

Abstract
Hypertension is a risk factor for renal impairment. While treatment of hypertension provides significant renal protection, there is still an unmet need requiring further exploration of additional pathogenetic mechanisms. We have found that the hypertension-related, calcium-regulated gene (HCaRG/COMMD5) is involved in renal repair. HCaRG is a small intracellular protein of 225 amino acids and its gene expression is negatively regulated by extracellular calcium concentrations. HCaRG is mostly expressed in the kidneys, with higher levels found in the spontaneously hypertensive rat than in normotensive rats. In an acute kidney injury model, HCaRG expression decreases immediately after injury but increases above baseline during the repair phase. In cell cultures, HCaRG has been shown to facilitate differentiation and to inhibit cell proliferation via p21 transactivation through the p53-independent signaling pathway. Induction of p21 independently of p53 is also observed in transgenic mice overexpressing HCaRG during the repair phase after ischemia/reperfusion injury, resulting in their improved renal function and survival with rapid re-differentiation of proximal tubular epithelial cells. In addition, transgenic mice recover rapidly from the inflammatory burst most likely as a result of maintenance of the tubular epithelial barrier. Recent studies indicate that facilitating re-differentiation and cell cycle regulation in injured renal proximal tubules might be important functions of HCaRG. We have proposed that HCaRG is a component of differential genetic susceptibility to renal impairment in response to hypertension.
Keywords: Acute kidney injury, Copper metabolism MURR1 domain-containing proteins, Hypertension-related calcium-regulated gene, p21, Re-differentiation, Renal repair
Introduction
High blood pressure is a risk factor for renal failure. It is well known that malignant hypertension leads to kidney injury and that mild to moderate chronic hypertension can accelerate the progression of renal disease. Paradoxically, the most used animal model of human hypertension, the spontaneously hypertensive rat (SHR), is relatively resistant to renal damage not only when compared to other hypertensive rat models, such as the Dahl salt-sensitive rat or the Munich hypertensive strain, but also in comparison to normotensive rats such as the Brown–Norway (BN) rat, suggesting that genetic factors may affect the susceptibility to hypertension-induced or -accelerated renal disease. This has been well demonstrated by elegant studies of Theodore W. Kurtz’s group when they transplanted a kidney of the BN rat to the uninephrectomized SHR: it resulted in more severe kidney damage to the BN donor kidney than to the SHR kidney [1]. Our review focuses here on a hypertension-related gene, hypertension-related, calcium-regulated gene (HCaRG/COMMD5) that is more expressed in kidneys of SHR than of normotensive rats, and could explain in part the resistance of the SHR kidney by its accelerating effects on renal repair.
Discovery of HCaRG
HCaRG was initially discovered by our group in the parathyroid glands of the SHR and shown to be negatively regulated by extracellular calcium concentrations. It encodes a small intracellular protein of 225 amino-acids [2]. Incubation of parathyroid cells under the low extracellular calcium condition increases HCaRG expression in the same manner as parathyroid hormone. Other hormones, such as natriuretic peptides and vasopressin, have also been shown to respond to low extracellular calcium concentrations, but HCaRG is the first intracellular protein reported to have this property, and all these genes, including HCaRG, possess a negative calcium-responsive element in their promoters [3].
Hypertension is one of the risk factors not only for kidney injuries but also for renal cell carcinoma (RCC) [4–7]. Genetic hypertension is characterized at birth by suppression of apoptosis and increment of cell proliferation, leading to neonatal heart and kidney hyperplasia [8]. During the period of hypertension development, apoptosis is increased in response to the increment of blood pressure but so is proliferation, resulting in acceleration of cell turnover. The HCaRG gene exists in one copy on human chromosome 8q24.3 with paralogs on syntenic regions of rat chromosome 7 and mouse chromosome 15. This locus is associated with kidney weight (KW), urinary calcium excretion in rats and oncogenesis in humans [9, 10]. Chromosome 8 abnormalities, which might be of particular pathogenic importance, can be detected in 15 % of patients with acute myeloid leukemia [11, 12]. Gains and high-level gene amplification at 8q have been reported in renal cell carcinoma (RCC), metastatic prostate cancers, sporadic colorectal carcinomas and metastatic gastrointestinal stromal tumors [13–18]. Chromosome 8 anomalies in certain cancers could disrupt the HCaRG gene. In fact, HCaRG levels are decreased in various cancer cell lines [2]. HCaRG levels are more abundant in the kidneys, heart and adrenal glands. HCaRG is mainly located in renal proximal tubules (RPTs) of kidneys. We became interested in the possible roles played by HCaRG in the process of kidney development and renal repair after injury.
HCaRG as a member of the COMMD protein family
HCaRG has been assigned to a novel copper metabolism MURR1 domain-containing (COMMD) protein family, based on the identification of a well-conserved and unique domain at its carboxy-terminal end the COMM domain. The COMMD protein family has 10 members, and HCaRG corresponds to COMMD5. The length of the COMM domain varies between 70 to 85 amino acids across members of the family. The prototype of the COMMD protein family, COMMD1, exerts the function of blocking nuclear factor of kappa light polypeptide gene enhancer in B-cells (NF-κB)-chromatin interaction which, in turn, is induced by various stimuli, including tumor necrosis factor (TNF) and interleukin (IL)-1β [19, 20]. It has been determined that all members of the COMMD family are capable of inhibiting NF-κB transcriptional activity to various degrees and that the COMM domain is critical in the process. Recently, we reported that transgenic (Tg) mice overexpressing HCaRG/COMMD5 in RPTs recovered rapidly from the inflammatory burst that increased TNF-α and IL-1β as well as infiltration of macrophages after acute kidney injury (AKI) [21].
The COMM domain, which defines all COMMD proteins and is well conserved among members of this protein family, provides a critical interface for protein–protein interactions. It has been established that COMMD proteins are highly interactive and that the COMM domain mediates COMMD1-COMMD1 homodimerization as well as binding to other COMMD proteins [22]. While the COMM domains of COMMD1 and HCaRG are highly conserved, 87 % of amino acids differ between HCaRG and COMMD1 outside this domain, suggesting that members of the COMMD protein family could have different functions (Fig. 1).
Fig. 1.
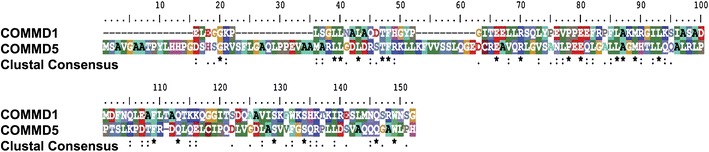
Alignment of human HCaRG/COMMD5 and COMMD1 proteins, excluding the COMM domain. While COMM domains of the COMMD protein family are highly conserved, HCaRG has only 13 % homology with COMMD1. Asterisks indicate positions with a single, fully-conserved residue. Colons depict strongly similar properties—scoring >0.5 in the Gonnet PAM 250 matrix. Periods indicate weak similarity—scoring ≤0.5 in the Gonnet PAM 250 matrix. HCaRG hypertension-related calcium-regulated gene, COMM copper metabolism MURR1
HCaRG as cell cycle regulator
Renal proximal tubules repair after injury requires de-differentiation, proliferation and migration of surviving tubular cells to replace dead cells [23, 24]. The process of tubular repair is similar to renal development in the fetal stage and includes a high cell proliferation rate, soaring apoptosis and a specific gene expression pattern. Interestingly, Joseph V. Bonventre’s group has demonstrated that epithelial cell cycle arrest at G2/M during the injury phase results in the activation of c-jun NH2-terminal kinase signaling, which acts to up-regulate the production of fibrogenic cytokines, thus accelerating progression of chronic kidney disease (CKD) [25]. In contrast, inhibition of cell proliferation with cycle arrest at G1 mediated by rosiglitazone, which has been used to decrease insulin resistance by activating peroxisomal proliferator-activated receptor, facilitates the recovery of proximal tubular epithelial cells (PTECs) after cisplatin-induced injury through the down-regulation of extracellular signal-regulated kinase and Akt signaling [26].
The effect of HCaRG on cell proliferation was demonstrated in two kidney cell lines, human embryonic kidney (HEK)-293 cells and Madin–Darby canine kidney (MDCK)-C7 cells [27]. The cells were stably transfected with either control plasmid (Neo) or HCaRG expression plasmid (HCaRG). HCaRG-overexpressing cells presented a much lower proliferation rate (Fig. 2a) and DNA synthesis than control cells. In addition, HCaRG delayed the cell cycle with accumulation of cells at G2/M, without arrest [28].
Fig. 2.
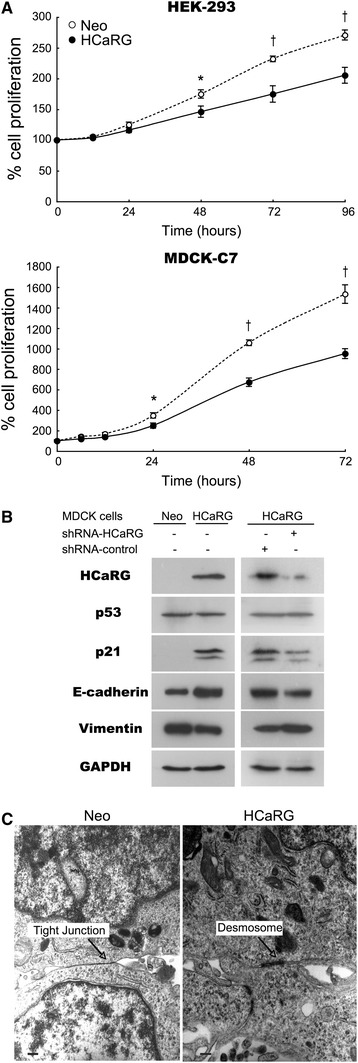
HCaRG controls cell proliferation and facilitates cellular differentiation. a Cell proliferation in Neo- or HCaRG-plasmid-transfected HEK-293 and MDCK-C7 cells. HCaRG inhibited cell proliferation in both kidney cell lines *p < 0.05, †p < 0.01. b Protein expression of HCaRG, p53, p21 and differentiation phenotype markers (E-cadherin and vimentin) in Neo- and HCaRG-plasmid-transfected MDCK-C7 cells with or without shRNA. HCaRG was detected only in HCaRG-MDCK cells. HCaRG-MDCK cells showed more mature gene expression patterns than Neo-MDCK cells. HCaRG induced p21 transactivation without p53 induction. shRNA-HCaRG treatment lowered HCaRG levels compared to a non-target control. HCaRG reduction led to lower p21 expression and abolition of differentiation in HCaRG-MDCK cells. Data from Matsuda et al. [21]. c Electron microscopy of Neo- and HCaRG-plasmid-transfected HEK-293 cells. HCaRG-HEK-293 cells had mature desmosome-like junctions instead of the tight junctions of Neo-control cells. Scale bars 300 nm. Figure from Devlin et al. [28]
The gene expression profiles of 111 cell cycle regulatory genes in stably-transfected cells have been analyzed [28]. HCaRG overexpression markedly induced p21 and down-regulated p27 in HEK-293 cells. HCaRG overexpression also augmented p21 expression independently of p53 expression in MDCK-C7 cells (Fig. 2b) [21]. This up-regulation was diminished by HCaRG knockdown with short-hairpin RNA (shRNA). p21, accompanied by p53 up-regulation, has been demonstrated to be induced in cells undergoing either p53-dependent G1 growth arrest or apoptosis [29]. p21 levels can also rise rapidly by differentiation inducers through a p53-independent pathway. In that case, p21 induction is coupled with the expression of early differentiation markers [30]. p21, independently of p53, could act as an inducible growth inhibitor that contributes to slow down the cell cycle and facilitates differentiation [31].
HCaRG fosters cellular maturation
HCaRG has been associated with changes in cell morphology and the appearance of differentiated phenotypes, namely, HCaRG-overexpressing HEK-293 cells present features of mature epithelial cells, such as higher protein content, cell size and volume [28]. Transmission electron microscopy ultrastructural analysis has revealed the presence of more differentiated junctions (desmosome-like) in HCaRG-overexpressing cells than in Neo controls, and these cells demonstrate features consistent with junctional (glandular-like) complex formation and numerous microvilli (Fig. 2c). These cellular changes are concordant with several modifications in the levels of specific epithelial/mesenchymal markers. E-cadherin, a marker of epithelial integrity [32], is higher in HCaRG-overexpressing cells than in control cells (Fig. 2b) [21]. In contrast, vimentin, an intermediate filament protein that is only expressed in mesenchymal cells [33], is lower in HCaRG-overexpressing cells. These observations indicate that HCaRG could accelerate differentiation and maturation via p21 transactivation through a p53-independent pathway in kidney cells, resulting in delay of the cell cycle with G2/M accumulation.
HCaRG stimulates renal cell migration
Acute kidney injury evokes actin cytoskeleton disruption and aggregation by induction of cofilin and, consequently, causes the breakdown of PTEC apical membrane microvilli during the early stage after ischemia [34]. Aquaporin-1, which is widely expressed in epithelial and endothelial cell membranes and facilitates transepithelial water transport, accelerates the migration of PTECs by actin re-organization, resulting in decreased tubular injury after ischemia [35]. Such cytoskeletal organization and migration of PTECs are key events occurring during tubular injury and repair.
As described previously, HCaRG accelerates differentiation and maturation via p21 transactivation. On the other hand, HCaRG overexpression increases the motility of kidney cells through the secretion of transforming growth factor-α in the wound-healing assay (Fig. 3a) [36]. We recently examined the intracellular location of HCaRG and actin in migrating MDCK-C7 cells during wound healing (Fig. 3b). In non-migrating cells, we observed HCaRG localization in the perinuclear space and clearly visible actin fibers with no signs of interactions between the two proteins. In migrating cells, intracellular actin fibers disappeared, and short filamentous (F)-actin accumulation was seen between the nucleus and elongated tip. HCaRG was also seen concentrated with short F-actin between the nucleus and elongated tip. It is known that in migrating fibroblasts, centrosomes reorient between the nucleus and elongated tip, which in turn re-positions the Golgi apparatus that is thought to establish and maintain cell polarity during migration [37–40]. We postulate that HCaRG could play a role in stimulating cell migration and wound healing without de-differentiation, by directly involving actin re-organization.
Fig. 3.
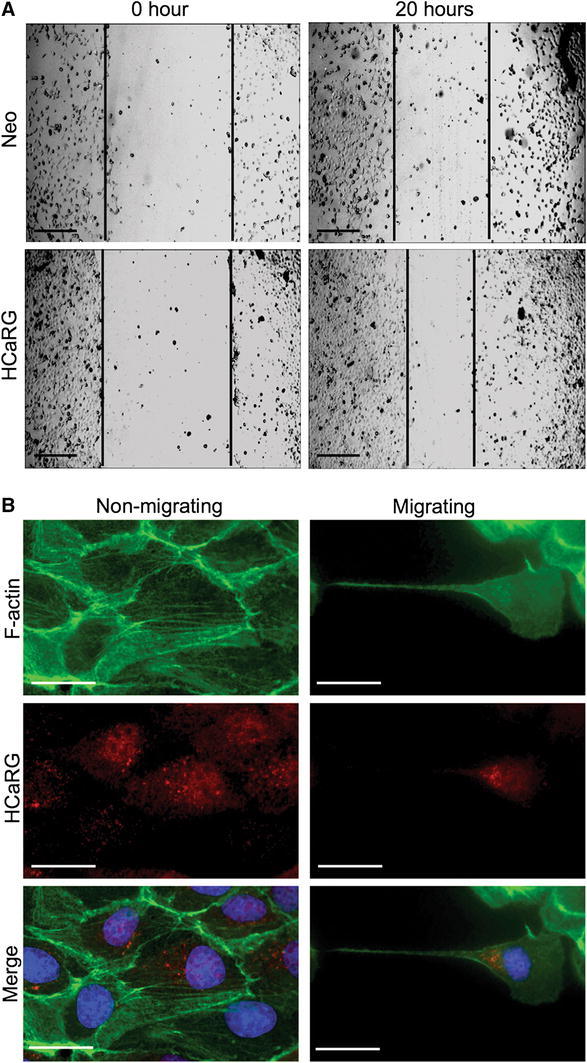
HCaRG stimulates renal cell migration. a Wound healing assay in Neo- or HCaRG-plasmid-transfected HEK-293 cells. HCaRG markedly accelerated wound healing after 20 h compared to Neo-controls. Scale bars 50 μm. b Intracellular location of F-actin (green) and HCaRG (red) in migrating MDCK cells during the wound-healing process. In non-migrating cells, HCaRG is located in the perinuclear space, and organized actin fibers are clearly visible. In the migrating cell, intracellular actin fibers are de-polymerized and short F-actin filaments are observed in the perinuclear space. HCaRG localizes with short F-actin filaments between the nucleus and elongated tip. Nuclei are stained by DAPI (blue). Scale bars 25 μm (color figure online)
HCaRG accelerates renal tubular repair after injury
Acute kidney injury
Ischemia/reperfusion injury (IRI) causes acute tubular damage, one of the most common forms of human AKI. After renal ischemia with renal hypoperfusion and hypoxia, PTECs lose their structural integrity through a sequence of events that include the disruption of brush border with blebbing of the apical membrane, fragmentation and internalization, and a rapid change in cell polarity. During the recovery phase, surviving PTECs proliferate and replace the irreversibly injured PTECs, migrate to cover the exposed areas of the basement membrane and re-differentiate to restore tubular integrity [23]. In a rat IRI model, we determined that HCaRG mRNA levels decreased soon after reperfusion and reached their lowest levels after 3–6 h, when PTECs were de-differentiated and proliferated. HCaRG mRNA levels then rose steadily to higher than baseline 48 h after reperfusion, corresponding to the regeneration phase [2].
Tg mice overexpressing HCaRG in RPTs were generated to test the hypothesis that HCaRG could stimulate renal repair [21]. Exogenous HCaRG was inserted into the kidney androgen-regulated protein (KAP) promoter plasmid. KAP was identified as an abundant protein under androgen control and expressed in RPTs [41, 42]; therefore, exogenous HCaRG was expressed in RPTs of Tg male mice under androgen control.
In an AKI model, IRI caused severe tubular damage that resulted in pronounced renal dysfunction in both non-Tg and Tg mice [21]. The survival rate 7 days after IRI injury was 64 % in Tg compared to only 25 % in non-Tg mice (p = 0.0249). In addition, morphological tubular damage and renal function in Tg mice improved significantly faster in comparison to non-Tg mice after 2 days (Table 1). We concluded that HCaRG overexpression significantly enhances renal function and survival after ischemia.
Table 1.
Renal function 2 days after ischemia/reperfusion injury (IRI)
| Genotype | Blood urea nitrogen (mmol/l) | Creatinine (μmol/l) |
|---|---|---|
| Non-Tg mice | 105.0 ± 25.7 (n = 10) | 206.1 ± 99.1 (n = 10) |
| Tg mice | 77.4 ± 33.5 (n = 10)* | 114.4 ± 63.6 (n = 10)* |
Data are shown as mean ± SD. *p < 0.05
Tubular epithelial cells and cells within the interstitial space, which are associated with vascular network components and resident fibroblasts, account for about 80 % of kidney volume, and increased KW could be due to edema, hypertrophy and cell proliferation after tubular injury [43]. HCaRG overexpression did not affect renal hypertrophy by uninephrectomy, but significantly reduced the increase of KW 2 days after reperfusion (Fig. 4a). Such KW reduction could have resulted from stimulation of cell death or inhibition of cell growth. However, we demonstrated that HCaRG had no effect on apoptotic cell death after IRI. A large number of proliferating cell nuclear antigen (PCNA)-positive cells were present in tubules on day 2 (Fig. 4b). HCaRG overexpression decreased only the number of proliferating tubular cells, while the number of proliferating cells in the interstitium was comparable between non-Tg and Tg mice. This inhibition of PTEC proliferation was accompanied by p53-independent p21 transactivation. It is known that p21 transactivation by a p53-independent pathway occurs in RPTs but not in the glomerulus or interstitium during the renal recovery phase, and might thus have contributed to cell differentiation [44–46]. In p21 knockout mice, AKI causes more rapid onset of renal dysfunction, and induces more severe morphological damage with a threefold higher mortality rate than in normal mice [45, 47]. Miyaji et al. [46] reported that cisplatin-induced AKI evoked 2 peaks of increased p21. The first peak was accompanied by up-regulation of p53 and PCNA, possibly reflecting G1 arrest and DNA repair. The second p21 peak occurred through a p53-independent pathway likely contributing to cell differentiation. These studies indicate that p53-independent p21 up-regulation could be crucial in controlling epithelial proliferation and morphogenesis in RPTs. Actually, in HCaRG Tg mice, E-cadherin levels recovered more rapidly, while vimentin induction after IRI disappeared faster than in non-Tg mice.
Fig. 4.
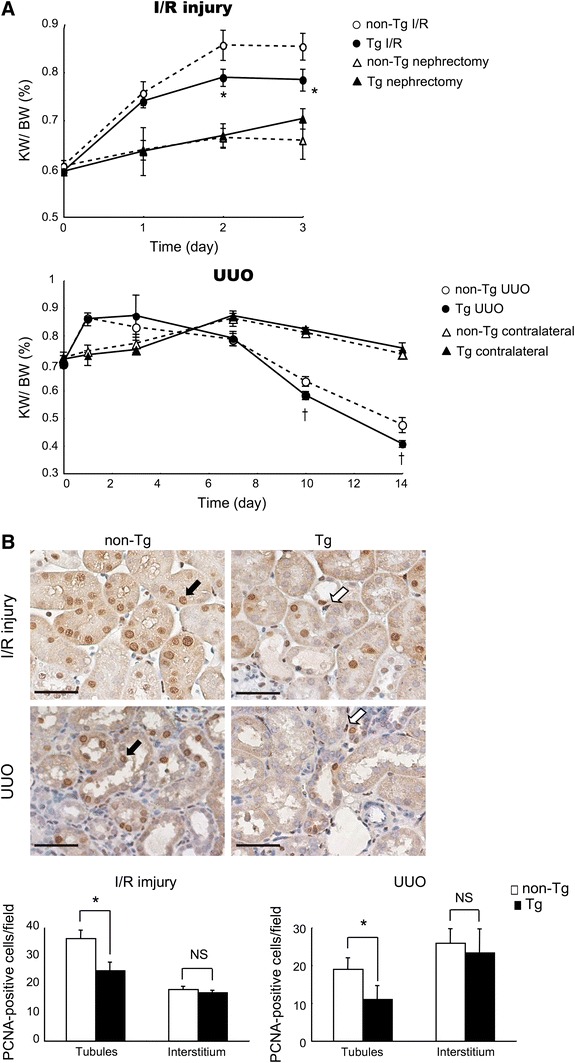
HCaRG-Tg mice exhibit lower tubular cell proliferation after ischemia/reperfusion injury (IRI). a Changes in the kidney weight (KW)/body weight (BW) ratio after IRI and unilateral ureteral obstruction (UUO). IRI increased KW compared to nephrectomized controls, and this increment was significantly reduced (*p < 0.05) in Tg mice after 2 days. In the UUO model, KW increased with hydronephrosis caused by ureteral obstruction for up to 3 days. After day 7, KW started to decline, and HCaRG elicited a significantly more rapid reduction of KW (†p < 0.05). b Localization of proliferating cell nuclear antigen (PCNA)-positive cells on day 2 after IRI and on day 10 after UUO. The number of PCNA-positive cells was counted in tubular and interstitial regions, respectively. PCNA-positive cells in tubules were lower in Tg than in non-Tg mice. The black arrow indicates PCNA-positive tubular cells, and the white arrow depicts PCNA-positive interstitial cells. *p < 0.05. Scale bars 50 μm. Data from Matsuda et al. [21]
Chronic kidney disease
Tubulointerstitial fibrosis is recognized to be a common endpoint of human CKD caused by hypertension, diabetes, and nephrotic syndrome. Unilateral ureteral obstruction (UUO) is used as an animal model of progressive tubulointerstitial fibrosis. UUO also leads to many pathophysiological events of obstructive nephropathy, such as cellular infiltration and proliferation, tubular de-differentiation, apoptosis and atrophy, fibroblasts differentiation and excessive extracellular matrix deposition [43, 48].
KW is increased 3 days after obstruction because UUO produces tubular dilatation and interstitial edema (Fig. 4a) [21]. HCaRG overexpression does not reduce the initial edema in Tg mice. Contralateral KW gain until 7 days after obstruction is mainly due to hypertrophy, and HCaRG overexpression does not affect this hypertrophy, as in the uninephrectomized kidneys. Ten days after surgery, UUO results in gradual KW reduction, and HCaRG overexpression causes more rapid KW diminution in Tg mice compared to non-Tg mice. This KW reduction also derives from the imbalance of cell growth and loss. In the obstructed kidneys after 10 days of UUO, HCaRG overexpression decreases only the number of proliferating tubular cells, as after IRI (Fig. 4b).
The results in both IRI and UUO models can be summarized as follows: (1) HCaRG overexpression does not reduce initial edema, hypertrophy and injury; (2) HCaRG overexpression leads to faster KW diminution in Tg than in non-Tg mice; (3) these KW decreases are due to inhibition of cell proliferation increment in tubules but not in the interstitium; (4) HCaRG elicits re-differentiation via p21 induction after injury. In conclusion, HCaRG does not reduce initial tubular injury, but accelerates repair of RPTs which improves survival by facilitating re-differentiation of resident PTECs and controlling proliferation via p21 induction through its p53-independent pathway.
Finally, slow-cycling renal progenitor-like cells provide regenerating cells that replace injured cells during the repair phase after AKI [49, 50]. At an early phase of tubular regeneration, their daughter cells after multiple cell division show mesenchymal phenotypes such as active proliferation and migration in the interstitium, and eventually differentiate into PTECs. HCaRG could play a role in the differentiation and survival of resident renal progenitor-like cells.
Conclusion
Acute kidney injury occurs in various clinical settings, including renal ischemia arising from septic shock and major cardiovascular surgery, or acute drug or toxic exposure. It is a common clinical problem with increasing incidence and mortality, poor prognosis, and unsatisfactory therapeutic options [51–54]. Despite better knowledge acquired during the last decade of the pathophysiological pathways underlying kidney diseases at both the basic research and clinical levels, the progression of renal dysfunction and the number of hemodialyzed patients is still increasing steadily every year. Repair after AKI involves the proliferation of PTECs as well as migration and re-differentiation. HCaRG has shown beneficial effects in facilitating re-differentiation and controlling proliferation through p21 induction via the p53-independent signaling pathway during the renal repair phase (Fig. 5). HCaRG also might stimulate the migration of PTECs during the repair phase directly involving actin re-organization. Moreover, IRI rapidly activates inflammatory responses, resulting in endothelial dysfunction in the cortex [55]. HCaRG rapidly reduces inflammatory mediators and infiltration of macrophages after reperfusion [21]. Sepsis-induced AKI has a distinct pathophysiology which involves intrarenal hemodynamic changes, vascular endothelial dysfunction, infiltration of inflammatory cells in the renal parenchyma, obstruction of tubules with apoptotic and necrotic cells, and tubular epithelial mitochondrial dysfunction [56, 57]. HCaRG may have the potential to diminish these inflammatory responses and to prevent mitochondrial dysfunction through maintenance of the tubular epithelial barrier not only after IRI but also in sepsis-induced AKI. In addition, lack of HCaRG might lead to uncontrolled cell proliferation and migration, thus increasing the risk of oncogenesis of RCC. In fact, HCaRG levels are significantly decreased in tumors of brain, kidney and liver compared to normal adjacent tissues [2] and rosiglitazone suppressed the growth of gastric cancer by up-regulating HCaRG [58]. Tubular regenerating functions mediated by HCaRG are most important for improved renal function and survival after AKI. Accelerated healing by HCaRG could serve as a new therapeutic approach to AKI.
Fig. 5.
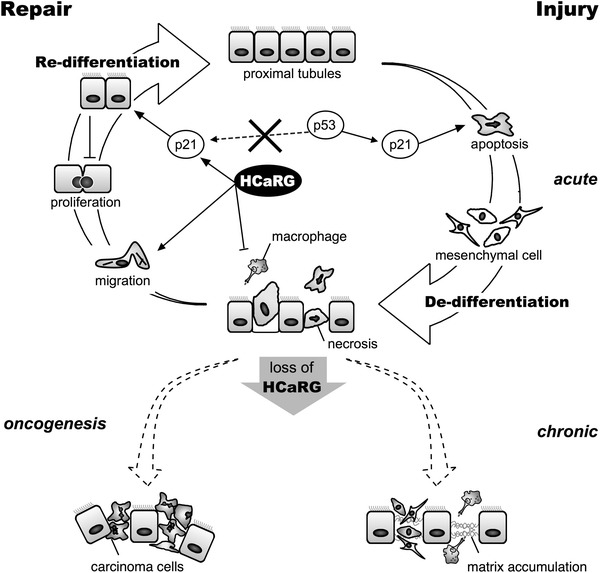
Scheme depicting the putative role of HCaRG in tubular repair after acute kidney injury (AKI). AKI mainly damages proximal tubular epithelial cells and causes cell death, resulting in necrosis and apoptosis. The induction of cell death is associated with p21 up-regulation dependent on the p53 pathway. Sublethally-injured proximal tubular epithelial cells (PTECs) de-differentiate into mesenchymal cells. De-differentiated cells proliferate and migrate to repair denuded areas. In the repair phase, HCaRG promotes cell migration, and controls PTEC proliferation by acceleration of their re-differentiation via p21 transactivation through the p53-independent pathway, resulting in improved renal function and survival. Lack of HCaRG might lead to uncontrolled cell proliferation and migration of PTECs with de-differentiated phenotypes, resulting in increased risk of chronic kidney diseases and oncogenesis of renal cell carcinoma
Acknowledgments
The authors thank Suzanne Cossette, Catherine Michel, Alison M. Devlin, Gilles Godefroid, Nicolas Solban, Carlos El Hader, Christian Croisetière and Dr. Junzheng Peng for their expert technical assistance and advice, and gratefully acknowledge manuscript editing by Ovid Da Silva. The work described in this review was supported by Canadian Institutes of Health Research Grant MOP-43859 (to J.T.).
Conflict of interest
None.
Contributor Information
Hiroyuki Matsuda, Email: hiroyuki.matsuda@med.nihon-u.ac.jp.
Johanne Tremblay, Email: johanne.tremblay@umontreal.ca.
References
- 1.Churchill PC, Churchill MC, Bidani AK, et al. Genetic susceptibility to hypertension-induced renal damage in the rat. Evidence based on kidney-specific genome transfer. J Clin Investig. 1997;100:1373–1382. doi: 10.1172/JCI119657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solban N, Jia HP, Richard S, et al. HCaRG, a novel calcium-regulated gene coding for a nuclear protein, is potentially involved in the regulation of cell proliferation. J Biol Chem. 2000;275:32234–32243. doi: 10.1074/jbc.M001352200. [DOI] [PubMed] [Google Scholar]
- 3.Okazaki T, Ando K, Igarashi T, Ogata E, Fujita T. Conserved mechanism of negative gene regulation by extracellular calcium. Parathyroid hormone gene versus atrial natriuretic polypeptide gene. J Clin Invest. 1992;89:1268–1273. doi: 10.1172/JCI115711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamet P. Cancer and hypertension: a potential for crosstalk? J Hypertens. 1997;15:1573–1577. doi: 10.1097/00004872-199715120-00058. [DOI] [PubMed] [Google Scholar]
- 5.Choi MY, Jee SH, Sull JW, Nam CM. The effect of hypertension on the risk for kidney cancer in Korean men. Kidney Int. 2005;67:647–652. doi: 10.1111/j.1523-1755.2005.67137.x. [DOI] [PubMed] [Google Scholar]
- 6.Setiawan VW, Stram DO, Nomura AM, Kolonel LN, Henderson BE. Risk factors for renal cell cancer: the multiethnic cohort. Am J Epidemiol. 2007;166:932–940. doi: 10.1093/aje/kwm170. [DOI] [PubMed] [Google Scholar]
- 7.Wang G, Hou J, Ma L, et al. Risk factor for clear cell renal cell carcinoma in Chinese population: a case–control study. Cancer Epidemiol. 2012;36:177–182. doi: 10.1016/j.canep.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Moreau P, Tea BS, Dam TV, Hamet P. Altered balance between cell replication and apoptosis in hearts and kidneys of newborn SHR. Hypertension. 1997;30:720–724. doi: 10.1161/01.HYP.30.3.720. [DOI] [PubMed] [Google Scholar]
- 9.Hamet P, Pausova Z, Dumas P, et al. Newborn and adult recombinant inbred strains: a tool to search for genetic determinants of target organ damage in hypertension. Kidney Int. 1998;53:1488–1492. doi: 10.1046/j.1523-1755.1998.00938.x. [DOI] [PubMed] [Google Scholar]
- 10.Solban N, Dumas P, Gossard F, et al. Chromosomal mapping of HCaRG, a novel hypertension-related, calcium-regulated gene. Folia Biol (Praha) 2002;48:9–14. [PubMed] [Google Scholar]
- 11.Batanian JR, Ma E, Huang Y, Gadre B. Co-existence of alternative forms of 8q gain in cytogenetic clones of three patients with acute myeloid leukemia, pointing to 8q22 approximately 8qter as a region of biologic significance. Cancer Genet Cytogenet. 2001;126:20–25. doi: 10.1016/S0165-4608(00)00385-X. [DOI] [PubMed] [Google Scholar]
- 12.Kaune KM, Baumgart M, Schmitke E, et al. Papular exanthem discloses acute myeloid leukaemia: interphase fluorescence in situ hybridization revealed deletion of p53 and gain at 8q22/8q24/tel8q without trisomy 8. Clin Exp Dermatol. 2010;35:160–164. doi: 10.1111/j.1365-2230.2009.03224.x. [DOI] [PubMed] [Google Scholar]
- 13.Barnabas N, Xu L, Savera A, Hou Z, Barrack ER. Chromosome 8 markers of metastatic prostate cancer in African American men: gain of the MIR151 gene and loss of the NK3-1 gene. Prostate. 2011;71:857–871. doi: 10.1002/pros.21302. [DOI] [PubMed] [Google Scholar]
- 14.El Gammal AT, Bruchmann M, Zustin J, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin Cancer Res. 2010;16:56–64. doi: 10.1158/1078-0432.CCR-09-1423. [DOI] [PubMed] [Google Scholar]
- 15.El Gedaily A, Bubendorf L, Willi N, et al. Discovery of new DNA amplification loci in prostate cancer by comparative genomic hybridization. Prostate. 2001;46:184–190. doi: 10.1002/1097-0045(20010215)46:3<184::AID-PROS1022>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 16.El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res. 2000;60:3899–3903. [PubMed] [Google Scholar]
- 17.Sayagues JM, Abad Mdel M, Melchor HB, et al. Intratumoural cytogenetic heterogeneity of sporadic colorectal carcinomas suggests several pathways to liver metastasis. J Pathol. 2010;221:308–319. doi: 10.1002/path.2712. [DOI] [PubMed] [Google Scholar]
- 18.Klatte T, Kroeger N, Rampersaud EN, et al. Gain of chromosome 8q is associated with metastases and poor survival of patients with clear cell renal cell carcinoma. Cancer. 2012;118:5777–5782. doi: 10.1002/cncr.27607. [DOI] [PubMed] [Google Scholar]
- 19.Burstein E, Hoberg JE, Wilkinson AS, et al. COMMD proteins, a novel family of structural and functional homologs of MURR1. J Biol Chem. 2005;280:22222–22232. doi: 10.1074/jbc.M501928200. [DOI] [PubMed] [Google Scholar]
- 20.Maine GN, Burstein E. COMMD proteins and the control of the NF kappa B pathway. Cell Cycle. 2007;6:672–676. doi: 10.4161/cc.6.6.3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuda H, Lavoie JL, Gaboury L, Hamet P, Tremblay J. HCaRG accelerates tubular repair after ischemic kidney injury. J Am Soc Nephrol. 2011;22:2077–2089. doi: 10.1681/ASN.2010121265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Bie P, van de Sluis B, Burstein E, et al. Characterization of COMMD protein–protein interactions in NF-kappaB signalling. Biochem J. 2006;398:63–71. doi: 10.1042/BJ20051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol. 2003;14(Suppl 1):S55–S61. doi: 10.1097/01.ASN.0000067652.51441.21. [DOI] [PubMed] [Google Scholar]
- 24.Nony PA, Schnellmann RG. Mechanisms of renal cell repair and regeneration after acute renal failure. J Pharmacol Exp Ther. 2003;304:905–912. doi: 10.1124/jpet.102.035022. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi IJ, Kim SY, Kwon CH, Kim YK. Rosiglitazone inhibits proliferation of renal proximal tubular cells via down-regulation of ERK and Akt. Ren Fail. 2010;32:103–111. doi: 10.3109/08860220903391259. [DOI] [PubMed] [Google Scholar]
- 27.Gaush CR, Hard WL, Smith TF. Characterization of an established line of canine kidney cells (MDCK) Proc Soc Exp Biol Med. 1966;122:931–935. doi: 10.3181/00379727-122-31293. [DOI] [PubMed] [Google Scholar]
- 28.Devlin AM, Solban N, Tremblay S, et al. HCaRG is a novel regulator of renal epithelial cell growth and differentiation causing G2M arrest. Am J Physiol Renal Physiol. 2003;284:F753–F762. doi: 10.1152/ajprenal.00252.2002. [DOI] [PubMed] [Google Scholar]
- 29.el-Deiry WS, Harper JW, O’Connor PM, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 30.Steinman RA, Hoffman B, Iro A, Guillouf C, Liebermann DA, el-Houseini ME. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene. 1994;9:3389–3396. [PubMed] [Google Scholar]
- 31.Parker SB, Eichele G, Zhang P, et al. P53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest. 1994;93:2175–2188. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashworth SL, Sandoval RM, Hosford M, Bamburg JR, Molitoris BA. Ischemic injury induces ADF relocalization to the apical domain of rat proximal tubule cells. Am J Physiol Renal Physiol. 2001;280:F886–F894. doi: 10.1152/ajprenal.2001.280.5.F886. [DOI] [PubMed] [Google Scholar]
- 35.Hara-Chikuma M, Verkman AS. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J Am Soc Nephrol. 2006;17:39–45. doi: 10.1681/ASN.2005080846. [DOI] [PubMed] [Google Scholar]
- 36.El Hader C, Tremblay S, Solban N, et al. HCaRG increases renal cell migration by a TGF-alpha autocrine loop mechanism. Am J Physiol Renal Physiol. 2005;289:F1273–F1280. doi: 10.1152/ajprenal.00103.2005. [DOI] [PubMed] [Google Scholar]
- 37.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/S0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 38.Gomes ER, Jani S, Gundersen GG. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell. 2005;121:451–463. doi: 10.1016/j.cell.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 39.Mellor H. Cell motility: Golgi signalling shapes up to ship out. Curr Biol. 2004;14:R434–R435. doi: 10.1016/j.cub.2004.05.038. [DOI] [PubMed] [Google Scholar]
- 40.Palazzo AF, Joseph HL, Chen YJ, et al. Cdc42, dynein, and dynactin regulate MTOC reorientation independent of rho-regulated microtubule stabilization. Curr Biol. 2001;11:1536–1541. doi: 10.1016/S0960-9822(01)00475-4. [DOI] [PubMed] [Google Scholar]
- 41.Meseguer A, Catterall JF. Mouse kidney androgen-regulated protein messenger ribonucleic acid is expressed in the proximal convoluted tubules. Mol Endocrinol. 1987;1:535–541. doi: 10.1210/mend-1-8-535. [DOI] [PubMed] [Google Scholar]
- 42.Toole JJ, Hastie ND, Held WA. An abundant androgen-regulated mRNA in the mouse kidney. Cell. 1979;17:441–448. doi: 10.1016/0092-8674(79)90170-3. [DOI] [PubMed] [Google Scholar]
- 43.Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol. 2002;283:F861–F875. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- 44.Megyesi J, Andrade L, Vieira JM, Jr, Safirstein RL, Price PM. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001;60:2164–2172. doi: 10.1046/j.1523-1755.2001.00044.x. [DOI] [PubMed] [Google Scholar]
- 45.Megyesi J, Udvarhelyi N, Safirstein RL, Price PM. The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol. 1996;271:F1211–F1216. doi: 10.1152/ajprenal.1996.271.6.F1211. [DOI] [PubMed] [Google Scholar]
- 46.Miyaji T, Kato A, Yasuda H, Fujigaki Y, Hishida A. Role of the increase in p21 in cisplatin-induced acute renal failure in rats. J Am Soc Nephrol. 2001;12:900–908. doi: 10.1681/ASN.V125900. [DOI] [PubMed] [Google Scholar]
- 47.Prozialeck WC, Lamar PC, Appelt DM. Differential expression of E-cadherin, N-cadherin and beta-catenin in proximal and distal segments of the rat nephron. BMC Physiol. 2004;4:10. doi: 10.1186/1472-6793-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bascands JL, Schanstra JP. Obstructive nephropathy: insights from genetically engineered animals. Kidney Int. 2005;68:925–937. doi: 10.1111/j.1523-1755.2005.00486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maeshima A, Yamashita S, Nojima Y. Identification of renal progenitor-like tubular cells that participate in the regeneration processes of the kidney. J Am Soc Nephrol. 2003;14:3138–3146. doi: 10.1097/01.ASN.0000098685.43700.28. [DOI] [PubMed] [Google Scholar]
- 50.Yamashita S, Maeshima A, Nojima Y. Involvement of renal progenitor tubular cells in epithelial-to-mesenchymal transition in fibrotic rat kidneys. J Am Soc Nephrol. 2005;16:2044–2051. doi: 10.1681/ASN.2004080681. [DOI] [PubMed] [Google Scholar]
- 51.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 52.Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223–228. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 54.Ympa YP, Sakr Y, Reinhart K, Vincent JL. Has mortality from acute renal failure decreased? A systematic review of the literature. Am J Med. 2005;118:827–832. doi: 10.1016/j.amjmed.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 55.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 56.Zarjou A, Agarwal A. Sepsis and acute kidney injury. J Am Soc Nephrol. 2011;22:999–1006. doi: 10.1681/ASN.2010050484. [DOI] [PubMed] [Google Scholar]
- 57.Parikh SM. Therapeutic targeting of the mitochondrial dysfunction in septic acute kidney injury. Curr Opin Crit Care. 2013;16:554–559. doi: 10.1097/MCC.0000000000000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen BL, Yu J, Zeng ZR, et al. Rosiglitazone suppresses gastric carcinogenesis by up-regulating HCaRG expression. Oncol Rep. 2008;20:1093–1097. [PubMed] [Google Scholar]