

 This work is licensed under a
This work is licensed under a Abstract
Patients with critical illness or hepatic failure exhibit impaired cortisol responses to ACTH, a phenomenon known as ‘relative adrenal insufficiency’. A putative mechanism is that elevated bile acids inhibit inactivation of cortisol in liver by 5α-reductases type 1 and type 2 and 5β-reductase, resulting in compensatory downregulation of the hypothalamic–pituitary–adrenal axis and adrenocortical atrophy. To test the hypothesis that impaired glucocorticoid clearance can cause relative adrenal insufficiency, we investigated the consequences of 5α-reductase type 1 deficiency in mice. In adrenalectomised male mice with targeted disruption of 5α-reductase type 1, clearance of corticosterone was lower after acute or chronic (eightfold, P<0.05) administration, compared with WT control mice. In intact 5α-reductase-deficient male mice, although resting plasma corticosterone levels were maintained, corticosterone responses were impaired after ACTH administration (26% lower, P<0.05), handling stress (2.5-fold lower, P<0.05) and restraint stress (43% lower, P<0.05) compared with WT mice. mRNA levels of Nr3c1 (glucocorticoid receptor), Crh and Avp in pituitary or hypothalamus were altered, consistent with enhanced negative feedback. These findings confirm that impaired peripheral clearance of glucocorticoids can cause ‘relative adrenal insufficiency’ in mice, an observation with important implications for patients with critical illness or hepatic failure, and for patients receiving 5α-reductase inhibitors for prostatic disease.
Keywords: glucocorticoids, 5α-reductases, adrenal insufficiency, HPA axis
Introduction
Activation of the hypothalamic–pituitary–adrenal (HPA) axis is a vital component of the stress response, driving production of glucocorticoid hormones (cortisol in humans, corticosterone in rodents) that mediate essential adaptations of the immune, metabolic and cardiovascular systems in response, for example, to infection or injury. Paradoxically, low cortisol concentrations have been observed in patients with critical illness, particularly after administration of synthetic adrenocorticotrophic hormone (ACTH); this phenomenon has been termed ‘relative adrenal insufficiency’ and has led patients being treated with hydrocortisone (Annane et al. 2002). Similarly impaired cortisol responses to synthetic ACTH have been observed in patients with liver disease (Harry et al. 2002, Fernandez et al. 2006, Tsai et al. 2006). The mechanisms involved in these altered cortisol responses are complex, with abnormalities observed in central HPA axis drive, adrenal steroidogenesis and plasma protein binding (Cooper & Stewart 2009). In addition, recent observations have indicated that a key contribution results from alterations in the enzymatic clearance of cortisol. In liver disease, inactivation of cortisol by the enzymes 5β-reductase (Akr1d1) and 11β-hydroxysteroid dehydrogenase type 2 (Hsd11b2; Frey 2006, McNeilly et al. 2010) is impaired. In critical illness, we have recently reported profoundly impaired clearance of cortisol, which was attributed to impaired 5α-reductase and 5β-reductase activity in liver and Hsd11b2 in kidney (Boonen et al. 2013). We also demonstrated an association between impaired cortisol clearance and a reduced cortisol response to synthetic ACTH. As endogenous ACTH levels were paradoxically low in critically ill patients, we proposed the hypothesis that substantially slower clearance of cortisol results in enhanced negative feedback suppression of ACTH and hence compensatory downregulation of cortisol production to maintain cortisol levels (Boonen et al. 2013). As a result, adrenal atrophy ensues and the incremental response to exogenous ACTH is impaired. However, in disease settings, it is difficult to confirm the primary cause of changes in HPA axis function.
An influence of peripheral steroid metabolism on HPA responses has been demonstrated in mice unable to regenerate or inactivate active glucocorticoid by Hsd11b1 or Hsd11b2 respectively (Kotelevtsev et al. 1997, 1999, Harris et al. 2001, Carter et al. 2009). Patients with impaired cortisol clearance due to Hsd11b2 deficiency have reduced total cortisol production, presumably through suppression of the HPA axis (Stewart et al. 1988), which is mirrored in Hsd11b2 knockout mice (Kotelevtsev et al. 1999). In contrast, mice unable to regenerate tissue glucocorticoids due to deletion of Hsd11b1 or hexose-6-phosphate dehydrogenase exhibit adrenal hypertrophy (Kotelevtsev et al. 1997, Harris et al. 2001, Lavery et al. 2006, Carter et al. 2009). This again is consistent with the clinical scenario, where enhanced cortisol clearance in patients with rare Hsd11b1 or hexose-6-phosphate dehydrogenase deficiency results in increased ACTH drive to the adrenals, and hence elevated adrenal androgens (Taylor et al. 1984, Phillipov et al. 1996, Jamieson et al. 1999, Draper et al. 2003).
5α-Reductases are responsible for one-third to a half of total peripheral glucocorticoid clearance, but their participation in the regulatory feedback loops mediated via the HPA axis has not been studied. Upregulation of 5α-reductase has been invoked to explain adrenal androgen excess in polycystic ovary syndrome (Stewart et al. 1990, Fassnacht et al. 2003, Tsilchorozidou et al. 2003). Although congenital deficiency of 5α-reductase is rare (Imperato-McGinley et al. 1974), an increasing number of men are prescribed 5α-reductase inhibitors for benign prostatic hyperplasia. The consequences, if any, for the HPA axis are unknown. We tested, in a mouse model, whether deficiency of 5α-reductase 1 (Srd5a1) activity causes relative adrenal insufficiency.
Materials and methods
Chemicals were obtained from Sigma unless otherwise stated.
In vivo protocols
Mouse embryos with targeted disruption of Srd5a1 (Mahendroo et al. 1996, 1997) (C57Bl6/SvEv/129 mixed background; Jackson Laboratory, Bar Harbor, ME, USA), were re-derived (University of Edinburgh) and heterozygote offspring crossed to generate homozygote male ‘WT’ and ‘knock-out’ (Srd5a1-KO) mice that were used for study at 4–5 months of age. All experiments were carried out under UK Home Office license and mice were housed singly for 1 week prior to investigations. Mice were maintained with ad libitum access to standard chow (Special Diet Services, Witham, UK) under regulated conditions of light and darkness (lights on from 0700 to 1900 h). Following killing by decapitation, trunk blood was collected and one adrenal gland and the thymus gland were removed and formalin-fixed. The hypothalamus, remaining brain, pituitary and other adrenal were frozen on soft dry ice. All samples were stored at −80 °C.
Glucocorticoid clearance
Weight matched (approximately 30 g) male WT and Srd5a1-KO mice were adrenalectomised under isoflurane anaesthesia to remove endogenous glucocorticoids (n=8/group). After 2 weeks recovery, mice received corticosterone (2 μg, in 10% ethanol with 0.025% β-cyclodextrin) by s.c. bolus injection and blood samples were obtained by tail-tip bleed after 0, 15, 30, 60 and 90 min.
In another group of mice, osmotic minipumps were implanted subcutaneously during adrenalectomy to administer corticosterone for 3 weeks before mice were killed (100 μg/day corticosterone in 1:1 DMSO:propylene glycol; Alzet model 2004, Charles River UK Ltd, Margate, Kent, UK). Clearance of corticosterone was calculated as the infusion rate divided by the steady-state plasma concentrations determined in trunk blood samples collected at killing.
Further intact WT and Srd5a1-KO mice (n=12/group) received corticosterone (50 μg/day) for 2 weeks by minipump as indicated earlier. Mice were killed by decapitation and trunk blood was collected.
Responses to stress
Mice (n=8/group) were killed 10 min following cage disturbance and handling. Trunk blood was collected and brains were harvested.
The remaining mice (n=12–18/genotype) were removed from their home cage at 0800 h with minimum disruption and blood was obtained rapidly (within 30 s) by tail-nick. The animals were subsequently restrained within Plexiglas restraint tubes for 15 min and another set of blood samples was taken. They were returned to their home cages and a further blood samples obtained either at 30, 60 or 90 min following the start of the restraint stress (n=6/time point). Blood samples were collected at only one time point following the end of the restraint stress to avoid confounding effects of repeated stress.
Responses to dexamethasone and ACTH
Male mice (n=9–10/genotype) received an injection of dexamethasone (not a substrate for Srd5a1 (McGuire et al. 1960), 10 μg/kg body weight intra-peritoneally) at 1300 h to achieve partial (approximately 50%) suppression of the HPA axis. Blood samples were obtained by tail-nick at 1500 h, after which mice received injections of either vehicle (saline) or ACTH (Synacthen diluted in saline, Alliance, Chippenham, UK, 0.1 μg/kg; EC50) and blood samples were again collected after 30 min.
Laboratory protocols
Morphometric analysis of adrenal glands
Fixed adrenal glands (n=8–14) were sectioned (5 μm), stained with haematoxylin and eosin and examined by light microscopy (Zeiss Axioscope, 40× magnification). The cells were photographed using a Cool Snap Photometrics camera and the number of cells in a given area (∼160 μm2 for the inner and outer zona fasciculata and 40 μm2 for the zona glomerulosa and zona reticularis) from each adrenal was counted using MCID 7.0 software. The adrenals were analysed in a randomised order by an observer blinded to the genotype. Two areas were examined in two slices from each adrenal and the mean values were calculated. Cryosections (10 μm) of frozen adrenals were stained with Oil Red O to visualise lipid distribution and examined by microscopy.
Quantification of mRNAs by real-time quantitative PCR
Adrenals (n=11–14/genotype), hypothalami and pituitaries (n=6/genotype) were homogenised using a QIAshredder column (Qiagen Ltd) and total RNA was extracted from snap-frozen tissue samples using the Qiagen RNeasy system, and 500 ng total RNA were reverse transcribed into cDNA with random primers using the QuantiTect DNase/reverse Transcription Kit. cDNA (equivalent to 1 ng total RNA) was incubated in triplicate with gene-specific primers and fluorescent probes (Table 1: Applied Biosystems or the Universal Probe Library, Roche Diagnostics) in 1× Roche LightCycler 480 Probes mastermix. PCR cycling and detection of fluorescent signal were carried out using a Roche LightCycler480. A standard curve was constructed for each set of primer probes using a serial dilution of cDNA pooled from all samples. The results were corrected for the mean of abundance of reference genes (Gapdh and Tbp in the hypothalamus, Rn18s, Tbp and Actb mRNAs for adrenal with Ppia in addition for pituitary); the respective means of the reference genes did not differ between groups.
Table 1.
Assay details for real-time PCR. ABI assay numbers: cyclophilin (Ppia), Mm02342429_g1; corticotrophin-releasing hormone receptor 1 (Crhr1), Mm00432670_m1; 11β-hydroxylase (Cyp11b1), Mm01204952_m1
| Forward primer | Reverse primer | UPL probe no. | |
|---|---|---|---|
| 18S ribosomal RNA (Rn18s) | ctcaacacgggaaacctcac | cgctccaccaactaagaacg | 77 |
| β-actin (Actb) | ctaaggccaaccgtgaaaag | accagaggcatacagggaca | 64 |
| 5α-reductase 1 (Srd5a1) | gggaaactggatacaaaataccc | ccacgagctccccaaaata | 41 |
| 5α-reductase 2 (Srd5a2) | cgcacattacttccacagga | cagaaagatcaccgctgataaa | 34 |
| 5β-reductase (Akr1d1) | gaaaagatagcagaagggaaggt | gggacatgctctgtattccataa | 79 |
| 11β-hydroxysteroid dehydrogenase 1 (Hsd11b1) | tctacaaatgaagagttcagaccag | gccccagtgacaatcacttt | 1 |
| Glucocorticoid receptor (Nr3c1) | gacgtgtggaagctgtaaagt | catttcttccagcacaaaggt | 56 |
| Arginine vasopressin (Avp) | gctgccaggaggagaactac | aaaaccgtcgtggcactc | 84 |
| TATA box-binding protein (Tbp) | gggagaatcatggaccagaa | gatgggaattccaggagtca | 97 |
| GAPDH | gggttcctataaatacggactgc | ccattttgtctacgggacga | 52 |
Quantification of mRNAs by in situ hybridisation
Cryosections (10 μm) of brains (n=8–9/group) were mounted onto electrostatic slides. Antisense and sense riboprobes for Nr3c1, Nr3c2 and Crh transcripts were prepared as described previously (Harris et al. 2001). Tissues were processed and hybridised, probes visualised by autoradiography and quantified using a microcomputer imaging system operated by Zeiss KS300 3.0 computer software. Nr3c1 and Nr3c2 transcripts were quantified in the dentate gyrus (DG), CA1, CA2, CA3 and CA4 regions of the hippocampus and Crh transcripts in the paraventricular nucleus of the hypothalamus PVN, by counting of the number of silver grains in each region by a blinded observer, reporting the mean counts from six randomly selected areas (radius=43 μm) minus background count.
Biochemical assays
Velocities of hepatic Akr1d1 and Hsd11b1 were quantified in tissue homogenates as reported previously (Livingstone et al. 2009). Plasma corticosterone was measured by an in-house RIA, without significant cross-reactivity with dexamethasone (Holmes et al. 1995), and ACTH by RIA (MP Biomedical, California, CA, USA).
To quantify tissue corticosterone, liver (approximately 300 mg) was homogenised (3 ml, 7:2 methanol:water) and enriched with 125 ng epi-cortisol as internal standard. The homogenates were shaken (15 min) and centrifuged (3200 g, 45 min at 4 °C), the supernatant was reduced to dryness under oxygen-free nitrogen (OFN, 60 °C) and then dissolved in 1:1 methanol:hexane (20 ml). The methanolic layer was reduced to dryness under OFN; the residue was dissolved in water (400 μl) and steroids were extracted with ethyl acetate (ten volumes). Organic extracts were dried under OFN; the residue was dissolved in 30% methanol (5 ml) and then applied to Megabond columns (2 g, C18, Varian, Oxford, UK) and steroids were eluted with methanol (5 ml). The eluate was dissolved in mobile phase (methanol:water 50:50 containing 0.1% formic acid) and steroids were analysed by liquid chromatography tandem mass spectrometry using a Surveyor pump and TSQ Quantum Discovery triple quadropole mass spectrometer (Thermo Electron, Hemel Hempstead, UK). Steroids were separated using a Kinetex PFP column (100×3 mm, 2.6 μm, 15 °C; Phenomenex, Macclesfield, UK) using gradient elution; initial conditions of methanol:water 50:50, each with 0.1% formic acid, were maintained for 1 min and then programmed to achieve 35:65 (1–2.5 min), 30:70 (2.5–6 min) and 10:90 (6–6.5 min). The mass spectrometer was operated in a positive electrospray ionisation mode with selected reactions monitoring (transition, collision energy, tube lens: corticosterone; m/z 347→121, 27 V, 103 V epi-cortisol m/z 363→121, 30 V, 95 V). The peak areas were integrated using Xcalibur software (Thermo Electron) and corticosterone was quantified as a proportion of epi-cortisol as internal standard against a calibration curve. The limit of detection was less than 1 ng corticosterone. Corticosterone concentrations are presented corrected for total tissue weight. For brain steroid measurement, half brains (sagitally sectioned) were homogenised in 1 ml ethyl acetate:ethanol (1:1 v/v), the homogenate was dripped into 10 ml ice-cold ethanol:acetic acid:water (95:3:2 v/v) and incubated at −20 °C overnight. The samples were then processed in the same way as for liver homogenates.
Statistical analysis
Data are mean±s.e.m. and were compared by Student's t-test or repeated measure ANOVA with Fisher's post hoc test as appropriate. Area under the curve was calculated using Kinetica software (Thermo Electron). P<0.05 was considered statistically significant.
Results
Srd5a1-KO mice have decreased clearance of corticosterone
Clearance of corticosterone was slower in Srd5a1-KO mice compared with WT mice (3.42±0.61 vs 30.15±11.75 ml/min; P<0.05), demonstrated by higher circulating corticosterone concentrations after both an acute bolus (Fig. 1A) and chronic infusion (Fig. 1B) of corticosterone in adrenalectomised mice. Residual corticosterone was detectable in adrenalectomised mice, but the levels were not different between genotypes. Slower clearance was also reflected in higher liver (Fig. 1C) and brain (Fig. 1D) corticosterone concentrations in Srd5a1-KO mice following chronic corticosterone infusion. The velocities of hepatic Hsd11b1 and Akr1d1 were not different between Srd5a1-KO and WT mice (11.7±0.8 vs 13.4±0.5 nmol/mg per h, 244±22 vs 299±32 pmol/mg per h, respectively), and this was also reflected in the abundances of the transcript for Hsd11b1 (0.86±0.16 vs 1.05±0.11), although mRNA encoding Akr1d1 was present at lower levels in Srd5a1-KO mice than in WT controls (0.54±0.03 vs 0.90±0.15; P=0.04).
Figure 1.
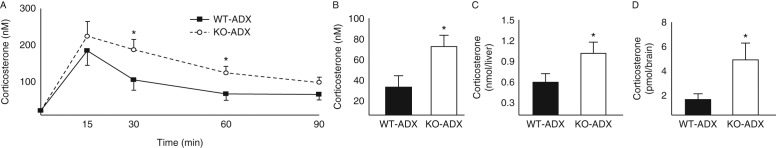
Clearance of corticosterone in Srd5a1-KO and WT mice. Plasma corticosterone concentrations following (A) corticosterone bolus injection and (B) chronic infusion were higher in adrenalectomised mice deficient in 5α-reductase 1 (KO, open circles/white bars) than in WT mice (WT, black squares/bars). Amounts of corticosterone in (C) liver and (D) brain were higher in KO than in WT mice following chronic infusion of corticosterone. *P<0.05 versus WT.
Resting plasma corticosterone levels are maintained in intact Srd5a1-KO mice
There were no differences between Srd5a1-KO and WT mice in basal (unstressed) circulating corticosterone at the diurnal nadir (Fig. 2A). Intact Srd5a1-KO mice adapted appropriately to corticosterone infused at sub-physiological replacement dose, maintaining the same total circulating corticosterone concentrations as WT mice (Fig. 2B). The ACTH level measured in trunk blood was not different between Srd5a1-KO and WT mice (208.4±49.4 vs 125.5±24.3 pg/ml; P=0.13, n=17–18).
Figure 2.
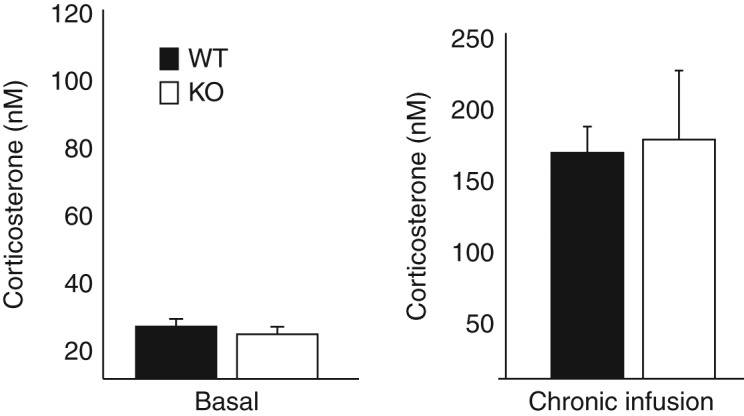
Plasma corticosterone in Srd5a1-KO and WT mice. Basal plasma corticosterone concentrations were the same in (A) unstressed mice of both genotypes and (B) after chronic infusion of corticosterone. n=6–8/group per treatment compared by Student's t-tests. WT (white bars); KO, Srd5a1-deficient mice (black bars).
Weights of the adrenal and thymus glands were not different between genotypes (Table 2). Gross morphometric examination of the adrenal glands did not reveal any differences (Fig. 3A and B). Neither the number of cells per unit area (Fig. 3C, D, E, F, G, H, and K), the distribution of lipid by Oil Red O staining (Fig. 3I and J), nor the abundance of adrenal mRNA encoding Cyp11b1 (0.46±0.05 vs 0.43±0.05) differed between genotypes.
Table 2.
Characteristics of Srd5a1-KO and WT mice. Data are mean±s.e.m. n=12–16/group
| WT | 5αR1-KO | |
|---|---|---|
| Body weight (g) | 26.3±0.85 | 27.0±0.76 |
| Adrenal weight (mg) | 2.2±0.08 | 2.1±0.09 |
| Adrenal weight/body weight (mg/g) | 0.17±0.01 | 0.16±0.01 |
| Thymus weight (mg) | 40.4±3.3 | 39.2±3.5 |
Srd5a1-KO, 5α-reductase 1 knockout.
Figure 3.

Morphological analysis of adrenal glands from WT and 5αR1-KO mice. Representative sections (5 μm) of adrenal glands stained with haematoxylin and eosin. Gross histological (10× magnification) differences between (A) WT mice and (B) those deficient in 5α-reductase 1 (KO) were not observed. Individual zones are shown at higher magnification (40×): (C and F) zona glomerulosa; (D and G) zona fasciculata and (E and H) zona reticularis in WT and KO mice respectively. Panels I (WT) and J (KO) show representative sections (5 μm) of frozen adrenal glands stained with Oil red O and photographed at 10× magnification; differences in lipid accumulation were not observed. Panel K shows the number of cells per zone of the adrenal gland in WT (black bars) and KO (white bars) mice. Significant changes in the number of cells in any of the adrenal zones were not observed between genotypes. M, adrenal medulla; ZG, zona glomerulosa; ZF, zona fasciculata; IZF, inner zona fasciculata; OZF, outer zona fasciculata; ZR, zona reticularis. n=6–8/group per treatment.
5αR1-KO mice exhibit ‘relative adrenal insufficiency’ during stimulation by stress or ACTH
Corticosterone levels were suppressed to a similar degree in response to a sub-maximal dose of dexamethasone (Fig. 4A). Following exogenous ACTH administration, the increase in plasma corticosterone concentrations was attenuated in Srd5a1-KO mice compared with WT (Fig. 4A). Circulating corticosterone levels were lower in Srd5a1-KO than in WT mice, in response to both minor stress (cage disturbance and handling; Fig. 4B) and after restraint stress (Fig. 4C; area under the curve for plasma corticosterone (10.3±2.2 vs 18.0±2.0 μM.min, P=0.003).
Figure 4.
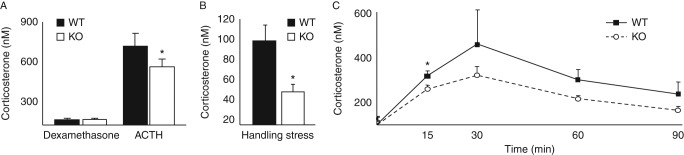
Dynamic responses of the hypothalamic–pituitary–adrenal axis to stimulation and suppression. (A) Circulating concentrations of corticosterone were not different between mice deficient in Srd5a1 (KO, open circles/white bars) and WT mice (WT, black squares/bars), following partial suppression of the hypothalamic–pituitary–adrenal axis with dexamethasone, but the corticosterone response to ACTH stimulation was attenuated in KO mice compared with WT (by two-way ANOVA, P<0.01 for effect of ACTH and P<0.05 for effect of genotype). (B) Circulating corticosterone levels were lower in KO mice following mild handling stress than in WT mice. (C) The increase in corticosterone in response to 15 min acute restraint stress (indicated by dark bar on graph) was attenuated in KO mice (open circles) compared with WT mice (black squares) (n=12–18/genotype at t=0 and 15 and n=6 at other time points; P<0.01 for change with time and P<0.05 for genotype effect by repeated measures ANOVA). Unless stated, n=6–8/group per treatment. *P<0.05 vs WT.
The molecular control of the HPA axis compensates for altered corticosterone clearance in Srd5a1-KO mice
Pituitary and brain transcript abundances are shown in Fig. 5. In Srd5a1-KO mice, glucocorticoid receptor (Nr3c1) transcripts were more abundant in the pituitary and hypothalamus but not in any regions of the hippocampus (Fig. 5A, C, and D). Mineralocorticoid receptor (Nr3c2) transcripts in the hippocampus were also unaffected by genotype (Fig. 5E). Corticotrophin-releasing hormone receptor 1 (Crh-r1) transcript abundance in pituitary was not different between genotypes (Fig. 5A); however, Crh mRNA in the PVN (Fig. 5B) and arginine–vasopressin (Avp; Fig. 5C) mRNAs in the hypothalamus were both lower in abundance in 5αR1-KO than in WT mice. Representative images of the in situ analysis are shown in Supplementary Figure 1, see section on supplementary data given at the end of this article.
Figure 5.

Transcript abundances of genes regulating the hypothalamic–pituitary–adrenal axis. (A) In pituitary, abundance of transcripts for glucocorticoid receptor (GR) but not corticotrophin releasing hormone receptor 1 (Crhr1) was higher in mice deficient in Srd5a1 (KO; black bars) compared with WT controls (WT; white bars). In the paraventricular nucleus of the hypothalamus, the transcripts of (B) Crh and (C) arginine–vasopressin (Avp) were lower in KO mice compared with WT, while Nr3c1 transcript was higher. In the CA1, CA2, CA3, CA4 regions or dentate gyrus (DG) of the hippocampus, differences were not observed between genotypes in abundances of transcripts of (D) Nr3c1 or (E) mineralocorticoid receptors (Nr3c2). Data are mean±s.e.m., HK, housekeeping genes; n=6–9/group. *P<0.05 vs WT.
Discussion
These data confirm that the enzyme Srd5a1 contributes in large part to glucocorticoid clearance in mice, and that its targeted disruption is sufficient to induce a phenotype analogous to ‘relative adrenal insufficiency’ in patients with impaired cortisol clearance. Although baseline plasma ACTH and corticosterone are normal, mice lacking Srd5a1 exhibit not only impaired corticosterone responses to ACTH but also impaired responses to mildly and moderately stressful stimuli. Moreover, this occurred in the absence of any demonstrable primary histological abnormality in the adrenal glands and was associated with alterations in neuroendocrine signalling pathways in the hypothalamus and pituitary that are most readily explained by enhanced negative feedback by corticosterone resulting from its impaired peripheral clearance and causing a compensatory downregulation of the HPA axis.
Srd5a1 is present in high abundance in murine liver, similar to the situation in humans, and therefore deficiency at this site predicts impaired peripheral metabolism, as observed in the experiment involving chronic infustion of corticosterone. However 5α-reductases are expressed also in brain (Lephart 1993a), specifically in the hypothalamus (Thigpen et al. 1993, Lund et al. 2006) (where Srd5a1 is more abundant than Srd5a2 (Lephart 1993b)) and to a lesser extent in pituitary (Lephart 1993b). Deficiency or inhibition of 5αR1 at these sites may attenuate local inactivation of glucocorticoids, and thereby enhance negative feedback suppression of the HPA axis independently of the altered peripheral clearance. Following chronic infusion of corticosterone, we did not find a disproportionate elevation in brain corticosterone levels in Srd5a1-KO mice; the elevation in brain corticosterone levels was of a similar magnitude to the higher plasma corticosterone levels in Srd5a1-KO mice. Furthermore, the observation that plasma ACTH in unstressed animals is not suppressed by 5αR1 deficiency indicates that the primary driver for altered HPA axis function lies in altered peripheral corticosterone clearance rather than failure of corticosterone inactivation in the brain.
The modulatory role of ligand concentrations on glucocorticoid and mineralocorticoid receptors (Nr3c1 and Nr3c2) at feedback sites within the HPA axis has been studied in other rodent models. In the extreme scenario of adrenalectomy, mice have dramatically higher ACTH levels, along with greater Nr3c1 abundance at feedback sites (Han et al. 2007), the receptor apparently auto-regulating in response to absence of ligand (Herman & Spencer 1998, Ing 2005). The converse is true with chronic stress associated with elevated glucocorticoids, suppressing GR levels in the PVN but not in the hippocampus (Makino et al. 1995). However, in models of more subtle modulation of ligand, e.g. in mice with increased peripheral clearance induced by Hsd11b1 deficiency, changes in Nr3c1 expression may not auto-regulate in the same manner. On a 129/MF1 genetic background, mice lacking Hsd11b1 develop a greater degree of adrenal hypertrophy (approximately 70%) (Harris et al. 2001) than on a C57BL/6 background (approximately 20%) (Carter et al. 2009). This appears to be attributable to C57BL/6 mice being better able than 129/MF1 mice to upregulate Nr3c1 at central feedback sites, contributing to resetting of the HPA axis (Carter et al. 2009). Specifically, in the mixed strain 129/MF1 Hsd11b1-deficient mice with more pronounced HPA abnormalities, exaggerated peripheral glucocorticoid clearance was accompanied by suppressed Nr3c1 expression in the PVN. Against this background, our findings are consistent with impaired peripheral glucocorticoid metabolism in Srd5a1-KO mice bred on a mixed genetic background (comprising C57Bl6/SvEv/129), with an upregulation of Nr3c1 transcript in central feedback sites (Mahendroo et al. 1997). Increased Nr3c1 action may be associated with downregulation of transcription of Crh (Malkoski & Dorin 1999) and Avp (Burke et al. 1997) mRNAs in the hypothalamus, together mediating a compensatory downregulation of stress-induced ACTH secretion. Similar downregulation of hypothalamic Crh transcripts has been demonstrated in vivo in mice with increased gene dosage of Nr3c1 (Reichardt et al. 2000). However, any change in Nr3c1-mediated signalling with manipulation of Srd5a1 appears too subtle to be detected by dexamethasone suppression testing in mice. Furthermore, basal circulating corticosterone levels were unaltered by disruption of Srd5a1, and intact mice adapted appropriately to the infusion of low physiological levels of glucocorticoid. Overall these data indicate that a centrally driven primary abnormality of the HPA axis causing relative adrenal insufficiency is unlikely and that the phenotype is most probably driven by impaired peripheral metabolic clearance of corticosterone.
Although our investigations focused on glucocorticoids, 5α-reductases also metabolise a wide range of other steroid hormones, some of which may have important effects on the CNS. Androgens can repress CRH (Bao et al. 2006), but lack of the most potent androgen 5α-dihydrotestosterone (DHT) would predict a more, rather than less, dynamic HPA axis with deficiency of Srd5a1. Reduction of testosterone to DHT by Srd5a1 is also thought to regulate the feedback responses of GNRH neurons to androgens (Tobyn & Canny 1998), which we did not investigate here. 5α-Reduced neurosteroids (e.g. allopregnanolone) have been implicated in the attenuation of behavioural responses to anxiety, acting via the GABA-A receptors to reduce neuronal excitability; this may impinge upon the HPA axis and attenuate stress responses, as is most apparent during late pregnancy (Russell & Brunton 2006).
These findings may be relevant to clinical practice. It is notable that in Srd5a1-KO mice corticosterone clearance was reduced by approximately 80% compared with that of WT mice; this was attributed to changes in 5α-reduction as the velocities of hepatic Akr1d1 and Hsd11b1 were unaltered (despite a modest reduction in Akr1d1 mRNA). Findings in Srd5a1-KO mice were therefore rather larger in magnitude than the approximately 50% reduction in cortisol clearance we demonstrated in critically ill patients during infusion of deuterated-cortisol (Boonen et al. 2013), especially considering that critically ill patients have ‘extra’ loss of cortisol-clearing capacity attributed to reduced activity of Akr1d1 and Hsd11b2. It may be that Srd5a1 contributes more to glucocorticoid clearance in mice than it does in humans, which is plausible as 5α-reductase type 1 is the only isozyme expressed in mouse liver (Mahendroo et al. 1996, 1997) whereas human liver contains both 5α-reductase type 1 and type 2 (Evans & Goa 2003).
These data indicate that there may be a risk of inadequate cortisol responses to stress in men taking 5α-reductase inhibitors for prostatic disease, particularly when the dual 5α-reductase type 1 plus type 2 inhibitor, dutasteride, is prescribed in preference to the selective type 2 inhibitor, finasteride. Previous investigations following inhibition of 5α-reductases in humans indicate that women, who are sometimes treated with finasteride for hirsutism, do exhibit changes in the HPA axis (Fruzzetti et al. 1994), at least for a short-term period, with decreased basal plasma cortisol and an impaired cortisol response to exogenous ACTH. However, studies in men have shown no change in basal cortisol (Rittmaster et al. 1994, Lewis et al. 1997, Uygur et al. 1998) or in response to high dose ACTH (Rittmaster et al. 1994), although a lower, potentially more discriminatory dose has not been tested. Administration of 5α-reductase inhibitors to men suppresseses daily total glucocorticoid production rates by approximately 20% (Upreti et al. 2014), but the proportion of cortisol excreted in urine as 5α-reduced metabolites is substantially higher in women than in men (Andrew et al. 1998), consistent with 5α-reductases making a greater contribution to clearance of cortisol. In women, this may be attributable to Srd5a2 rather than Srd5a1 in liver, because effects on the HPA axis consistent with altered cortisol clearance were observed with the selective type 2 inhibitor finasteride (Fruzzetti et al. 1994). Thus, women may be more susceptible than men to ‘relative adrenal insufficiency’ if given a 5α-reductase inhibitor.
In conclusion, our findings emphasise that peripheral steroid metabolism has potentially potent effects on the HPA axis. In mice with life-long deficiency of Srd5a1, adrenal responsiveness to stress is substantially impaired. This may have implications for individuals receiving 5α-reductase inhibitors chronically, and particularly for women in whom the 5α-reductase pathway of cortisol metabolism is thought to be of greater significance than in men. Our data confirm the hypothesis that ‘relative adrenal insufficiency’ can result from impaired glucocorticoid clearance, which appears to be relevant in many clinical scenarios, including hepatic failure and critical illness.
Supplementary data
This is linked to the online version of the paper at http://dx.doi.org/10.1530/JOE-13-0563.
Author contribution statement
The authors have made the following declarations about their contributions: D E W L designed and performed experiments, analysed and interpreted data and prepared the manuscript; E M D, C Y, L E C, J A M, M K and K A H, performed experiments and analysed and interpreted data; C J K, B R W, R A analysed and interpreted data and prepared the manuscript.
Acknowledgements
The authors express their gratefulness to Dr Mala Mahendroo for her advice and support. They thank Carolynn Cairns, Karen French, Rachel McDonnell, Sanjay Kothiya and Jill Harrison for excellent technical support, and the Genetic Screening and Intervention Technologies, University of Edinburgh for rederivation services.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the Wellcome Trust (072217/Z/03/Z), the British Heart Foundation (FS/08/065), Medical Research Council (MRC)-Hutchison Whampoa Dorothy Hodgkin (C Y) and Carnegie Trust and Samuel Leonard Travelling Fellowships (R A).
References
- Andrew R, Phillips DIW, Walker BR. Obesity and gender influence cortisol secretion and metabolism in man. Journal of Clinical Endocrinology and Metabolism. 1998;83:1806–1809. doi: 10.1210/jcem.83.5.4951. [DOI] [PubMed] [Google Scholar]
- Annane D, Sebille V, Charpentier C, Bollaert P-E, Francois B, Korach J-M, Capellier G, Chen Y, Azoulay E, Troche G, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. Journal of the American Medical Association. 2002;288:862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- Bao AM, Fischer DF, Wu YH, Balesar R, Unmehopa UA, Zhou JN, Swaab DF. A direct androgenic involvement in the expression of human corticotropin-releasing hormone. Molecular Psychiatry. 2006;11:567–576. doi: 10.1038/sj.mp.4001800. [DOI] [PubMed] [Google Scholar]
- Boonen E, Vervenne H, Meersseman P, Andrew R, Mortier L, Declercq PE, Vanwijngaeden Y-M, Spriet I, Wouters PJ, Vader Perre S, et al. Reduced cortisol metabolism during critical illness. New England Journal of Medicine. 2013;368:1477–1488. doi: 10.1056/NEJMoa1214969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke ZD, Ho MY, Morgan H, Smith M, Murphy D, Carter D. Repression of vasopressin gene expression by glucocorticoids in transgenic mice: evidence of a direct mechanism mediated by proximal 5′ flanking sequence. Neuroscience. 1997;78:1177–1185. doi: 10.1016/S0306-4522(96)00603-3. [DOI] [PubMed] [Google Scholar]
- Carter RN, Paterson JM, Tworowska U, Stenvers DJ, Mullins JJ, Seckl JR, Holmes MC. Hypothalamic–pituitary–adrenal axis abnormalities in response to deletion of 11β-HSD1 is strain-dependent. Journal of Neuroscience. 2009;21:879–887. doi: 10.1111/j.1365-2826.2009.01899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MS, Stewart PM. 11β-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus–pituitary–adrenal axis, metabolic syndrome, and inflammation. Journal of Clinical Endocrinology and Metabolism. 2009;94:4645–4654. doi: 10.1210/jc.2009-1412. [DOI] [PubMed] [Google Scholar]
- Draper N, Walker EA, Bujalska IJ, Tomlinson JW, Chalder SM, Arlt W, Lavery GG, Bedendo O, Ray DW, Laing I, et al. Mutations in the genes encoding 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nature Genetics. 2003;34:434–439. doi: 10.1038/ng1214. [DOI] [PubMed] [Google Scholar]
- Evans HC, Goa KL. Dutasteride. Drugs & Aging. 2003;20:905–916. doi: 10.2165/00002512-200320120-00005. [DOI] [PubMed] [Google Scholar]
- Fassnacht M, Schlenz N, Schneider SB, Wudy SA, Allolio B, Arlt W. Beyond adrenal and ovarian androgen generation: increased peripheral 5α-reductase activity in women with polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism. 2003;88:2760–2766. doi: 10.1210/jc.2002-021875. [DOI] [PubMed] [Google Scholar]
- Fernandez J, Escorell A, Zabalza M, Felipe V, Navasa M, Mas A, Lacy AM, Gines P, Arroyo V. Adrenal insufficiency in patients with cirrhosis and septic shock. Hepatology. 2006;44:1288–1295. doi: 10.1002/hep.21352. [DOI] [PubMed] [Google Scholar]
- Frey FJ. Impaired 11β-hydroxysteroid dehydrogenase contributes to renal sodium avidity in cirrhosis: hypothesis or fact? Hepatology. 2006;44:795–801. doi: 10.1002/hep.21381. [DOI] [PubMed] [Google Scholar]
- Fruzzetti F, de Lorenzo D, Parrini D, Ricci C. Effects of finasteride, a 5α-reductase inhibitor, on circulating androgens and gonadotropin secretion in hirsute women. Journal of Clinical Endocrinology and Metabolism. 1994;79:831–835. doi: 10.1210/jcem.79.3.8077369. [DOI] [PubMed] [Google Scholar]
- Han F, Ozawz H, Matsude K-I, Lu H, de Kloet ER, Kawata M. Changes in the expression of corticotrophin-releasing hormone, mineralocorticoid receptor and glucocorticoid receptor mRNAs in the hypothalamic paraventricular nucleus induced by fornix transection and adrenalectomy. Journal of Neuroendocrinology. 2007;19:229–238. doi: 10.1111/j.1365-2826.2006.01519.x. [DOI] [PubMed] [Google Scholar]
- Harris HJ, Kotelevtsev Y, Mullins JJ, Seckl JR, Holmes MC. Intracellular regeneration of glucocorticoids by 11β-hydroxysteroid dehydrogenase (11β-HSD)-1 plays a key role in regulation of the hypothalamic–pituitary–adrenal axis: analysis of 11β-HSD-1-deficient mice. Endocrinology. 2001;142:114–120. doi: 10.1210/endo.142.1.7887. [DOI] [PubMed] [Google Scholar]
- Harry R, Auzinger G, Wendon J. The clinical importance of adrenal insufficiency in acute hepatic dysfunction. Hepatology. 2002;36:395–402. doi: 10.1053/jhep.2002.34514. [DOI] [PubMed] [Google Scholar]
- Herman JP, Spencer R. Regulation of hippocampal glucocorticoid receptor gene transcription and protein expression in vivo. Journal of Neuroscience. 1998;18:7462–7473. doi: 10.1523/JNEUROSCI.18-18-07462.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MC, French KL, Seckl JR. Modulation of serotonin and corticosteroid receptor gene expression in the rat hippocampus with circadian rhythm and stress. Molecular Brain Research. 1995;28:186–192. doi: 10.1016/0169-328X(94)00207-U. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5α-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science. 1974;186:1213–1215. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- Ing NH. Steroid hormones regulate gene expression posttranscriptionally by altering the stabilities of messenger RNAs. Biology of Reproduction. 2005;72:1290–1296. doi: 10.1095/biolreprod.105.040014. [DOI] [PubMed] [Google Scholar]
- Jamieson A, Wallace AM, Walker BR, Andrew R, Fraser R, White PC, Connell JMC. Apparent cortisone reductase deficiency: a functional defect in 11β-hydroxysteroid dehydrogenase type 1. Journal of Clinical Endocrinology and Metabolism. 1999;84:3570–3574. doi: 10.1210/jcem.84.10.6031. [DOI] [PubMed] [Google Scholar]
- Kotelevtsev YV, Holmes MC, Burchell A, Houston PM, Scholl D, Jamieson PM, Best R, Brown RW, Edwards CRW, Seckl JR, et al. 11β-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid inducible responses and resist hyperglycaemia on obesity and stress. PNAS. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotelevtsev YV, Brown RW, Fleming S, Edwards CRW, Seckl JR, Mullins JJ. Hypertension in mice caused by inactivation of 11β-hydroxysteroid dehydrogenase type 2. Journal of Clinical Investigation. 1999;103:683–689. doi: 10.1172/JCI4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery GG, Walker EA, Draper N, Jeyasuria P, Marcos J, Shakleton CHL, Parker KL, White PC, Stewart PM. Hexose-6-phosphate dehydrogenase knock-out mice lack 11β-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. Journal of Biological Chemistry. 2006;10:6546–6551. doi: 10.1074/jbc.M512635200. [DOI] [PubMed] [Google Scholar]
- Lephart ED. Brain 5α-reductase: cellular, enzymatic, and molecular perspectives and implications for biological function. Molecular and Cellular Neuroscience. 1999a;4:473–484. doi: 10.1006/mcne.1993.1059. [DOI] [PubMed] [Google Scholar]
- Lephart ED. Pituitary and brain 5α-reductase messenger RNA levels in control, castrated and dihydrotestosterone treated rats. Molecular and Cellular Neurosciences. 1993b;4:526–531. doi: 10.1006/mcne.1993.1064. [DOI] [PubMed] [Google Scholar]
- Lewis JG, George PM, Elder PA. Plasma androsterone/epiandrosterone sulfates as markers of 5α-reductase activity: effect of finasteride in normal men. Steroids. 1997;62:632–635. doi: 10.1016/S0039-128X(97)00048-2. [DOI] [PubMed] [Google Scholar]
- Livingstone DEW, Grassick S, Currie GL, Walker BR, Andrew R. Dysregulation of glucocorticoid metabolism in murine obesity: comparable effects of leptin resistance and deficiency. Journal of Endocrinology. 2009;201:211–218. doi: 10.1677/JOE-09-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TD, Hinds LR, Handa RJ. The androgen 5α-dihydrotestosterone and its metabolite 5α-androstan-3β, 17β-diol inhibit the hypothalamo–pituitary–adrenal response to stress by acting through estrogen receptor β-expressing neurons in the hypothalamus. Journal of Neuroscience. 2006;26:1448–1456. doi: 10.1523/JNEUROSCI.3777-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahendroo MS, Cala KM, Russell DW. 5α-Reduced androgens play a key role in murine parturition. Molecular Endocrinology. 1996;10:380–392. doi: 10.1210/mend.10.4.8721983. [DOI] [PubMed] [Google Scholar]
- Mahendroo MS, Cala KM, Landrum CP, Russell DW. Fetal death in mice lacking 5α-reductase type 1 caused by estrogen excess. Molecular Endocrinology. 1997;11:917–927. doi: 10.1210/mend.11.7.9933. [DOI] [PubMed] [Google Scholar]
- Makino S, Smith MA, Gold PW. Increased expression of corticotropin-releasing hormone and vasopressin messenger ribonucleic acid (mRNA) in the hypothalamic paraventricular nucleus during repeated stress: association with reduction in glucocorticoid receptor mRNA levels. Endocrinology. 1995;136:3299–3309. doi: 10.1210/endo.136.8.7628364. [DOI] [PubMed] [Google Scholar]
- Malkoski SP, Dorin RI. Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Molecular Endocrinology. 1999;13:1629–1644. doi: 10.1210/mend.13.10.0351. doi:10nd.13.10.0351 [DOI] [PubMed] [Google Scholar]
- McGuire JS, Hollis VW, Tomkins GM. Some characteristics of the microsomal steroid reductases (5α) of rat liver. Journal of Biological Chemistry. 1960;235:3112–3117. [Google Scholar]
- McNeilly AD, MacFarlane DP, O'Flaherty EN, Livingstone DEW, MacKenzie SM, Mitic T, McConnell KM, Davies E, Reynolds RM, Thiesson HC, et al. Bile acids modulate glucocorticoid metabolism and the hypothalamic–pituitary–adrenal axis in obstructive jaundice. Journal of Hepatology. 2010;52:705–711. doi: 10.1016/j.jhep.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipov G, Palermo M, Shackleton CHL. Apparent cortisone reductase deficiency: a unique form of hypercortisolism. Journal of Clinical Endocrinology and Metabolism. 1996;81:3855–3860. doi: 10.1210/jcem.81.11.8923828. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Umland T, Bauer A, Kretz O, Schutz G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Molecular and Cellular Biology. 2000;20:9009–9017. doi: 10.1128/MCB.20.23.9009-9017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittmaster RS, Antonian L, New MI, Stoner E. Effect of finasteride on adrenal steroidogenesis in men. Journal of Andrology. 1994;15:298–301. doi: 10.1002/j.1939-4640.1994.tb00453.x. [DOI] [PubMed] [Google Scholar]
- Russell JA, Brunton PJ. Neuroactive steroids attenuate oxytocin stress responses in late pregnancy. Neuroscience. 2006;138:879–889. doi: 10.1016/j.neuroscience.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Stewart PM, Corrie JET, Shackleton CHL, Edwards CRW. Syndrome of apparent mineralocorticoid excess: a defect in the cortisol–cortisone shuttle. Journal of Clinical Investigation. 1988;82:340–349. doi: 10.1172/JCI113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PM, Shackleton CHL, Beastall GH, Edwards CRW. 5α-reductase activity in polycystic ovarian syndrome. Lancet. 1990;335:431–433. doi: 10.1016/0140-6736(90)90664-q. doi:10REF43=10.1172/JCI116665 [DOI] [PubMed] [Google Scholar]
- Taylor NF, Bartlett WA, Dawson DJ, Enoch BA. Cortisone reductase deficiency: evidence for a new inborn error in metabolism of adrenal steroids. Journal of Endocrinology. 1984;102(Suppl):90. doi: 10.1677/joe.0.102S00NP. [DOI] [Google Scholar]
- Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, Russell DW. Tissue distribution and ontogeny of steroid 5α-reductase isozyme expression. Journal of Clinical Investigation. 1993;92:903–910. doi: 10.1172/JCI116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobyn VA, Canny BJ. The regulation of gonadotropin-releasing hormone-induced calcium signals in male rat gonadotrophs by testosterone is mediated by dihydrotestosterone. Endocrinology. 1998;139:1038–1045. doi: 10.1210/endo.139.3.5796. [DOI] [PubMed] [Google Scholar]
- Tsai MH, Peng YS, Cheng YC, Liu NJ, Ho YP, Fang JT, Lien JM, Yang C, Chen P-C, Wu C-S. Adrenal insufficiency in patients with cirrhosis, severe sepsis and septic shock. Hepatology. 2006;43:673–681. doi: 10.1002/hep.21101. [DOI] [PubMed] [Google Scholar]
- Tsilchorozidou T, Honour JW, Conway GS. Altered cortisol metabolism in polycystic ovary syndrome: insulin enhances 5α-reduction but not the elevated adrenal steroid production rates. Journal of Clinical Endocrinology and Metabolism. 2003;88:5907–5913. doi: 10.1210/jc.2003-030240. [DOI] [PubMed] [Google Scholar]
- Upreti R, Hughes KA, Livingstone DEW, Gray CD, Minns FC, MacFarlane DP, Marshall I, Stewart LH, Walker BR, Andrew R. 5α-Reductase type 1 modulates insulin sensitivity in men. Journal of Clinical Endocrinology and Metabolism. 2014 doi: 10.1210/jc.2014-1395. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uygur MC, Arik AI, Altug U, Erol D. Effects of the 5α-reductase inhibitor finasteride on serum levels of gonadal, adrenal, and hypophyseal hormones and its clinical significance: a prospective clinical study. Steroids. 1998;63:208–213. doi: 10.1016/S0039-128X(98)00005-1. [DOI] [PubMed] [Google Scholar]