clostridium genus have well-established pathogenic properties and are capable of producing potent toxins (Lopetuso et al., 2013; Songer, 2004; Taur and Pamer, 2013), which may contribute to pulmonary alveolar endothelial damage and vascular permeability (Songer, 2004). Indeed, administration of bacterial toxin such as those from C. sordellii in mice causes similar lethal pulmonary damage as reported in this study (Bourke et al., 1994; Geny et al., 2007; Pusterla et al., 2008; Saeed et al., 2012). Whether certain members of the Clostridium genus are responsible for pulmonary damage in DKO mice remains to be demonstrated. A better understanding of the host-microbiota interactions will be essential for future efforts to target specific subsets of gut microbiota as a therapeutic means. The conclusion of this study may be important for patients with immunodeficiency or immune suppression, particularly those on chemotherapy or radiotherapy, where gut microbiota-caused conditions are often life-threatening (Taur and Pamer, 2013).
EXPERIMENTAL PROCEDURES
Summary of animal experiments
WT C57BL/6 mice were purchased from the Jackson Laboratory (#000664) and bred in our own colony. TLR2/TLR4 DKO mice on the C57BL/6 background (backcrossed over 6 generations) were generated by Shizuo Akira (Osaka University) and provided to us by Dr. Lynn Hajjar (University of Washington) via Dr. David Russell (Cornell University) (Darveau et al., 2004). The DKO and WT control mice were bred, reared and housed separately in our specific pathogen-free facility. Of note, we recently generated a transgenic mouse line on B6 background in our colony and reported that they survived well beyond 1 year on HFD (Sha et al., 2014), excluding the possibility of the existence of a peculiar pathogen in our facility. Mice were bred, reared and housed in separated microisolators (i.e. single-housed by the genotype) in our brand-new pathogen-free facility. In cohousing experiments, 3-week-old DKO and WT pups were weaned into the same cages, 4–5 mice per cage, and were fed a 60% HFD when they reached 6 weeks of age. In the antibiotic administration experiments, mice were given metronidazole (1g/L; RPI), ampicillin (1g/L; Fisher), vancomycin (0.5g/L; VWR), and neomycin (1g/L; Fisher) in the drinking water throughout the period of HFD feeding. Fresh drinking water containing antibiotics was supplied every 4–7 days. Bone marrow transplantation was carried out as previously described (Sun et al., 2012b).
Briefly, 6-week-old recipient male mice were sublethally irradiated (10 Gy) and transplanted with 5 million bone marrow cells via i.v. injection. Two weeks before irradiation, mice were treated with water containing 0.2 mg/ml ciprofloxacin and 1 mg/ml metronidazole and maintained for a total of 3 weeks. After a 6-week recovery, recipient mice were fed HFD or LFD. For the cohousing BMT experiments, recipients were cohoused WT and DKO mice since weaning prior to the BMT as recipients. For fecal Transplantation, feces from eight WT or DKO mice on 17-week HFD were pooled together, grounded in PBS and filtered through a 70μm nylon mesh (Fisher). The concentration of filtrate was adjusted to 300mg feces per ml and recipient mice were gavaged with 500 μl filtrate each, twice a week for a total of three weeks. The two cohorts of recipient mice were separately housed and put on HFD concurrently with the first gavage. For i.p. challenge of filter-sterilized water-soluble gut luminal content, all intestinal luminal contents from both small intestine and colon were collected from one moribund DKO and healthy WT mice on HFD for 35 weeks. Supernatants were filtered with a 70μm nylon mesh (Fisher) followed by a 0.45 μm filter (Millipore). The solutions were collected and adjusted to 1 ml with PBS and injected i.p. into 8-week-old WT mice on LFD at 500 μl per mice. 16 h later, mice were sacrificed and lungs were collected and subjected for histological analysis.
In all of the studies, mice were carefully monitored for body weight, activities and overall health. Moribund mice with labored breathing and lethargy were euthanized and subjected to complete postmortem and histological examination of all major organs. For mice that were found dead without obvious clinical signs, postmortem examination was carried out to identify the cause of death. Only age-matched male mice were used in the survival studies. Mice were fed a LFD, composed of 13% fat, 67% carbohydrate and 20% protein from Harlan Teklad (#2914). For chronic HFD feeding, 6-week-old males were fed a 60% HFD with 60% fat, 20% carbohydrate and 20% protein from Research Diets Inc. (D12492) for up to a year. Fatty acid composition of the diets has been previously described (Ji et al., 2012b). All animal procedures were approved by the Cornell IACUC.
H&E histology and disease diagnosis
Tissues were fixed in 10% neutral buffered formalin, and processed by the Cornell Histology Core Facility for sectioning and H&E staining on a fee-for-service basis. Tissue section examination was performed by a board-certified veterinary pathologist (G.E.D.).
Fecal bacterial analyses
Stool samples were collected from mice, snap frozen, and stored in −80 °C until use. Total DNA was extracted using PowerSoil DNA isolation kit (MOBIO Laboratories) with a bead-beating method per supplier’s protocol. Stool DNA samples from cohorts treated with or without antibiotics were subjected to qPCR analysis targeting the conserved region of the 16S rRNA gene for all groups of bacteria as previously described (Fierer et al., 2005). Primers were Eub338, ACTCCTACGGGAGGCAGCAG and Eub518, ATTACCGCGGCTGCTGG. Bacterial 16S rRNA genes were amplified using primers for the V4 region (515F and barcoded 806R) with 5PRIME Hot Start Master Mix (Fisher Scientific). Primer sequences can be found at the Earth Microbiome Project (http://www.earthmicrobiome.org/emp-standard-protocols/16s/). Duplicate PCR reactions were performed for each sample, combined, and purified with Mag-Bind E-Z Pure magnetic beads (Omega). After quantification with the Quant-iT PicoGreen assay (Invitrogen), the PCR products were sequenced using paired-en 250 with Illumina MiSeq platform at Cornell Biotechnology Resource Center Genomics Facility. Mate-pairs were merged using the fastq-join command in the ea-utils software package (Aronesty, 2011), and the merged sequences were analyzed using the software QIIME. Closed-reference OTU picking at 97% identity was done against the October 2012 Greengenes database, and the UniFrac beta-diversity metric was calculated on the resulting OTU abundances after rarefaction to 27,233 sequences per sample. PCoA was performed on the unweighted UniFrac distance matrix. Over/under represented OTUs in a given category (e.g. HFD vs LFD, WT vs. DKO) were identified using the Predictive Analysis of Microarrays (PAM) package under the R software, which implements a “nearest shrunken centroid” classification method.
Statistical analysis
Results are expressed as mean ± s.e.m. Comparisons between groups were made using either unpaired two-tailed Student t-test of the EXCEL software for two-group comparisons or the one-way ANOVA test with the Tukey Post-tests of the PRISM software for multi-group comparisons. Statistical analyses for survival curves were performed using the Log-rank (Mantel-cox) test.
Supplementary Material
HIGHLIGHTS.
Chronic intake of HFD causes lethal pulmonary damage in TLR2/TLR4-null (DKO) mice
Mortality is transmissible to WT mice upon cohousing and fecal transplantation
HFD and innate deficiency synergistically influence gut microbiota composition
Hyperendotoxemia and increased pulmonary cell death were observed in DKO mice on HFD
Acknowledgments
We thank Drs. Shizuo Akira, Lynn Hajjar and David Russell for the DKO mice and other members of Qi lab for comments and suggestions. Supported by a David and Lucile Packard Foundation Fellowship (R.E.L.), National Science Foundation Graduate Fellowship (J.K.G.), R21AI085332 (G.D.), R01DK082582, Juvenile Diabetes Research Foundation 47-2012-767, American Diabetes Association (ADA) 1-12-CD-04 (L.Q.). S.S. is an International Student Research Fellow of the Howard Hughes Medical Institute (59107338). L.Q. is the recipient of the Junior Faculty and Career Development Awards from ADA.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTION
Y.J., S.S. and L.Q. designed the experiments; Y.J and S.S. performed most of the experiments; J.G., A.C.P., and R.L. assisted S.S. perform microbiota sequencing and performed data analysis of gut microbiota; H.K. helped collect tissues and measure endotoxin levels; G.E.D. performed histological examination of the tissue; Y.J. and L.Q. wrote the manuscript; everybody edited and approved the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- Aronesty E. ea-utils: “Command-line tools for processing biological sequencing data”. 2011 ( http://code.google.com/p/ea-utils)
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- Blainey PC, Milla CE, Cornfield DN, Quake SR. Quantitative analysis of the human airway microbial ecology reveals a pervasive signature for cystic fibrosis. Sci Transl Med. 2012;4:153ra130. doi: 10.1126/scitranslmed.3004458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke WJ, O’Connor CM, FitzGerald MX, McDonnell TJ. Pseudomonas aeruginosa exotoxin A induces pulmonary endothelial cytotoxicity: protection by dibutyryl-cAMP. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 1994;7:1754–1758. doi: 10.1183/09031936.94.07101754. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi A, Vieira P, Camara NO, Curi R, Carvalheira JB, Saad MJ. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011;9:e1001212. doi: 10.1371/journal.pbio.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, Cotter PD. The gut microbiota and its relationship to diet and obesity: new insights. Gut microbes. 2012;3:186–202. doi: 10.4161/gmic.20168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, Gaillot O, Schreiber S, Lemoine Y, Ryffel B, Hot D, Nunez G, Chen G, Rosenstiel P, Chamaillard M. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care. 2010;33:861–868. doi: 10.2337/dc09-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, Martinez FJ, Huffnagle GB. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE. 2011;6:e16384. doi: 10.1371/journal.pone.0016384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer N, Jackson JA, Vilgalys R, Jackson RB. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Applied and environmental microbiology. 2005;71:4117–4120. doi: 10.1128/AEM.71.7.4117-4120.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geny B, Khun H, Fitting C, Zarantonelli L, Mazuet C, Cayet N, Szatanik M, Prevost MC, Cavaillon JM, Huerre M, Popoff MR. Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability. Am J Pathol. 2007;170:1003–1017. doi: 10.2353/ajpath.2007.060583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, Moffatt MF, Cookson WO. Disordered microbial communities in asthmatic airways. PLoS ONE. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, Medzhitov R. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340:1230–1234. doi: 10.1126/science.1233632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short Term High Fat Diet Challenge Promotes Alternative Macrophage Polarization in Adipose Tissue via Natural Killer T Cells and Interleukin-4. J Biol Chem. 2012a;287:24378–24386. doi: 10.1074/jbc.M112.371807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, Qi L. Activation of Natural Killer T Cells Promotes M2 Macrophage Polarization in Adipose Tissue and Improves Systemic Glucose Tolerance via Interleukin-4 (IL-4)/STAT6 Protein Signaling Axis in Obesity. J Biol Chem. 2012b;287:13561–13571. doi: 10.1074/jbc.M112.350066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Flavell RA. Innate sensors of pathogen and stress: Linking inflammation to obesity. The Journal of allergy and clinical immunology. 2013;132:287–294. doi: 10.1016/j.jaci.2013.06.022. [DOI] [PubMed] [Google Scholar]
- Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M. Claudin heterogeneity and control of lung tight junctions. Annu Rev Physiol. 2013;75:551–567. doi: 10.1146/annurev-physiol-030212-183809. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Commun. 2009;388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- Lammers AJ, de Porto AP, de Boer OJ, Florquin S, van der Poll T. The role of TLR2 in the host response to pneumococcal pneumonia in absence of the spleen. BMC infectious diseases. 2012;12:139. doi: 10.1186/1471-2334-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut pathogens. 2013;5:23. doi: 10.1186/1757-4749-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose Y, Maruyama A, Iwasaki T, Miyamoto Y, Itoh K. 16S rRNA gene sequence-based analysis of clostridia related to conversion of germfree mice to the normal state. Journal of applied microbiology. 2009;107:2088–2097. doi: 10.1111/j.1365-2672.2009.04431.x. [DOI] [PubMed] [Google Scholar]
- Murray JF. Pulmonary edema: pathophysiology and diagnosis. The international journal of tuberculosis and lung disease: the official journal of the International Union against Tuberculosis and Lung Disease. 2011;15:155–160. i. [PubMed] [Google Scholar]
- Perina DG. Noncardiogenic pulmonary edema. Emergency medicine clinics of North America. 2003;21:385–393. doi: 10.1016/s0733-8627(03)00020-8. [DOI] [PubMed] [Google Scholar]
- Pusterla N, Jones ME, Mohr FC, Higgins JK, Mapes S, Jang SS, Samitz EM, Byrne BA. Fatal pulmonary hemorrhage associated with RTX toxin producing Actinobacillus equuli subspecies haemolyticus infection in an adult horse. Journal of veterinary diagnostic investigation: official publication of the American Association of Veterinary Laboratory Diagnosticians, Inc. 2008;20:118–121. doi: 10.1177/104063870802000127. [DOI] [PubMed] [Google Scholar]
- Saeed AI, Rieder SA, Price RL, Barker J, Nagarkatti P, Nagarkatti M. Acute lung injury induced by Staphylococcal enterotoxin B: disruption of terminal vessels as a mechanism of induction of vascular leak. Microscopy and microanalysis: the official journal of Microscopy Society of America, Microbeam Analysis Society, Microscopical Society of Canada. 2012;18:445–452. doi: 10.1017/S1431927612000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H, Yang L, Liu M, Xia S, Liu Y, Liu F, Kersten S, Qi L. Adipocyte spliced form of x-box-binding protein 1 promotes adiponectin multimerization and systemic glucose homeostasis. Diabetes. 2014;63:867–879. doi: 10.2337/db13-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrett SJ, Wilson CB, Liggitt HD, Hajjar AM. Redundant Toll-like receptor signaling in the pulmonary host response to Pseudomonas aeruginosa. American journal of physiology. Lung cellular and molecular physiology. 2007;292:L312–322. doi: 10.1152/ajplung.00250.2006. [DOI] [PubMed] [Google Scholar]
- Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, Geuking MB, Beutler B, Tedder TF, Hardt WD, Bercik P, Verdu EF, McCoy KD, Macpherson AJ. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Dore J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- Songer JG. Enteric Clostridia. In: Gyles CL, Prescott JF, Songer JG, Thoen CO, editors. Pathogenesis of bacterial infections in animals. Oxford, United Kingdom: Blackwell Publishing; 2004. pp. 131–142. [Google Scholar]
- Sun S, Ji Y, Kersten S, Qi L. Mechanisms of inflammatory responses in obese adipose tissue. Annu Rev Nutr. 2012a;32:261–286. doi: 10.1146/annurev-nutr-071811-150623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Xia S, Ji Y, Kersten S, Qi L. The ATP-P2X7 Signaling Axis Is Dispensable for Obesity-Associated Inflammasome Activation in Adipose Tissue. Diabetes. 2012b;61:1471–1478. doi: 10.2337/db11-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Taur Y, Pamer EG. The intestinal microbiota and susceptibility to infection in immunocompromised patients. Current opinion in infectious diseases. 2013;26:332–337. doi: 10.1097/QCO.0b013e3283630dd3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton SC, Waters KM, Karin NJ, Webb-Robertson BJ, Zangar RC, Lee KM, Bigelow DJ, Pounds JG, Corley RA. Diet-induced obesity reprograms the inflammatory response of the murine lung to inhaled endotoxin. Toxicol Appl Pharmacol. 2013;267:137–148. doi: 10.1016/j.taap.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009a;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009b;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, Pamer EG. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445–1456. doi: 10.1084/jem.20120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Russell SL, Finlay BB. Shifting the balance: antibiotic effects on host-microbiota mutualism. Nat Rev Microbiol. 2011;9:233–243. doi: 10.1038/nrmicro2536. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, Flavell RA. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol. 2013;190:5306–5312. doi: 10.4049/jimmunol.1300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.