

Abstract
Breast cancer remains the leading cause of cancer-related mortality in women. Comprehensive genomics, proteomics, and metabolomics studies are emerging that offer an opportunity to model disease biology, prognosis, and response to specific therapies. Although many biomarkers have been identified through advances in data mining techniques, few have been applied broadly to make patient-specific decisions. Here, we review a selection of breast cancer prognostic indicators and their implications. Our goal is to provide clinicians with a general evaluation of emerging computational methodologies for outcome prediction.
Keywords: computational model, precision prognosis, tumor
Introduction
One in eight women develops breast cancer, the most common cause of malignancy in females. Although the majority of patients now survive for many years after initial diagnosis and therapy, a significant subpopulation remains at risk of metastatic relapse. These women have a median survival time of less than 1.5 years at the time of relapse,1 with only 10% expected to survive 10 years after diagnosis.2
The ability to classify patients into clinically relevant subgroups to allow for precise therapy is urgently needed. Traditional tumor stratification is based largely on morphology, but is relevant in less than one-quarter of invasive breast carcinomas.3 However, novel computational models are emerging as powerful tools to address this deficiency. Recent work has integrated critical observations in the biology of breast cancer, including gene deletions, translocations, and locus amplification;4,5 biomarkers from high-throughput “-omics” technologies such as genomics, proteomics, and metabolomics; and long recognized outcome variables such as tumor size, histologic grade, axillary nodal status, and estrogen receptor (ER) status. We anticipate that these biomarkers will emerge as an effective molecular classification or guide for the determination of prognosis and the development of tailored therapy (reviewed by Gruver et al3).
However, much work regarding validation of these approaches is still required. For example, single biomarkers have not proven highly informative in most women. Somatic mutations with the potential ability to act as a biomarker were noted to occur in single genes (eg, TP53, PIK3CA, and GATA3) in <10% of 825 patients with primary breast cancers.6 Similarly, only three multi-gene signatures are incorporated into current clinical practice.7–9 Effective and clinically applicable prognostic indicators will also require the ability to address the emerging importance of the intra-tumoral heterogeneity of breast cancer.10
This review will not address specific clinical prognostic variables as they have been reviewed extensively by others.11–14 Instead, we have chosen to evaluate emergent prognostic indicators for breast cancer, focusing on computational methodologies and strategies to identify a molecular indicator (a biomarker or set of biomarkers). We are interested particularly in statistical and computational methodologies based on cutting-edge “-omics” technologies, moving from the conventional single oncogene/tumor suppressor gene strategy to one that exploits advances in systems biology. To achieve our goal, we performed a systematic search of all the English language literature regarding computational breast cancer prognosis, using a MEDLINE search for the period from January 2005 to July 2013. This review will begin with the utility of single molecular biomarkers and multi-gene signatures, followed by an evaluation of novel system-biology based analyses that infer information from high-throughput “-omics” data. Importantly, we will discuss exciting possibilities in the development of new integrative methodologies.
Identification of prognostic molecular signatures
Examination of genetic variation, genomic association, or transcriptomic alterations in large study cohorts allows the selection of single or multiple prognostic markers that can stratify patients into groups showing distinct outcomes (Table S1). It may also allow the specific tailoring of care. Sample size and biological hypotheses are distinguishing features of prognostic marker studies.15,16 In terms of sample size, a practical clinically based prognostic index for patients with metastatic breast cancer was recently proposed by a retrospective analysis of 2,322 patients after primary treatment.2 In parallel, others have focused on using biological determinants. For example, the Cancer Genome Atlas Network has incorporated the four major breast cancer subtypes with five genetic and epigenetic factors (genomic DNA copy numbers, DNA methylation, exome and messenger RNA expression, microRNA sequencing, and reverse-phase protein expression).6 The results are promising for understanding the biological underpinning of subtype-specific prognostic indicators. However, there remains a critical gap between an integrative model of “omics”-data and clinical variables. Describing biological pathways underlining clinical biomarkers in a large sample size could provide the opportunity to significantly improve our understanding of the biology and heterogeneity of breast cancers. For instance, the enrichment patterns of gene sets associated with embryonic stem cell (ESC) identity in the expression profiles of various human tumor types is associated with poor outcome in breast cancer.17
Single gene prognostic determinants
For specific tumor subtypes, the expression of single critical genes can serve as prognostic indicators (reviewed by Adam Maciejczyk).18 Germ-line mutations of BRCA1 are extensively used for early detection of familial breast cancer, and are predictive for 15%–20% of women with a family history of breast cancer, and 60%–80% of patients with combined breast and ovarian cancer.19 Additional prognostic factors include enhanced RAD21 and cohesin expression, which has been associated with resistance to chemotherapy in high-grade luminal, basal, and HER2 breast cancers.20 Transcription factor muscle segment homeobox 2 (Msx2) expression has been implicated in an increased likelihood of tumor cell death via apoptosis in invasive breast cancers.21 Enhanced expression of the anterior gradient-2 (AGR2) protein, which occurs in the presence of ER antagonist tamoxifen, confers poor prognosis in ER-positive breast cancers.22
The most common computational method to assess these critical biomarkers is the hazard Cox regression model.23 Given a time after diagnosis for values of the predictor variables, the model produces a survival function for the probability that the binary event of interest (eg, death or survival at the endpoint) occurs. In this context, additional computational models to improve the prediction have been proposed, including Bayesian network analysis evaluating probabilistic relationships among candidate genes24 and support vector machine methodologies.25 A review of standard survival analyses and the use of the outstanding Bioconductor tool suite is available at http://cran.r-project.org/web/views/Survival.html.
Current status of multi-gene prognostic determinants
Multi-gene signatures are most often derived from transcriptomic microarray and sequence data.26–28 These signatures not only have potential for classification and prognosis,29 but they can also predict specific tumor sub-phenotypes such as resistance to radiation.30,31 Generally, these multi-gene models can classify patients into subgroups with either distinct outcomes or diverse treatment responses in an unsupervised manner. As an example, the DNA content of breast adenocarcinomas can be classified as either stable, conferring good prognosis, or unstable, conferring poor prognosis.32
To identify multi-gene signatures or pathways, computational pattern learning algorithms have been successfully applied to transcript or mass spectrometry profiles. These algorithms can be roughly categorized into three groups: 1) unsupervised data mining, eg, hierarchical clustering,7,33 topographic projection,34 and other methodologies; 2) supervised classifiers, eg, decision trees,35 and Prediction Analysis of Microarray (PAM);9 and 3) semi-supervised learning models.36 In supervised algorithms, sample labels such as good- or poor-outcome are required to train the model before making decisions. In contrast, unsupervised algorithms are data-driven. In a semi-supervised model, the algorithm makes decisions based on both the raw data and the input labels, allowing partial labeling. Which method to choose depends on study purpose – for example, if a certain phenotype is known to be an important factor in identifying the multi-gene signature, one should select supervised methodologies to make use of that information.
Clinical application of prognostic determinants
In terms of single determinants, the expression levels of four genes (ER, PR, HER2, and Ki67), and combinations thereof, have been shown to have a strong prognostic impact (reviewed by Gökmen-Polar et al37). Missense mutations, eg, within the tumor suppressor gene p53 or increased levels of urokinase-type plasminogen activator (uPA) and/or plasminogen activator inhibitor-1 (PAI-1), which indicates poor clinical outcome, have been included by the American Society of Clinical Oncology 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer.38 Other single-gene determinants with somatic mutations are sensitive to targeted therapy; for instance, active ESR1 mutations in ER-positive metastatic breast cancer.39
Several multi-gene–based commercial prognostic testing methodologies are now available. The MammaPrint® 70-gene signature assay (Agendia, Inc., Irvine, CA, USA) stratifies patients’ outcomes and allows personalized therapeutic prediction.7,40,41 Two additional clinical decision-making assays, the Oncotype DX 21-gene assay42 (Genomic Health, Inc. (Redwood City, CA, USA) and a clinically updated version of the intrinsic subtype PAM50 assay43 (ARUP Laboratories, Salt Lake City, UT, USA), have been compared. Predicting prognostic intrinsic subtypes among 151 patients, investigators observed good agreement between the 21-gene and PAM50 assays for high and low prognostic risk assignment.44
Overall, molecular prognostic determinants have been successfully developed as adjuvant tools for innovative diagnostic, prognostic, and therapeutic approaches. As shown by Albain et al,45 changes in treatment decision after review of multi-gene signatures occurred in 30% of individuals predominantly from chemotherapy plus endocrine therapy to endocrine therapy alone with an associated diminution in treatment-related toxicity.
However, there are several limitations in the utility of either single gene or multi-gene prognostic signatures in routine practice, including requirement of fresh or frozen tissues to measure uPA/PAI-1 and deliberate measurement compared with a gold standard or “housekeeping genes” for single gene determinants. Another limitation for both single and multi-gene signatures is that they are positive in only a specific subtype of breast cancer. For instance, the 70-gene assay can only identify potential chemotherapy benefits in high-risk patients.45 In another study, the discordance rate was approximately 30% between the clinicopathologic risk categories given by the 70-gene assay and the 21-gene assay.45 Both assays are particularly limited in assigning high-risk status to ER-negative patients.46
How do we overcome these challenges?
Key issues with current multi-gene signature methodologies include specificity of prediction and standardization across diagnostic platforms. Li et al have reported that many randomly selected genes show predictive power for cancer prognosis in one dataset, while losing predictive power in other datasets.47 Therefore, an evaluation using independent datasets is necessary to assess a prognostic model or indicator. Recently, Venet et al28 compared 47 published breast cancer outcome signatures to mimic signatures made of random genes. Surprisingly, they found that 60% of reported gene signatures of identical size were equally predictive of “mimic” gene signatures. Of particular concern, 23% of the signatures were worse than random, and more than 90% of mimic signatures with 100 genes were significant outcome predictors.28 Thus, the deficiencies associated with the reproducibility of multi-gene–based prediction prevent effective usage at this time.
The complexity and discrepancy of single- or multi-gene signatures also precludes easy extraction of biologically and therapeutically relevant information. A clue may be provided by observations that suggest that although genetic alterations between patients differ, they frequently involve common pathways. It is therefore critical to identify relevant pathways involved in breast cancer progression and detect the corresponding indicators that are prognostic in different tumor subtypes or patients.
Novel strategies for prognostic indicator development
Incorporation of network or pathway information into prognostic biomarker discovery could significantly improve prediction performance. For this purpose, investigators have used a large number of supervised machine-learning approaches (ie, inferring knowledge from data with labeled samples) to take advantage of prior knowledge. For example, data mining on gene expression profiling reveals an association between hyperactivity in the PI3K pathway, lowered ER levels, and resistance to endocrine therapy.48 Unfortunately, there is no single “one-size-fits-all” algorithm, integrating accuracy, stability, and interpretability of gene selection.49 Table 1 lists methods to identify prognostic determinants based on gene expression in breast cancer. Two recent reviews discuss prognostication of tumor mutations using pathway and network analysis.50,51
Table 1.
Methods to identify prognostic determinants based on gene expression in breast cancer
| Data source | Method | Description |
|---|---|---|
| Genome-wide gene expression | RXA-GSP53 | Summarizes the individualized relative expression between biological experiment-defined gene-set pairs, thus tolerating the diverse noise and differences observed from multiple technologies and laboratories. |
| LDS36 | This semi-supervised approach successfully employed unlabeled gene expression data and achieved significant performance in gene expression-based outcome prediction for cancer patients. | |
| MSS47 | Identifies prognostic markers that can be used in combination to stratify breast cancer patients into groups of different risk ranks with high accuracy. | |
| Correlation56 | Correlation between two biomarkers is a more useful prognostic factor than their individual expressions. | |
| BCRSVM25 | Uses modern machine-learning method SVM to train six clinical prognostic variables (histological grade, tumor size, number of metastatic lymph nodes, estrogen receptors, lymphovascular invasion, local invasion of tumor, and number of tumors) into a prognostic model. | |
| PGL34 | A literature-proposed predictive gene list for breast cancer is benchmarked against a separate gene list to construct nonlinear topographic projection maps for prognosis. | |
| PAM9 | PAM together with other conventional methods was used to define gene expression-based “intrinsic” subtypes that showed prognosis. | |
| Cox proportional-hazards regression modeling, gene-set enrichment analysis17 | Based on a careful gene-set enrichment analysis, multiple gene-set signatures stratify samples into prognostic subgroups. | |
| Bayesian network analysis24 | Bayesian probability was employed in neural networks to model censored data. | |
|
| ||
| Gene expression, experiment-based gene signatures | Expression levels relative to a baseline condition, hierarchical clustering, “leave-one-out” cross-validation7 | Top genes were selected to distinguish subtypes of breast cancers that show prognosis. |
|
| ||
| Gene expression, text mining | eScience–Bayesian59 | Permits coherent integration of prior information and multiple data sources, such as gene expression and information derived from literature. |
|
| ||
| Gene expression, clinical and genetic markers | I-RELIEF57, an iterative method based on the feature selection algorithm called RELIEF | Integrated clinical variables with gene expression or biological pathway. |
|
| ||
| Gene expression, copy number | iCluster58 | A likelihood-based, joint latent variable model for integrative clustering samples. |
|
| ||
| Gene expression, copy number, pathway | PARADIGM6,60 | Integrates copy number, mRNA expression, and pathway interaction data into a personalized pathway-by-sample matrix that clusters patients into distinct prognostic subgroups. |
Abbreviations: BCRSVM, breast cancer recurrence prediction based on SVM; LDS, low density separation; MSS, multiple survival screening; PAM, prediction analysis of microarray; PARADIGM, pathway recognition algorithm using data integration on genomic models; PGL, predictive gene lists; RXA-GSP, relative expression analysis of gene set pair; SVM, support vector machine.
Using biological hypothesis-based gene selection and interpretation, we and others have identified transcriptional prognostic indicators (Table 1). As prognostic biomarkers, these models calculate a score per sample and use a simple threshold value of 1 to dichotomize patients into prognostic groups. Pitroda et al piloted a tumor endothelium-derived inflammatory signature consisting of six genes that were associated with poor outcome in multiple cancers.52 Similarly, in multiple adult tumors, we found that histologically poorly differentiated tumors display an ESC-associated expression imbalance, ie, preferential overexpression of targets of ESC-associated transcription factors (NANOG, OCT, SOX2, and MYC) combined with underexpression of Polycomb-regulated genes.53 This finding is in agreement with the known significance of survival difference between patients with the expression pattern of these ESC-like signatures and other patients.17 More recently, the coactivation of the ESC marker MYC and oncogene HER2 was associated with an acquisition of a self-renewal phenotype that is associated with poor outcome.54
However, the lack of standards for measurement across various studies, assays, and diagnostic platforms makes it difficult to translate individual findings into clear clinical applications. A popular solution is to compare the median scores of a calculated indicator in a measured cohort,52 whereas adding new patients changes the estimation of median score and thus the final decision.
Relative expression analysis of gene-set pairs (RXA-GSP), is a novel methodology that we have pioneered to translate prognostic gene targets for individualized treatment planning.53 RXA-GSP is built on three principles: 1) each individual has both favorable and unfavorable prognostic factors for breast cancer. It is the imbalance that determines the individual outcome (Figure 1). For example, NANOG expression is an indicator of poor prognosis stimulating the growth and metastasis of breast cancer cells, whereas KLF4 is a favorable prognostic indicator inhibiting these processes.55 2) The correlation between two factors is, at least in some cases, more significant than the overall expression of either. For example, SIRT1 and DBC1 are both overexpressed in breast tumor tissue, but the correlation between their levels of expression is diminished.56 3) We employ a hypothesis-based and experimentally derived approach to the identification of candidates.
Figure 1.
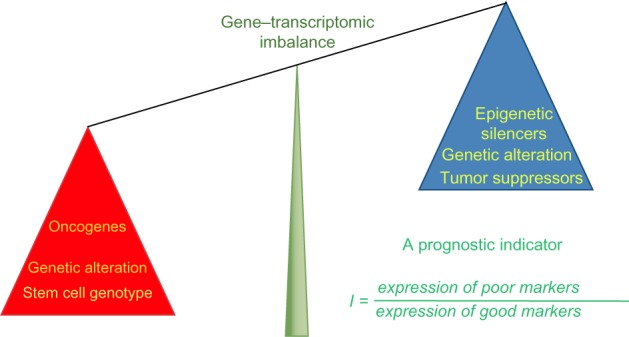
Illustration of RXA-GSP method.
Notes: This prognostic indicator is the ratio between scores (eg, expression values) of poor prognostic markers versus that of good prognostic markers. It has the ability to integrate different scales of data, bridging cancer biology with the clinic by employing both hypothesis-based and experimentally derived gene-set selection.
Abbreviation: RXA-GSP, relative expression analysis with gene-set pairs.
Using RXA-GSP, a resultant indicator is computed as the optimal combination of candidate genes that was trained from several large cohorts, and validated in independent cohorts.53 Considering the heterogeneity of breast cancer in each individual patient along with the relative expression of gene-set pairs and the correlations between them, RXA-GSP allows the computationally driven derivation of individualized prognostic indicators, bridging cancer biology with the clinic through gene expression analysis.
Next generation integrative prognostic methods
To date, the majority of studies focused on prognosis have integrated clinical variables with gene expression or biological pathway data.53,57,58 However, attractive opportunities are now available to integrate comprehensive genotype, metabolomics, and phenotype data. Success should facilitate robust prognostic and therapeutic prediction for each patient. Indeed, such success in this endeavor will have significant impact across biomedicine.
However, this potential is limited by the challenges of the sheer size and complexity of high-throughput data resources, often resulting in significant imprecision in data usage. Classical statistical models are frequently insufficient for integrating “omics”-data with other resources, so specific problems are dealt with on a case-by-case basis due to the lack of a coherent overarching method of analysis. Indeed, we and others, have proposed the development of integrated breast cancer prognostic indicators to provide insights into significant variables.53,59,60
Eklund et al59 have addressed this problem through an eScience–Bayes approach, complementing a Bayesian probability models’ ability to incorporate gene expression with text mining, modeling highly complex problems with high performance computing. Applying this model to several sets of independent gene expression data results in consistently accurate prediction of breast cancer metastases across cohorts.59 Though such methods show promise for tailoring specific models to complex problems with high accuracy, their application is cumbersome, as they require manual selection of genes for which we have prior knowledge. This deficiency is related to a lack of a standard for displaying scientific text in formats that are computationally readable. Another obvious weakness is that a microarray represents a single snapshot of the patient.
New avenues of patient-specific tumor phenotyping are also facilitating prognosis prediction. These methodologies include the evaluation of risk associated with inherited genetic variation or genome-wide association studies (GWAS).61,62 Similarly, epigenetic changes in breast cancer cells (such as modifications in histone acetylation and DNA methylation at specific gene regulatory elements) have been implicated in breast oncogenesis.6,63 These observations, coupled with the emerging use of epigenetic-modifying agents suggest an era of genomics-based therapy selection.33,64,65 For example, stromal/microenvironmental elements affect the prognosis, modifying tumor invasion and/or drug targeting.66,67
Thus, the effectiveness of interpretations of multi-gene assays likely depends on adjustment for other factors. A breakthrough work, Pathway Recognition Algorithm using Data Integration on Genomic Models (PARADIGM),60 integrates copy number, mRNA expression, and pathway interaction data to a personalized pathway-by-sample matrix that clusters patients into distinct prognostic subgroups. This integration algorithm has been recently applied to depict comprehensive molecular portraits of human breast tumors.6
In summary, we believe biological hypothesis-driven computational models that integrate current significant variables in multiple facets, including transcriptomic alterations, chromatin modification, and stroma response, will provide significant insight not only into the biology of breast cancer, but also into determining the prognosis of individual patients.
Conclusion
Challenges remain for breast cancer prognosis due to its complexity and the lack of standardization among models and “omics”-datasets. Specifically, what impedes the convenience and simplicity necessary for clinical application is the lack of precision molecular indicators that help individualized therapeutic decisions. Ideally, a prognostic indicator performs in a specific, sensitive, inexpensive, and easy manner. We expect an integrative computational model of “omics”-data and clinical variables to significantly improve our understanding of the biology and heterogeneity of breast cancers. This is likely to be achieved through either enlarging the sample size or characterizing a specific subtype with given phenotypes. The latter has been recently recognized, though controversial, hypothesizing a collection of genuinely variable malignances that happen to originate from breast epithelium.68 In conclusion, a prognostic indicator should clearly delineate the risk group for an individual, in terms of tumor growth, invasion, and metastatic potential, and/or the likelihood of response to a given therapeutic modality.
Supplementary material
Table S1.
Selected instances from literature search
| 1st author | Year | Journal | Pubmed ID | Cancer biology underpinning | Related clinical indicator | Major contribution/conclusion |
|---|---|---|---|---|---|---|
| van ’t Veer1 | 2002 | Nature | 11823860 | Gene expression patterns | Lymph node status | Provides a powerful tool to tailor adjuvant systemic treatment that could greatly reduce the cost of BC treatment. |
| Rennstam2 | 2003 | Cancer Research | 14695203 | Chromosomal copy number aberrations | Patterns of copy number gains and losses define BCs with distinct clinicopathological features and patient prognosis. | |
| Paik3 | 2004 | New England Journal of Medicine | 15591335 | Gene expression patterns | Node-negative, ER-positive, tamoxifen treatment | A novel recurrence score based on 21 genes to quantify the likelihood of distant recurrence in patients as well as overall survival time. |
| Kronenwett4 | 2006 | Cancer Epidemiology, Biomarkers and Prevention | 16985023 | Genomic stability | Objective classification of BCs into stable and unstable subtypes that are a prognostic indicator independent of established clinical factors. | |
| Bacac5 | 2006 | PLoS One | 17183660 | Stromal cells | Human genes expressed in mouse stromal response to tumor invasion predicts BC patient survival. | |
| Suh6 | 2007 | Clinical Cancer Research | 17200346 | CLIC4 (chloride intracellular channel 4) | Reactivation and restoration of CLIC4 in tumor cells or the converse in tumor stromal cells could provide a novel approach to inhibit tumor growth. | |
| Conlin7 | 2007 | Molecular Diagnosis and Therapy | 18078353 | Oncotype DX recurrence score assay | Lymph node negative, ER-expressing BC | The Oncotype DX assay and others aim to help improve risk classification and recurrence prediction and optimize selection of patients for adjuvant chemotherapy. |
| Rodriguez8 | 2008 | Carcinogenesis | 18499701 | HOXB13 (homeobox B13) and IL17BR (interleukin 17 receptor B) | Estrogen signaling | Hypermethylation of HOXB13 is a later event of tumor progression and a prognostic indicator of advanced BC. |
| Wei9 | 2008 | Molecular Carcinogenesis | 18176935 | H3K27me3 | Loss of H3K27me3 is a predictor of poor outcome in BCs. | |
| Kim10 | 2008 | Annals of Oncology | 17956886 | CDKs | Patients recruited for study underwent mastectomy or breast-conserving surgery | Tumors with high CDK1SA and high CDK2SA showed significantly poorer 5-year relapse-free survival than those with low CDK1SA and low CDK2SA, respectively. |
| Han11 | 2008 | Nature | 18337816 | SATB1 (SATB homeobox 1) | SATB1 is a genome organizer that tethers multiple genomic loci and recruits chromatin-remodeling enzymes to regulate chromatin structure and gene expression. | |
| Ben-Porath12 | 2008 | Nature Genetics | 18443585 | Stem cell genetic expression signatures | Detailed characterization of the stem-cell regulatory networks active in cancer is likely to yield powerful diagnostic and prognostic markers. | |
| Parker13 | 2009 | Journal of Clinical Oncology | 19204204 | A 50-gene set (PAM50) | “Intrinsic” subtypes, pathologic staging, histologic grade | The intrinsic subtype and risk predictors based on the PAM50 gene set adds significant prognostic and predictive value. |
| Sung14 | 2010 | Cancer Science | 20412117 | SIRT1 (sirtuin 1) and CCAR2 (also known as DBC1, deleted in breast cancer 1, KIAA1967) | Luminal subtype, ER and PR expressions | Correlation between SIRT1 and DBC1 is a more useful prognostic factor than their individual expressions. Correlation between the two is decreased in tumor cells. |
| Gevensleben15 | 2010 | International Journal of Molecular Medicine | 21042777 | 70-gene expression profile MammaPrint® | Size, age, histological grade, hormone receptor status, peritumoral vascular invasion and HER2 status | Gene signature MammaPrint® is shown to provide additional independent prognostic information. |
| Lanigan16 | 2010 | Breast Cancer Research | 20682066 | Msx2 (msh homeobox 2) | Msx2 expression results in improved outcome for BCs, possibly by increasing the likelihood of tumor cell death by apoptosis. | |
| Creighton17 | 2010 | Breast Cancer Research | 20569503 | PI3K pathway | Luminal ER+ breast tumors | Luminal B tumors have hyperactive GFR/PI3K signaling associated with lower ER levels, which has been correlated with resistance to endocrine therapy. Targeting PI3K in these tumors may reverse loss of ER expression and signaling and restore hormonal sensitivity. |
| Xu18 | 2011 | Breast Cancer Research | 21255398 | RAD21 (RAD21 homolog [S. pombe]) | N/A | RAD21 expression confers poor prognosis and resistance to chemotherapy in high-grade luminal, basal, and HER2 BCs. |
| Littlepage19 | 2012 | Cancer Discovery | 22728437 | ZNF217 (zinc finger protein 217) | Amplification of the human chromosomal region 20q13 | ZNF217 (amplified in numerous cancers) is a poor prognostic indicator and therapeutic target in patients with BC and may be a strong biomarker of triciribine treatment efficacy in patients. |
| Pitroda20 | 2012 | PLoS One | 23056240 | Endothelial inflammatory pathways | The first prognostic cancer gene signature derived from an experimental model of tumor-associated endothelial inflammation. | |
| Kim21 | 2012 | Journal of Breast Cancer | 22807942 | Gene expression patterns | Histological grade, size, number of metastatic lymph nodes, ER, lymphovascular invasion, local invasion of tumor, and number of tumors | As the selected prognostic factors can be easily obtained in clinical practice, the proposed model might prove useful in the prediction of BR recurrence. |
| Faryna22 | 2012 | FASEB Journal | 22930747 | Aberrant DNA methylation | Low-grade ER- and/or PR-positive BC | Early methylation changes are frequent in the low-grade pathway of BC and may be useful in the development of prognostic markers. |
| Fasching23 | 2012 | Human Molecular Genetics | 22532573 | TOX3 (TOX high mobility group box family member 3) | With the exception of rs3803662 (TOX3), there was no evidence that any of the SNPs associated with BC susceptibility were associated with BC survival. | |
| Huang24 | 2013 | Cell and Bioscience | 23497677 | MACC1 (metastasis associated in colon cancer 1) | Clinicopathologic features | The over expression of MACC1 in BR is significantly correlated with adverse clinicopathological features, including metastasis and patient survival. |
| Yang25 | 2013 | PLoS One | 23441166 | Mediation of transcription factor GRHL2 (grainyhead-like 2 [Drosophila]) on its targets is prognostic in BC | Histological grade | Proposed the RXA-GSP (relative expression analysis with gene-set pairs) method, shows promise as both a valid prediction model as well as high potential for clinical utility. |
| Nagata26 | 2014 | Breast Cancer | 22528804 | Induced pluripotent stem cell inducing factors | Strong expression of NANOG is an indicator of poor prognosis for BC patients, whereas KLF4 is a favorable prognostic indicator. |
Note: Each key word (genomic, transcriptional, epigenetic, sequence, novel) respectively together with “breast cancer” and “prognostic indicator” was searched in PubMed, from Jan 2005–July 2013.
Abbreviations: BC, breast cancer; ER, estrogen receptor; PAM, prediction analysis of microarray; RXA-GSP, relative expression analysis of gene set pair; SNPs, single-nucleotide polymorphisms.
References
- 1.van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 2.Rennstam K, Ahlstedt-Soini M, Baldetorp B, et al. Patterns of chromosomal imbalances defines subgroups of breast cancer with distinct clinical features and prognosis. A study of 305 tumors by comparative genomic hybridization. Cancer Res. 2003;63(24):8861–8868. [PubMed] [Google Scholar]
- 3.Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 4.Kronenwett U, Ploner A, Zetterberg A, et al. Genomic instability and prognosis in breast carcinomas. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2006;15(9):1630–1635. doi: 10.1158/1055-9965.EPI-06-0080. [DOI] [PubMed] [Google Scholar]
- 5.Bacac M, Provero P, Mayran N, Stehle JC, Fusco C, Stamenkovic I. A mouse stromal response to tumor invasion predicts prostate and breast cancer patient survival. PLoS One. 2006;1:e32. doi: 10.1371/journal.pone.0000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suh KS, Crutchley JM, Koochek A, et al. Reciprocal modifications of CLIC4 in tumor epithelium and stroma mark malignant progression of multiple human cancers. Clin Cancer Res. 2007;13(1):121–131. doi: 10.1158/1078-0432.CCR-06-1562. [DOI] [PubMed] [Google Scholar]
- 7.Conlin AK, Seidman AD. Use of the Oncotype DX 21-gene assay to guide adjuvant decision making in early-stage breast cancer. Molecular diagnosis & therapy. 2007;11(6):355–360. doi: 10.1007/BF03256259. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez BA, Cheng AS, Yan PS, et al. Epigenetic repression of the estrogen-regulated Homeobox B13 gene in breast cancer. Carcinogenesis. 2008;29(7):1459–1465. doi: 10.1093/carcin/bgn115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Y, Xia W, Zhang Z, et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Molecular carcinogenesis. 2008;47(9):701–706. doi: 10.1002/mc.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SJ, Nakayama S, Miyoshi Y, et al. Determination of the specific activity of CDK1 and CDK2 as a novel prognostic indicator for early breast cancer. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2008;19(1):68–72. doi: 10.1093/annonc/mdm358. [DOI] [PubMed] [Google Scholar]
- 11.Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature. 2008;452(7184):187–193. doi: 10.1038/nature06781. [DOI] [PubMed] [Google Scholar]
- 12.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sung JY, Kim R, Kim JE, Lee J. Balance between SIRT1 and DBC1 expression is lost in breast cancer. Cancer Sci. 2010;101(7):1738–1744. doi: 10.1111/j.1349-7006.2010.01573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gevensleben H, Gohring UJ, Buttner R, et al. Comparison of MammaPrint and TargetPrint results with clinical parameters in German patients with early stage breast cancer. International journal of molecular medicine. 2010;26(6):837–843. doi: 10.3892/ijmm_00000532. [DOI] [PubMed] [Google Scholar]
- 16.Lanigan F, Gremel G, Hughes R, et al. Homeobox transcription factor muscle segment homeobox 2 (Msx2) correlates with good prognosis in breast cancer patients and induces apoptosis in vitro. Breast Cancer Res. 2010;12(4):R59. doi: 10.1186/bcr2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creighton CJ, Fu X, Hennessy BT, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12(3):R40. doi: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu H, Yan M, Patra J, et al. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011;13(1):R9. doi: 10.1186/bcr2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Littlepage LE, Adler AS, Kouros-Mehr H, et al. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer discovery. 2012;2(7):638–651. doi: 10.1158/2159-8290.CD-12-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pitroda SP, Zhou T, Sweis RF, et al. Tumor endothelial inflammation predicts clinical outcome in diverse human cancers. PLoS One. 2012;7(10):e46104. doi: 10.1371/journal.pone.0046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim W, Kim KS, Lee JE, et al. Development of novel breast cancer recurrence prediction model using support vector machine. Journal of breast cancer. 2012;15(2):230–238. doi: 10.4048/jbc.2012.15.2.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faryna M, Konermann C, Aulmann S, et al. Genome-wide methylation screen in low-grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. FASEB J. 2012;26(12):4937–4950. doi: 10.1096/fj.12-209502. [DOI] [PubMed] [Google Scholar]
- 23.Fasching PA, Pharoah PD, Cox A, et al. The role of genetic breast cancer susceptibility variants as prognostic factors. Human molecular genetics. 2012;21(17):3926–3939. doi: 10.1093/hmg/dds159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Zhang H, Cai J, et al. Overexpression of MACC1 and Its significance in human Breast Cancer Progression. Cell & bioscience. 2013;3(1):16. doi: 10.1186/2045-3701-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Vasudevan P, Parekh V, Penev A, Cunningham JM. Bridging cancer biology with the clinic: relative expression of a GRHL2-mediated gene-set pair predicts breast cancer metastasis. PLoS One. 2013;8(2):e56195. doi: 10.1371/journal.pone.0056195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagata T, Shimada Y, Sekine S, et al. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2012;21(1):96–101. doi: 10.1007/s12282-012-0357-y. [DOI] [PubMed] [Google Scholar]
Acknowledgments
This research was supported by NIH grant R21 CA167305-01A1. We appreciate Zakir Gowani for his careful proofreading.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ravnan MC, Ravnan SL, Walberg MP. Metastatic breast cancer: a review of current and novel pharmacotherapy. Formulary. 2011;46:130–146. [Google Scholar]
- 2.Puente J, López-Tarruella S, Ruiz A, et al. Practical prognostic index for patients with metastatic recurrent breast cancer: retrospective analysis of 2,322 patients from the GEICAM Spanish El Alamo Register. Breast Cancer Res Treat. 2010;122(2):591–600. doi: 10.1007/s10549-009-0687-4. [DOI] [PubMed] [Google Scholar]
- 3.Gruver AM, Portier BP, Tubbs RR. Molecular pathology of breast cancer: the journey from traditional practice toward embracing the complexity of a molecular classification. Arch Pathol Lab Med. 2011;135(5):544–557. doi: 10.5858/2010-0734-RAIR.1. [DOI] [PubMed] [Google Scholar]
- 4.Buness A, Kuner R, Ruschhaupt M, Poustka A, Sültmann H, Tresch A. Identification of aberrant chromosomal regions from gene expression microarray studies applied to human breast cancer. Bioinformatics. 2007;23(17):2273–2280. doi: 10.1093/bioinformatics/btm340. [DOI] [PubMed] [Google Scholar]
- 5.Rennstam K, Ahlstedt-Soini M, Baldetorp B, et al. Patterns of chromosomal imbalances defines subgroups of breast cancer with distinct clinical features and prognosis. A study of 305 tumors by comparative genomic hybridization. Cancer Res. 2003;63(24):8861–8868. [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van ’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 8.Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 9.Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roché H. Cancers du sein : évolution des grands concepts thérapeutiques [Evolving strategies over time for treatment of breast cancer] Bull Cancer. 2013;100(9):857–863. doi: 10.1684/bdc.2013.1805. French. [DOI] [PubMed] [Google Scholar]
- 12.Rakha EA. Pitfalls in outcome prediction of breast cancer. J Clin Pathol. 2013;66(6):458–464. doi: 10.1136/jclinpath-2012-201083. [DOI] [PubMed] [Google Scholar]
- 13.Rakha EA, Reis-Filho JS, Baehner F, et al. Breast cancer prognostic classification in the molecular era: the role of histological grade. Breast Cancer Res. 2010;12(4):207. doi: 10.1186/bcr2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Boer M, van Dijck JA, Bult P, Borm GF, Tjan-Heijnen VC. Breast cancer prognosis and occult lymph node metastases, isolated tumor cells, and micrometastases. J Natl Cancer Inst. 2010;102(6):410–425. doi: 10.1093/jnci/djq008. [DOI] [PubMed] [Google Scholar]
- 15.McGuire WL, Clark GM. Prognostic factors and treatment decisions in axillary-node-negative breast cancer. N Engl J Med. 1992;326(26):1756–1761. doi: 10.1056/NEJM199206253262607. [DOI] [PubMed] [Google Scholar]
- 16.Rogers CE, Loveday RL, Drew PJ, Greenman J. Molecular prognostic indicators in breast cancer. Eur J Surg Oncol. 2002;28(5):467–478. doi: 10.1053/ejso.2002.1258. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adam Maciejczyk A. New prognostic factors in breast cancer. Adv Clin Exp Med. 2013;22(1):5–15. [PubMed] [Google Scholar]
- 19.Cuda G, Cannataro M, Quaresima B, et al. Proteomic profiling of inherited breast cancer: identification of molecular targets for early detection, prognosis and treatment, and related bioinformatics tools. In: Apolloni B, Marinaro M, Tagliaferri R, editors. Neural Nets. Vol. 2859. Berlin, Germany: Springer; 2003. pp. 245–257. [Google Scholar]
- 20.Xu H, Yan M, Patra J, et al. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011;13(1):R9. doi: 10.1186/bcr2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanigan F, Gremel G, Hughes R, et al. Homeobox transcription factor muscle segment homeobox 2 (Msx2) correlates with good prognosis in breast cancer patients and induces apoptosis in vitro. Breast Cancer Res. 2010;12(4):R59. doi: 10.1186/bcr2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hrstka R, Nenutil R, Fourtouna A, et al. The pro-metastatic protein anterior gradient-2 predicts poor prognosis in tamoxifen-treated breast cancers. Oncogene. 2010;29(34):4838–4847. doi: 10.1038/onc.2010.228. [DOI] [PubMed] [Google Scholar]
- 23.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. Hoboken, NJ: John Wiley & Sons, Inc; 1980. [Google Scholar]
- 24.Lisboa PJ, Wong H, Harris P, Swindell R. A Bayesian neural network approach for modelling censored data with an application to prognosis after surgery for breast cancer. Artif Intell Med. 2003;28(1):1–25. doi: 10.1016/s0933-3657(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 25.Kim W, Kim KS, Lee JE, et al. Development of novel breast cancer recurrence prediction model using support vector machine. J Breast Cancer. 2012;15(2):230–238. doi: 10.4048/jbc.2012.15.2.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massagué J. Sorting out breast-cancer gene signatures. N Engl J Med. 2007;356(3):294–297. doi: 10.1056/NEJMe068292. [DOI] [PubMed] [Google Scholar]
- 27.Fu J, Jeffrey SS. Transcriptomic signatures in breast cancer. Mol Biosyst. 2007;3(7):466–472. doi: 10.1039/b618163e. [DOI] [PubMed] [Google Scholar]
- 28.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7(10):e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378(9805):1812–1823. doi: 10.1016/S0140-6736(11)61539-0. [DOI] [PubMed] [Google Scholar]
- 30.Gee JM, Nicholson RI. Expanding the therapeutic repertoire of epidermal growth factor receptor blockade: radiosensitization. Breast Cancer Res. 2003;5(3):126–129. doi: 10.1186/bcr584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lacombe J, Mange A, Azria D, Solassol J. Identification de marqueurs prédictifs de la réponse à la radiothérapie par approche protéomique [Identification of predictive biomarkers to radiotherapy outcome through proteomics approaches] Cancer Radiother. 2013;17(1):62–69. doi: 10.1016/j.canrad.2012.11.003. quiz 70, 72. French. [DOI] [PubMed] [Google Scholar]
- 32.Kronenwett U, Ploner A, Zetterberg A, et al. Genomic instability and prognosis in breast carcinomas. Cancer Epidemiol Biomarkers Prev. 2006;15(9):1630–1635. doi: 10.1158/1055-9965.EPI-06-0080. [DOI] [PubMed] [Google Scholar]
- 33.Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature. 2008;452(7184):187–193. doi: 10.1038/nature06781. [DOI] [PubMed] [Google Scholar]
- 34.Sivaraksa M, Lowe D. Predictive gene lists for breast cancer prognosis: a topographic visualisation study. BMC Med Genomics. 2008;1:8. doi: 10.1186/1755-8794-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anunciação O, Gomes BC, Vinga S, Gaspar J, Oliveira AL, Rueff J. A data mining approach for the detection of high-risk breast cancer groups. Adv Bioinformatics. 2010;74:43–51. [Google Scholar]
- 36.Shi M, Zhang B. Semi-supervised learning improves gene expression-based prediction of cancer recurrence. Bioinformatics. 2011;27(21):3017–3023. doi: 10.1093/bioinformatics/btr502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gökmen-Polar Y, Badve S. Breast cancer prognostic markers: where are we now? MLO Med Lab Obs. 2012;44(9):22, 24–25. [PubMed] [Google Scholar]
- 38.Harris L, Fritsche H, Mennel R, et al. American Society of Clinical Oncology American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007;25(33):5287–5312. doi: 10.1200/JCO.2007.14.2364. [DOI] [PubMed] [Google Scholar]
- 39.Segal CV, Dowsett M. Estrogen receptor mutations in breast cancer – new focus on an old target. Clin Cancer Res. 2014;20(7):1724–1726. doi: 10.1158/1078-0432.CCR-14-0067. [DOI] [PubMed] [Google Scholar]
- 40.Slodkowska EA, Ross JS. MammaPrint 70-gene signature: another milestone in personalized medical care for breast cancer patients. Exp Rev Mol Diagn. 2009;9(5):417–422. doi: 10.1586/erm.09.32. [DOI] [PubMed] [Google Scholar]
- 41.Gevensleben H, Göhring UJ, Büttner R, et al. Comparison of MammaPrint and TargetPrint results with clinical parameters in German patients with early stage breast cancer. Int J Mol Med. 2010;26(6):837–843. doi: 10.3892/ijmm_00000532. [DOI] [PubMed] [Google Scholar]
- 42.Conlin AK, Seidman AD. Use of the Oncotype DX 21-gene assay to guide adjuvant decision making in early-stage breast cancer. Mol Diagn Ther. 2007;11(6):355–360. doi: 10.1007/BF03256259. [DOI] [PubMed] [Google Scholar]
- 43.Prat A, Parker JS, Fan C, Perou CM. PAM50 assay and the three-gene model for identifying the major and clinically relevant molecular subtypes of breast cancer. Breast Cancer Res Treat. 2012;135(1):301–306. doi: 10.1007/s10549-012-2143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelly CM, Bernard PS, Krishnamurthy S, et al. Agreement in risk prediction between the 21-gene recurrence score assay (Oncotype DX®) and the PAM50 breast cancer intrinsic Classifier™ in early-stage estrogen receptor-positive breast cancer. Oncologist. 2012;17(4):492–498. doi: 10.1634/theoncologist.2012-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albain KS, Paik S, van’t Veer L. Prediction of adjuvant chemotherapy benefit in endocrine responsive, early breast cancer using multigene assays. Breast. 2009;18(Suppl 3):S141–S145. doi: 10.1016/S0960-9776(09)70290-5. [DOI] [PubMed] [Google Scholar]
- 46.Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med. 2009;360(8):790–800. doi: 10.1056/NEJMra0801289. [DOI] [PubMed] [Google Scholar]
- 47.Li J, Lenferink AE, Deng Y, et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun. 2010;1:34. doi: 10.1038/ncomms1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Creighton CJ, Fu X, Hennessy BT, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12(3):R40. doi: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cun Y, Fröhlich HF. Prognostic gene signatures for patient stratification in breast cancer: accuracy, stability and interpretability of gene selection approaches using prior knowledge on protein-protein interactions. BMC Bioinformatics. 2012;13:69. doi: 10.1186/1471-2105-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nat Methods. 2013;10(11):1108–1115. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raphael BJ, Dobson JR, Oesper L, Vandin F. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med. 2014;6(1):5. doi: 10.1186/gm524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitroda SP, Zhou T, Sweis RF, et al. Tumor endothelial inflammation predicts clinical outcome in diverse human cancers. PLoS One. 2012;7(10):e46104. doi: 10.1371/journal.pone.0046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang X, Vasudevan P, Parekh V, Penev A, Cunningham JM. Bridging cancer biology with the clinic: relative expression of a GRHL2-mediated gene-set pair predicts breast cancer metastasis. PLoS One. 2013;8(2):e56195. doi: 10.1371/journal.pone.0056195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nair R, Roden DL, Teo WS, et al. c-Myc and Her2 cooperate to drive a stem-like phenotype with poor prognosis in breast cancer. Oncogene. 2013 Sep 23; doi: 10.1038/onc.2013.368. Epub. [DOI] [PubMed] [Google Scholar]
- 55.Nagata T, Shimada Y, Sekine S, et al. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2014;21(1):96–101. doi: 10.1007/s12282-012-0357-y. [DOI] [PubMed] [Google Scholar]
- 56.Sung JY, Kim R, Kim JE, Lee J. Balance between SIRT1 and DBC1 expression is lost in breast cancer. Cancer Sci. 2010;101(7):1738–1744. doi: 10.1111/j.1349-7006.2010.01573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Y, Goodison S, Li J, Liu L, Farmerie W. Improved breast cancer prognosis through the combination of clinical and genetic markers. Bioinformatics. 2007;23(1):30–37. doi: 10.1093/bioinformatics/btl543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25(22):2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eklund M, Spjuth O, Wikberg JE. An eScience-Bayes strategy for analyzing omics data. BMC Bioinformatics. 2010;11:282. doi: 10.1186/1471-2105-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaske CJ, Benz SC, Sanborn JZ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26(12):i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fasching PA, Pharoah PD, Cox A, et al. The role of genetic breast cancer susceptibility variants as prognostic factors. Hum Mol Genet. 2012;21(17):3926–3939. doi: 10.1093/hmg/dds159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rafiq S, Tapper W, Collins A, et al. Identification of inherited genetic variations influencing prognosis in early-onset breast cancer. Cancer Res. 2013;73(6):1883–1891. doi: 10.1158/0008-5472.CAN-12-3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faryna M, Konermann C, Aulmann S, et al. Genome-wide methylation screen in low-grade breast cancer identifies novel epigenetically altered genes as potential biomarkers for tumor diagnosis. FASEB J. 2012;26(12):4937–4950. doi: 10.1096/fj.12-209502. [DOI] [PubMed] [Google Scholar]
- 64.Connolly R, Stearns V. Epigenetics as a therapeutic target in breast cancer. J Mammary Gland Biol Neoplasia. 2012;17(3–4):191–204. doi: 10.1007/s10911-012-9263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong P, Kaneuchi M, Konno Y, Watari H, Sudo S, Sakuragi N. Emerging therapeutic biomarkers in endometrial cancer. BioMed Res Int. 2013;2013:130362. doi: 10.1155/2013/130362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rundqvist H, Johnson RS. Tumour oxygenation: implications for breast cancer prognosis. J Intern Med. 2013;274(2):105–112. doi: 10.1111/joim.12091. [DOI] [PubMed] [Google Scholar]
- 67.Moisa A, Fritz P, Eck A, et al. Growth/adhesion-regulatory tissue lectin galectin-3: stromal presence but not cytoplasmic/nuclear expression in tumor cells as a negative prognostic factor in breast cancer. Anticancer Res. 2007;27(4B):2131–2139. [PubMed] [Google Scholar]
- 68.Pusztai L, Iwamoto T. Breast cancer prognostic markers in the post-genomic era. Breast Cancer Res Treat. 2011;125(3):647–650. doi: 10.1007/s10549-010-0932-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Selected instances from literature search
| 1st author | Year | Journal | Pubmed ID | Cancer biology underpinning | Related clinical indicator | Major contribution/conclusion |
|---|---|---|---|---|---|---|
| van ’t Veer1 | 2002 | Nature | 11823860 | Gene expression patterns | Lymph node status | Provides a powerful tool to tailor adjuvant systemic treatment that could greatly reduce the cost of BC treatment. |
| Rennstam2 | 2003 | Cancer Research | 14695203 | Chromosomal copy number aberrations | Patterns of copy number gains and losses define BCs with distinct clinicopathological features and patient prognosis. | |
| Paik3 | 2004 | New England Journal of Medicine | 15591335 | Gene expression patterns | Node-negative, ER-positive, tamoxifen treatment | A novel recurrence score based on 21 genes to quantify the likelihood of distant recurrence in patients as well as overall survival time. |
| Kronenwett4 | 2006 | Cancer Epidemiology, Biomarkers and Prevention | 16985023 | Genomic stability | Objective classification of BCs into stable and unstable subtypes that are a prognostic indicator independent of established clinical factors. | |
| Bacac5 | 2006 | PLoS One | 17183660 | Stromal cells | Human genes expressed in mouse stromal response to tumor invasion predicts BC patient survival. | |
| Suh6 | 2007 | Clinical Cancer Research | 17200346 | CLIC4 (chloride intracellular channel 4) | Reactivation and restoration of CLIC4 in tumor cells or the converse in tumor stromal cells could provide a novel approach to inhibit tumor growth. | |
| Conlin7 | 2007 | Molecular Diagnosis and Therapy | 18078353 | Oncotype DX recurrence score assay | Lymph node negative, ER-expressing BC | The Oncotype DX assay and others aim to help improve risk classification and recurrence prediction and optimize selection of patients for adjuvant chemotherapy. |
| Rodriguez8 | 2008 | Carcinogenesis | 18499701 | HOXB13 (homeobox B13) and IL17BR (interleukin 17 receptor B) | Estrogen signaling | Hypermethylation of HOXB13 is a later event of tumor progression and a prognostic indicator of advanced BC. |
| Wei9 | 2008 | Molecular Carcinogenesis | 18176935 | H3K27me3 | Loss of H3K27me3 is a predictor of poor outcome in BCs. | |
| Kim10 | 2008 | Annals of Oncology | 17956886 | CDKs | Patients recruited for study underwent mastectomy or breast-conserving surgery | Tumors with high CDK1SA and high CDK2SA showed significantly poorer 5-year relapse-free survival than those with low CDK1SA and low CDK2SA, respectively. |
| Han11 | 2008 | Nature | 18337816 | SATB1 (SATB homeobox 1) | SATB1 is a genome organizer that tethers multiple genomic loci and recruits chromatin-remodeling enzymes to regulate chromatin structure and gene expression. | |
| Ben-Porath12 | 2008 | Nature Genetics | 18443585 | Stem cell genetic expression signatures | Detailed characterization of the stem-cell regulatory networks active in cancer is likely to yield powerful diagnostic and prognostic markers. | |
| Parker13 | 2009 | Journal of Clinical Oncology | 19204204 | A 50-gene set (PAM50) | “Intrinsic” subtypes, pathologic staging, histologic grade | The intrinsic subtype and risk predictors based on the PAM50 gene set adds significant prognostic and predictive value. |
| Sung14 | 2010 | Cancer Science | 20412117 | SIRT1 (sirtuin 1) and CCAR2 (also known as DBC1, deleted in breast cancer 1, KIAA1967) | Luminal subtype, ER and PR expressions | Correlation between SIRT1 and DBC1 is a more useful prognostic factor than their individual expressions. Correlation between the two is decreased in tumor cells. |
| Gevensleben15 | 2010 | International Journal of Molecular Medicine | 21042777 | 70-gene expression profile MammaPrint® | Size, age, histological grade, hormone receptor status, peritumoral vascular invasion and HER2 status | Gene signature MammaPrint® is shown to provide additional independent prognostic information. |
| Lanigan16 | 2010 | Breast Cancer Research | 20682066 | Msx2 (msh homeobox 2) | Msx2 expression results in improved outcome for BCs, possibly by increasing the likelihood of tumor cell death by apoptosis. | |
| Creighton17 | 2010 | Breast Cancer Research | 20569503 | PI3K pathway | Luminal ER+ breast tumors | Luminal B tumors have hyperactive GFR/PI3K signaling associated with lower ER levels, which has been correlated with resistance to endocrine therapy. Targeting PI3K in these tumors may reverse loss of ER expression and signaling and restore hormonal sensitivity. |
| Xu18 | 2011 | Breast Cancer Research | 21255398 | RAD21 (RAD21 homolog [S. pombe]) | N/A | RAD21 expression confers poor prognosis and resistance to chemotherapy in high-grade luminal, basal, and HER2 BCs. |
| Littlepage19 | 2012 | Cancer Discovery | 22728437 | ZNF217 (zinc finger protein 217) | Amplification of the human chromosomal region 20q13 | ZNF217 (amplified in numerous cancers) is a poor prognostic indicator and therapeutic target in patients with BC and may be a strong biomarker of triciribine treatment efficacy in patients. |
| Pitroda20 | 2012 | PLoS One | 23056240 | Endothelial inflammatory pathways | The first prognostic cancer gene signature derived from an experimental model of tumor-associated endothelial inflammation. | |
| Kim21 | 2012 | Journal of Breast Cancer | 22807942 | Gene expression patterns | Histological grade, size, number of metastatic lymph nodes, ER, lymphovascular invasion, local invasion of tumor, and number of tumors | As the selected prognostic factors can be easily obtained in clinical practice, the proposed model might prove useful in the prediction of BR recurrence. |
| Faryna22 | 2012 | FASEB Journal | 22930747 | Aberrant DNA methylation | Low-grade ER- and/or PR-positive BC | Early methylation changes are frequent in the low-grade pathway of BC and may be useful in the development of prognostic markers. |
| Fasching23 | 2012 | Human Molecular Genetics | 22532573 | TOX3 (TOX high mobility group box family member 3) | With the exception of rs3803662 (TOX3), there was no evidence that any of the SNPs associated with BC susceptibility were associated with BC survival. | |
| Huang24 | 2013 | Cell and Bioscience | 23497677 | MACC1 (metastasis associated in colon cancer 1) | Clinicopathologic features | The over expression of MACC1 in BR is significantly correlated with adverse clinicopathological features, including metastasis and patient survival. |
| Yang25 | 2013 | PLoS One | 23441166 | Mediation of transcription factor GRHL2 (grainyhead-like 2 [Drosophila]) on its targets is prognostic in BC | Histological grade | Proposed the RXA-GSP (relative expression analysis with gene-set pairs) method, shows promise as both a valid prediction model as well as high potential for clinical utility. |
| Nagata26 | 2014 | Breast Cancer | 22528804 | Induced pluripotent stem cell inducing factors | Strong expression of NANOG is an indicator of poor prognosis for BC patients, whereas KLF4 is a favorable prognostic indicator. |
Note: Each key word (genomic, transcriptional, epigenetic, sequence, novel) respectively together with “breast cancer” and “prognostic indicator” was searched in PubMed, from Jan 2005–July 2013.
Abbreviations: BC, breast cancer; ER, estrogen receptor; PAM, prediction analysis of microarray; RXA-GSP, relative expression analysis of gene set pair; SNPs, single-nucleotide polymorphisms.