

Abstract
The inverted emulsion method is used to prepare giant liposomes by pushing water-in-oil droplets through the oil/water interface into an aqueous medium. Due to the high encapsulation efficiency of proteins under physiological conditions and the simplicity of the protocol, it has been widely used to prepare various cell models. However, the lamellarity of liposomes prepared by this method has not been evaluated quantitatively. Here, we prepared liposomes that were partially stained with a fluorescent dye, and analyzed their fluorescence intensity under an epifluorescence microscope. The fluorescence intensities of the membranes of individual liposomes were plotted against their diameter. The plots showed discrete distributions, which were classified into several groups. The group with the lowest fluorescence intensity was determined to be unilamellar by monitoring the exchangeability of the inner and the outer solutions of the liposomes in the presence of the pore-forming toxin α-hemolysin. Increasing the lipid concentration dissolved in oil increased the number of liposomes ∼100 times. However, almost all the liposomes were unilamellar even at saturating lipid concentrations. We also investigated the effects of lipid composition and liposome content, such as highly concentrated actin filaments and Xenopus egg extracts, on the lamellarity of the liposomes. Remarkably, over 90% of the liposomes were unilamellar under all conditions examined. We conclude that the inverted emulsion method can be used to efficiently prepare giant unilamellar liposomes and is useful for designing cell models.
Introduction
A liposome is a vesicle surrounded by a lipid bilayer, in which lipid molecules face their hydrophobic parts (tail regions) toward the interior of the bilayer and expose their hydrophilic parts (head regions) toward the surrounding aqueous medium (1). Because a lipid bilayer is the basic structure of the cell membrane (1), micrometer-sized giant liposomes serve as useful cell models (2–23).
Various methods have been proposed to prepare giant liposomes (24). The hydration method, which is also called the swelling method, is one of the most popular methods for vesicle preparation. In this method, lipids dissolved in organic solvent are spread on a substrate. As the solvent evaporates, a multilayered lipid bilayer film is formed. The dry lipid film is then filled with buffer solution. The lipid layers spontaneously peel from the film, enclose the solution, and transform into liposomes. This method is simple; thus, it is widely used (3,5–7). However, the efficiency of vesicle formation is sensitive to lipid composition and the hydration buffer. In particular, encapsulation of highly concentrated proteins under physiological buffer conditions is difficult (24), a feature that is crucial for the construction of cell models. Moreover, the lamellarity, which is defined as the number of bilayers surrounding the liposome, varies among these liposomes, resulting in contamination of the preparation by numerous multilamellar vesicles (24,25). Because cell membranes are unilamellar, this hydration method is not suited for developing cell models.
The electroformation method, in which an AC electric field is applied across a lipid film and the surrounding medium during the hydration process, is also extensively used (8–11); however, this method has the same limitations as the hydration method (24). The electric field applied during the vesicle formation process might also alter the activity of the enclosed enzymes (24). Although several improvements for these two methods have been reported (26–30), to our knowledge, their limitations in lipid composition, low encapsulation efficiency of highly concentrated proteins under physiological buffer conditions, and contamination with multilamellar liposomes have not been simultaneously solved. To overcome these problems, new methods such as the pulsed jetting (31–34) and transient membrane ejection (35) methods were proposed. However, these methods require expertise in microfluidics.
In contrast, the inverted emulsion method (36), which is also called the transfer method, involves spontaneous transformation of water-in-oil droplets into liposomes by passage through the water/oil interface (Fig. 1 A). This method enables encapsulation of cell extracts (15,21) or highly concentrated proteins (at close to intracellular concentrations) (19) into giant liposomes under various buffer conditions and lipid compositions in a simple and a unified way. In addition, membrane proteins are successfully reconstituted without loss of biochemical function (15,20,37). Because of the stepwise process of vesicle formation, the lipid composition of the inner and outer leaflets of the liposomes can be controlled independently (38–40). The orientation of a membrane protein can also be controlled (37). Due to the high encapsulation efficiency under physiological conditions and the simplicity of the protocol, the inverted emulsion method has been extensively used to prepare various cell models, including a self-replication model of protocells (14), model cells containing protein expression systems (15), and cytoskeletal networks such as an actin cortex (16,17), actomyosin gel (19,20), microtubule asters (21), and bacterial cytoskeletal filaments (22,23). Although it is assumed that the inverted emulsion method can be used to prepare unilamellar liposomes with high efficiency, and despite its broad use, the lamellarity of liposomes prepared by this method has not been investigated quantitatively.
Figure 1.
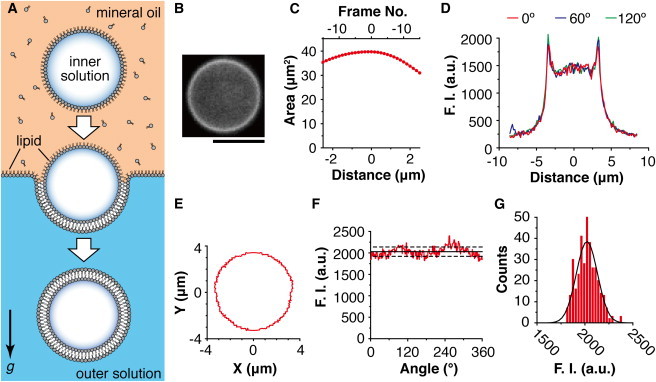
Measurement of the fluorescence intensity of individual liposomes prepared by the inverted emulsion method. (A) A schematic illustration of the inverted emulsion method. Water-in-oil droplets were transformed into liposomes by passage through the oil/water interface by centrifugation. (B) Epifluorescence image of a liposome across the equatorial plane. Scale bar: 5 μm. Data shown in (C–G) were obtained from this liposome. (C) Relationship between the cross-sectional areas of the liposome and the frame number scanned on the Z axis. The frame that had the maximum area was determined to be the equatorial plane, and is shown as frame number 0. (D) Fluorescence intensity (F.I.) profiles of a cross section of the liposome. The position at 0 μm indicates the center of mass. Measurements were performed radially from the center of the mass, every one degree. The intensity profiles at 0°, 60°, and 120° were shown as the examples. (E–G) The membrane fluorescence intensity profile of the liposome, obtained from (D). (E) The peak fluorescence intensity positions in the X-Y plane. The position at (0,0) indicates the center of mass. (F) The peak fluorescence intensity profile against the angle of the radial axis. The average value (black solid line) and the SD (black dashed line) are also shown. (G) Histogram of the membrane fluorescence intensities shown in (F) and the Gaussian fit (black solid line).
Here, we established a method to determine the lamellarity of individual liposomes under an optical microscope, and report the results of a quantitative analysis of liposomes prepared by the inverted emulsion method. The analysis method, which is rather simple, is based on a previous study by Akashi et al. (25). By analyzing the fluorescence intensity of the liposomal membranes, and by fitting a theoretical curve to the experimental data, the lamellarity of individual liposomes was quantitatively determined. We confirmed that the lamellarity was nearly independent of the lipid concentration dissolved in oil, lipid composition, and vesicle contents. Indeed, >90% of the liposomes were unilamellar under all preparation conditions tested.
Materials and Methods
Buffers
A50 buffer (50 mM HEPES-KOH pH 7.6, 50 mM KCl, 5 mM MgCl2, 1 mM EGTA) was used for all experiments except the actin polymerization assay. In the actin polymerization assay, G buffer (2 mM Tris-HCl pH 8.0, 0.05 mM CaCl2, 2 mM NaN3, 0.1 mM ATP, 0.5 mM 2-mercaptoethanol) was used to encapsulate G-actin in liposomes. For the inner solution of the liposomes, 150 mM sucrose and 350 mM glucose were added, and for the outer solution, 500 mM glucose was added so that the osmolality on each side of the membrane would be equivalent. To encapsulate Xenopus egg extracts, XB buffer (10 mM HEPES-KOH pH 7.7, 100 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, 50 mM sucrose) was used for the outer solution.
Proteins and lipids
Actin was purified from rabbit skeletal muscle (41), and labeled with Alexa Fluor 488 C5-maleimide (A-10254; Molecular Probes, Eugene, OR). Tetramethylrhodamine-bovine serum albumin (TMR-BSA) was prepared by labeling BSA (A3059; Sigma-Aldrich, St. Louis, MO) with tetramethylrhodamine-5-maleimide (T-6027; Molecular Probes). α-Hemolysin from Staphylococcus aureus was purchased from Toxin Technology (HT101; Sarasota, FL), dissolved in A50 buffer at a concentration of 5 mg/mL, snap frozen in liquid nitrogen, and stored at −84°C. Xenopus laevis egg extracts were prepared as described previously (42). L-α-phosphatidylcholine from chicken egg yolk (egg PC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phosphatidylglycerol (DOPG), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N lissamine rhodamine B sulfonyl ammonium salt (rhodamine PE) were purchased from Avanti Polar Lipids (Alabaster, AL). Oregon Green 488 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Oregon Green PE) was purchased from Invitrogen (Carlsbad, CA). Cholesterol was purchased from Wako (Osaka, Japan).
Liposome preparation
Giant liposomes were prepared according to the method of Noireaux and Libchaber (15) with slight modifications. Lipids were first dissolved in chloroform (034-02603; Wako), which was dehydrated with molecular sieves 4A, and put into 1.5 mL glass test tubes. These test tubes were placed inside a vacuum desiccator and evacuated over night to remove chloroform from the lipids completely. The lipid dry film was stored in a vacuum desiccator at room temperature under dark conditions and used within 2 weeks. The lipid dry film was mixed with 1 mL of mineral oil (23306-84; Nacalai Tesque, Kyoto, Japan), heated to 80°C and dissolved in the oil by vortexing. Heating and vortexing processes were repeated several times until the film disappeared completely. Next, the lipid-oil mixture was sonicated for 90 min in a bath sonicator at 60°C and a power of 60 W (AS12GTU; As One, Osaka, Japan). Immediately after the sonication, the lipid-oil mixture was vortexed, cooled to room temperature, and kept overnight under dark conditions to disperse lipid molecules completely. This lipid-oil mixture was stored at room temperature under dark conditions and used within 2 days.
Just before use, 200 μL of lipid-oil mixture was put into 1.5 mL sample tube and cooled on ice for >15 min. Next, 20 μL of inner-solution was added to the lipid-oil mixture and immediately emulsified by vortexing (power max; Vortex-Genie 2; Scientific Industries, Bohemia, NY) for 30 s. The sample tube was incubated on ice for 5 min to stabilize the emulsion by spontaneous alignment of lipid molecules at the inner-buffer/oil interface. Subsequently, 150 μL of the emulsion was gently placed on 1 mL of outer-solution in a 1.5 mL glass test tube and incubated on ice for 5 min to allow for the assembly of lipid monolayer at the oil/outer-solution interface. Finally, the glass test tube was centrifuged (12,000 × g, 30 min, 4°C) to push the emulsion droplets through the interface. After centrifugation, the oil layer and 500 μL of buffer solution were gentry drawn off from the top of the tube by using a suction aspirator. Finally, 500 μL of liposome solution remained at the bottom. For the encapsulation of Xenopus egg extracts, 10 μL of the extracts were mixed with 200 μL of lipid-oil mixture by gentle vortexing (power minimum; Vortex-Genie 2; Scientific Industries) for 10 s to prepare extracts-in-oil droplets. After 5 min incubation on ice, 150 μL of this emulsion was placed on 1 mL of XB buffer and incubated 5 min on ice. The sample was then gently centrifuged (100 × g, 15 min, 4°C) to transform droplets into liposomes.
Microscopy
Epifluorescence and bright-field images of liposomes were acquired with a custom-built inverted microscope equipped with a ×100 objective (PlanApo NA 1.40 oil; Olympus, Tokyo, Japan), an electron multiplying charge coupled device (EM-CCD) camera (iXon3 DU-897E-CS0-#BV; Andor Technology, Belfast, UK), a 100 W mercury lamp for the epifluorescence light source (U-ULS100HG; Olympus), and a custom-built XYZ motorized sample stage driven by stepping motors (SGSP-13ACT-BO; Sigma-Koki, Tokyo, Japan). The entire microscopy system was controlled by LabVIEW (National Instruments, Austin, TX).
Observation, image acquisition, and analysis
A flow chamber was assembled by placing two double-sided tapes (thickness 0.3 mm; PBW-20; 3M, Maplewood, MN) onto a silicone-coated coverslip (36 × 24 mm2, custom-ordered; Matsunami, Osaka, Japan) with another coverslip (18 × 18 mm2; Matsunami) on top. The inner distance between the two tapes was ∼8 mm, and the inner volume of the chamber was ∼40 μL. First, the flow chamber was coated with 60 μL of Pluronic-F127 (10 mg/ml dissolved in A50 buffer) for >1 min to prohibit nonspecific adsorption of liposomes. The flow chamber was then washed with 600 μL of the outer solution of liposomes. Just before use, liposome solution was mixed gently by pipetting to disperse liposomes homogeneously and 150 μL was perfused into the flow chamber. The flow chamber was sealed with Valap and kept horizontal for >30 min to settle down liposomes on the bottom coverslip. Liposomes that exhibited circular periphery and with the diameter larger than 2 μm were selected, and the whole fluorescence image of the liposomes was scanned from bottom to top at a constant speed of 5 μm/s, at 25 fps, and at 25 ± 1°C. The lamellarities of individual liposomes were analyzed with LabVIEW (National Instruments).
Actin polymerization assay
Liposomes containing 10 μM monomeric actin (10% Alexa Fluor 488-labeled), 10% (w/v) polyethylene glycol (20 k), 150 mM sucrose, and 350 mM glucose in G buffer were prepared. For the outer solution, 500 mM glucose in G buffer was used. The following was added to the outer solution: 7/20 volume of 142.9 mM HEPES-KOH pH 7.6, 142.9 mM KCl, 14.3 mM MgCl2, 2.9 mM EGTA, and 500 mM glucose containing 0.7 mg/mL α-hemolysin. The external medium contained final concentrations of 50 mM HEPES-KOH pH 7.6, 50 mM KCl, 5 mM MgCl2, 1 mM EGTA, 500 mM glucose, and 0.25 mg/mL α-hemolysin. This mixture was immediately perfused into a flow chamber, sealed with Valap to prevent flow, and then image acquisition was started.
Liposome counting
To count the number of liposomes, an inverted microscope (DIAPHOT 300; Nikon, Tokyo, Japan) equipped with ×60 objective (PlanApo NA 1.40 oil Ph4), CCD camera (CCD-300-RCX; Dage-MTI, Michigan, IN) and custom-built XYZ motorized sample stages driven by stepping motors (SGSP20-20 and SGSP60-10ZF; Sigma-Koki) was used. Phase-contrast images of liposomes were automatically scanned along the Z axis from –25 to 100 μm (0 μm corresponds to the position of the bottom coverslip) at the constant speed of 20 μm/s, 30 fps, 50 different fields of view, and at 24 ± 2°C. Clear spherical liposomes with the diameter larger than 2 μm were then automatically detected and the number was counted by LabVIEW (National Instruments).
Results
Lamellarity analysis of individual liposomes by epifluorescence microscopy
Giant liposomes were prepared by the inverted emulsion method (Fig. 1 A). For the phospholipids, we chose natural PC purified from chicken egg yolk, because PC is a major plasma membrane component (43); thus, egg PC is one of the phospholipids most commonly used to build cell models (7,10,12,16–21). For the encapsulating solution and external medium, we used biologically relevant buffers (see Materials and Methods). To measure the lamellarity of the liposomes, 0.1% (mol/mol) rhodamine PE was added to the egg PC.
Observations were performed under an epifluorescence microscope. Most of the liposomes exhibited a spherical shape (Fig. 1 B). The encapsulated solution had slightly higher densities than the external solution. Therefore, most of the liposomes settled down on the bottom coverslip within 30 min. After waiting for longer than 30 min, a spherical liposome on the bottom coverslip was scanned along the Z axis from the bottom to the top with a 0.2 μm interval per frame. As a result, an image of an entire liposome was obtained in every 0.2 μm slice.
We next analyzed the images. First, the background camera noise was subtracted from the cross-sectional images. Next, the unevenness of the illumination was corrected by taking an image of the homogeneous fluorescent solution (0.1 μM rhodamine B) and dividing the liposome image by this image. Cross-sectional areas of the liposome were then calculated in every frame (Fig. 1 C), and the frame that had the maximum area was selected for further analysis. In the selected frame (Fig. 1 B), the center of mass in the liposome was determined. The lines were drawn radially from the center of mass every one degree. The fluorescence intensity profiles were measured along these lines (Fig. 1 D), and the peak intensity value and its XY position were obtained for every angle (Fig. 1, E and F). If there were no lipid aggregates on the membrane and inside the liposome, the fluorescence intensity along the circumference of the liposome was almost homogeneous (Fig. 1 F), and this histogram was fitted well by a Gaussian function (Fig. 1 G). In contrast, if lipid aggregates were attached to the membrane (Fig. S1 A in the Supporting Material) or inside the liposome, the fluorescence intensity profile along the circumference of the liposome had a sharp peak(s) (Fig. S1 B). Such liposomes were not used in the lamellarity analysis described below.
We analyzed >100 liposomes in the same observation chamber, and the mean fluorescence intensities of individual liposomes along the circumference were plotted against their diameter. The plots showed discrete distributions and appeared to be classified into several groups (Fig. 2 A. See also Fig. S5 D, which had more data points in the higher fluorescence intensity groups).
Figure 2.
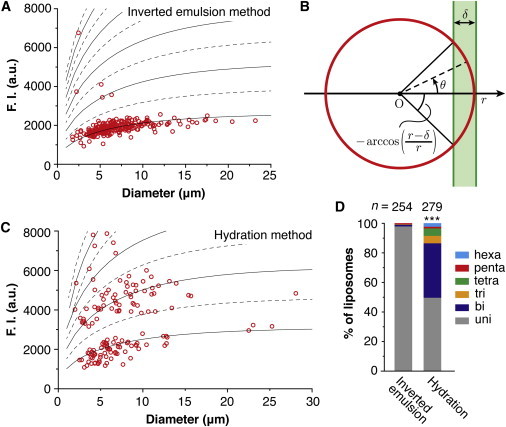
Lamellarity analysis and a comparison of the lamellarity of liposomes prepared by the inverted emulsion and hydration methods. (A) The membrane fluorescence intensities of individual liposomes prepared by the inverted emulsion method were plotted against their diameter. The lowest black solid line indicates the theoretical curve I(r) fitted to the lowest fluorescence intensity group of the plots. The upper four black solid lines are two-, three-, four-, and fivefold larger than the I(r). The black dashed lines are 1.5-, 2.5-, 3.5-, 4.5-, and 5.5-fold larger than the I(r). We determined that the liposomes with intensities that are under the lowest dashed line were unilamellar, whereas the liposomes that are between the lowest and second lowest dashed lines were bilamellar, and so on. Egg PC (1 mM) containing 0.1% (mol/mol) rhodamine PE dissolved in oil was used to prepare the liposomes. (B) A schematic illustration of the side view of a liposome under an epifluorescence microscope. The red circle represents the membrane of the liposome with radius r. The horizontal axis represents the focal plane. O: center of mass. δ: pixel size of the image. The fluorescence signal of the membrane out of the focal plane, illustrated as the green area, may contribute to the apparent fluorescence intensity measured in the focal plane. (C) Plots of the membrane fluorescence intensities of individual liposomes prepared by the hydration method. Egg PC containing 0.1% (mol/mol) rhodamine PE was used to prepare the liposomes. (D) Lamellarity distributions of the liposomes prepared by the inverted emulsion and hydration methods. Several independent experiments were performed and the total counts were plotted. The proportion of unilamellar liposomes prepared by the two methods were compared to each other with the χ2test and Fisher’s exact test. ∗∗∗p < 0.001.
The group that had the lowest fluorescence intensity was expected to be unilamellar. In the plots in this group, the intensity monotonically tended to increase with increasing vesicle diameter, which apparently plateaued at large diameters. This diameter dependency can be explained by the geometry of the observation setup (Fig. 2 B). Because we used an epifluorescence microscope, the focal depth was deep, and it was expected to be on the order of submicrons to several microns. Therefore, the contribution of the fluorescence signal from out of the focal plane had to be considered. If we assume that the magnitude of fluorescence contribution from out of the focal plane can be approximated by a Gaussian function , where z is the distance from the focal plane and σ is the width of the distribution corresponding to the focal depth, the apparent fluorescence intensity of the unilamellar membrane at the equatorial plane can be expressed as
where r is the radius of the liposome, I0 is the fluorescence intensity of the unilamellar membrane per unit surface area, and δ is the pixel size of the observed images.
We fitted I(r) to the lowest fluorescence intensity group via I0 and σ. The pixel size, δ, was fixed at 0.143 μm, which was measured with an objective micrometer. As a result, I(r) fitted well to the experimental data (Fig. 2 A, the bottommost black solid line). From the fitting, we obtained a reasonable value for the depth of fluorescence leakage: σ = 0.913 μm. The values for δ and σ should be dependent on the microscopy system, but independent of the sample to be measured. Therefore, we used the same values for all the experiments described below.
We next drew lines that were two-, three-, four-, and fivefold larger than I(r) (Fig. 2 A, the upper four black solid lines). Those lines overlapped with some of the data points that were separated from the lowest fluorescence intensity group, implying that those liposomes were multilamellar. To quantify the lamellarity distribution, we adopted the 1.5-, 2.5-, 3.5-, 4.5-, and 5.5-fold I(r) lines as the criteria (black dashed lines in Fig. 2 A), and defined the data points below the 1.5-fold line as unilamellar, those between the 1.5- and 2.5-fold lines as bilamellar, those between the 2.5- and 3.5-fold lines as trilamellar, and so on. Indeed, 98.0% of the liposomes were determined to be unilamellar (Fig. 2 D, Table S1).
We confirmed that the amount of rhodamine PE added with the egg PC hardly affected lamellarity (Fig. S2 B, Table S1). Additionally, replacement of rhodamine PE with Oregon Green PE did not significantly change the lamellarity (Fig. S2, A and B; Table S1). These results suggested that our lamellarity analysis method is robust against the type and amount of fluorescently labeled phospholipid used.
Confirmation of unilamellarity by α-hemolysin treatment
To confirm that the liposomes in the lowest fluorescence intensity group were unilamellar, the exchangeability of solutions through the liposomal membrane was examined by using the pore-forming toxin α-hemolysin (15). The pore size of α-hemolysin is ∼2.8 nm; therefore, small molecules such as ions can pass through the pore but proteins cannot (44). The stem domain of α-hemolysin that forms the transmembrane pore is ∼5.2 nm in length (44), which is approximately equal to the thickness of the lipid bilayer (45). Therefore, α-hemolysin can only make a pore in a unilamellar membrane, i.e., buffer exchange will be observed in unilamellar liposomes but not in multilamellar liposomes.
Exchangeability of solutions was determined by monitoring the actin polymerization reaction inside the liposomes. We prepared liposomes in which actin monomers were encapsulated, and then a high salt buffer containing α-hemolysin was added to the external medium. The osmotic pressure of this buffer was balanced with the inner solution of the liposomes. If the liposome is unilamellar, α-hemolysin will penetrate the membrane and make a transmembrane pore, inducing buffer exchange between the inside and outside of the liposome. The increase in the salt concentration inside the liposomes will initiate actin polymerization. To easily identify the polymerized actin filaments, polyethylene glycol (PEG) was also encapsulated inside the liposomes to induce actin filament bundling by depletion force (46).
Time-lapse microscopy showed that actin bundles began to appear an average of 30 min after the addition of α-hemolysin (Fig. 3 A; Movie S1), and the bundle formation was terminated ∼60 min after α-hemolysin was added (Fig. 3 B). After 120 min, the fluorescence intensities of the membranes of individual liposomes were measured, and a percentage of liposomes containing actin bundles in the lowest intensity group was compared to that in the higher intensity group (Fig. 3 C). In the lowest intensity group, 94.3% of liposomes contained actin bundles. In contrast, only 8.7% of liposomes in the higher intensity group contained actin bundles. We also confirmed that the addition of actin polymerization buffer without α-hemolysin to the liposomes did not induce actin bundle formation in both groups. These results indicate that buffer exchange after α-hemolysin treatment occurred in most liposomes in the lowest intensity group, but rarely occurred in the liposomes in the higher intensity group. It is worth noting that the percentage of liposomes that exchanged the solution could be higher than the percentage in which actin bundles were observed, because there is a possibility that the polymerized actin did not form visible bundles in some liposomes. Therefore, we conclude that the liposomes with the lowest fluorescence intensity were unilamellar, and thus over 90% of the liposomes prepared by the inverted emulsion method were unilamellar.
Figure 3.
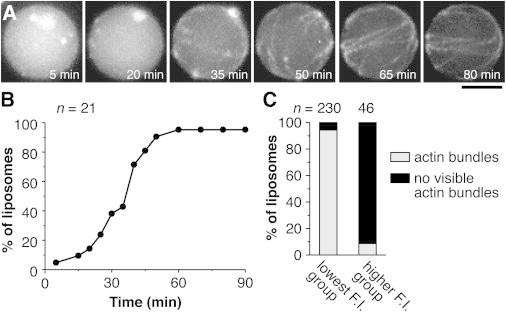
Actin polymerization inside liposomes induced by α-hemolysin treatment. (A) Time-lapse image series of the actin-encapsulated liposome, showing spontaneous polymerization and bundling of actin filaments. α-Hemolysin was added at 0 min. Actin was partially labeled with Alexa Fluor 488. Egg PC containing 0.1% (mol/mol) rhodamine PE was used to prepare the liposomes. Scale bar: 5 μm. (B) The percentage of liposomes in which actin bundles were assembled over time. (C) The percentages of actin bundle-assembled liposomes in the lowest and higher intensity groups were compared. We performed several independent experiments, and the total counts were plotted. We looked through many flow chambers to obtain a sufficient number of liposomes in the higher intensity group.
We found that actin bundles were formed in several multilamellar liposomes, presumably because these liposomes had portions that were unilamellar (Fig. S3), and α-hemolysin made pores in these portions. In addition, we tried to confirm the lamellarity by using the resonance transfer quencher QSY7, because it was reported that this quencher only quenched the fluorescence of the outer leaflet of a membrane exposed to solution containing the quencher (40). However, this quencher did not work as expected under our experimental conditions (Fig. S4).
Comparing the lamellarity of liposomes prepared by the inverted emulsion and hydration methods
We compared the lamellarity of liposomes prepared by the hydration method to that of those prepared by the inverted emulsion method. Although the plots of the fluorescence intensities of liposomes prepared by the hydration method showed a discrete distribution (Fig. 2 C), similar to those prepared by the inverted emulsion method (Fig. 2 A), the hydration method plots had many data points in the range of the higher intensity groups, indicating that there were many multilamellar liposomes. We quantified the lamellarity of these liposomes in the same way as the inverted emulsion method. The percentage of unilamellar liposomes prepared by the inverted emulsion and hydration methods were 98.0% and 49.8%, respectively, showing a significant difference (Fig. 2 D, Table S1).
High lipid concentrations promoted liposome formation but did not alter lamellarity
The effects of the concentration of lipid dissolved in oil were examined. On increasing the lipid concentration from 0.1 to 10 mM, the number of liposomes increased ∼100 times, and apparently saturated at 5 mM (Fig. 4 A). However, 92.1% of liposomes remained unilamellar even at 10 mM (Fig. 4 B; Fig. S5, A–D; Table S1). The size distribution of the liposomes was not significantly changed by differences in lipid concentration (Fig. S5 E). We conclude that it is easy to increase the liposome production efficiency when using the inverted emulsion method by increasing the lipid concentration without changing lamellarity or vesicle size.
Figure 4.
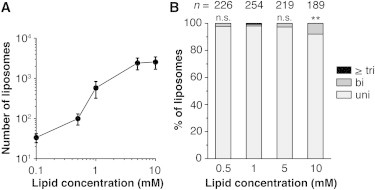
Effect of the lipid concentration dissolved in mineral oil on liposome lamellarity. In all experiments, egg PC containing 0.1% (mol/mol) rhodamine PE was used. (A) Average number of liposomes found in the observation chamber per 50 fields of view (0.031 mm2 × 50 = 1.55 mm2) was plotted against the lipid concentration dissolved in oil. Clear spherical-shaped liposomes with a diameter larger than 2 μm were counted. We performed at least three independent experiments, and averaged these results for each lipid concentration. Error bars indicate the SD. (B) Lamellarity distributions of the liposomes prepared at different lipid concentrations. The percentages of unilamellar liposomes in preparations containing 0.5, 5, and 10 mM lipid were compared with that prepared with 1 mM egg PC by the χ2test and Fisher’s exact test. n.s.: not significant (p > 0.05), ∗∗p < 0.01.
Lamellarity was independent of lipid composition and vesicle contents
We replaced a portion of the egg PC with DOPE, DOPG, or cholesterol, and performed the lamellarity analysis experiments. Phosphatidylethanolamine (PE) has a smaller head than phosphatidylcholine (PC). Therefore, it stabilizes the negative curvature of lipid bilayers and localizes at the outer leaflet of cleavage furrows in animal cells (47). Phosphatidylglycerol (PG) has a negative charge on its head, and it binds cations (48). Cholesterol changes the fluidity of the membrane by intercalating into the phospholipids (49). Because these lipids change the physical properties of lipid bilayers in membranes, we expected that they might affect the lamellarity of liposomes. However, over 95% of the liposomes were unilamellar under all conditions examined, and there were no significant differences among them (Fig. 5; Fig. S6; Table S1), suggesting that lamellarity was independent of lipid composition.
Figure 5.
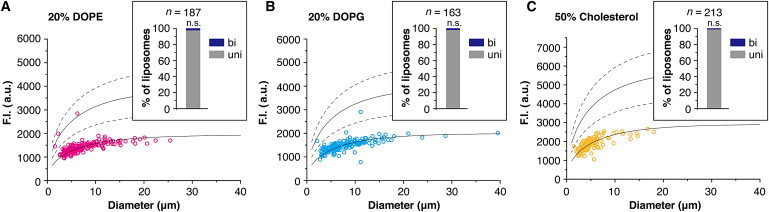
Effect of lipid composition on lamellarity. Lamellarity distributions of liposomes prepared with (A) egg PC and 20% (mol/mol) DOPE, (B) egg PC and 20% (mol/mol) DOPG, and (C) egg PC and 50% (mol/mol) cholesterol. In all experiments, the total lipid concentration in oil was fixed at 1 mM and 0.1% (mol/mol) rhodamine PE was added. We performed several independent experiments, and the data from all experiments are shown in the inset bar graphs. The percentages of unilamellar liposomes obtained from preparation using the different lipid compositions were compared to that obtained with 1 mM egg PC by the χ2test and Fisher’s exact test. In all conditions, the percentages did not differ significantly (n.s.: p > 0.05). We also confirmed that lamellarity of liposomes prepared with egg PC containing 5% or 10% DOPE, 5% or 10% DOPG, and 10% cholesterol did not differ significantly (Fig. S6).
To develop cell models, it is important to investigate whether lamellarity is independent of vesicle content. Here, we encapsulated highly concentrated proteins, specifically, 50 μM F-actin or 25 mg/mL BSA inside liposomes made of egg PC, and determined their lamellarity. Indeed, >98% of these liposomes were unilamellar (Fig. 6, A and B; Table S1), implying that the shape (globular or filamentous) and type of encapsulating protein did not affect lamellarity. Moreover, even when Xenopus egg extracts were encapsulated, 96.3% of the liposomes remained unilamellar (Fig. 6 C; Table S1).
Figure 6.
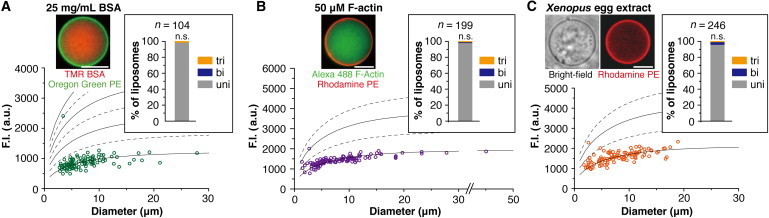
Effect of encapsulation of highly concentrated proteins. Lamellarity distributions and cross-sectional images of liposomes containing (A) 25 mg/mL TMR-labeled BSA, (B) 50 μM Alexa Fluor 488-labeled F-actin, or (C) Xenopus egg extracts. For the lipids, 1 mM egg PC containing 0.1% (mol/mol) Oregon Green PE (for BSA), 1 mM egg PC containing 0.1% (mol/mol) rhodamine PE (for F-actin), or 5 mM egg PC containing 50% (mol/mol) cholesterol and 0.1% (mol/mol) rhodamine PE (for Xenopus egg extract) dissolved in oil were used. We performed several independent experiments and all the data are shown in the inset bar graphs. The percentages of unilamellar liposomes prepared under each condition were compared to those prepared with 1 mM egg PC containing no vesicle contents by the χ2test and Fisher’s exact test. In all conditions, the percentages of unilamellar liposomes did not differ significantly (n.s.: p > 0.05). Scale bars: 5 μm.
Discussion
We showed that the inverted emulsion method generated giant unilamellar liposomes in remarkably high efficiency. One weak aspect of this method is the possibility of oil contamination inside the lipid bilayer (24). Although it has been reported that such contamination was less than the detection limit (36), the level of contamination depends on the type of oil used and the vesicle formation protocol. Therefore, further investigation is required to quantify this possibility. Recently, it was reported that the dynamic response of the membrane of liposomes prepared by the inverted emulsion method differed from that of liposomes prepared by the electroformation method, which might be due to low levels of oil contamination inside the lipid bilayer. However, interestingly, the viscoelastic properties of liposomes prepared by the inverted emulsion method were more similar to those of cellular membrane than that of liposomes prepared by the electroformation method (50). In this sense, the inverted emulsion method is well suited to modeling the physical properties of cell membranes.
Our findings showed that a small fraction of the liposomes prepared by the inverted emulsion method will be multilamellar liposomes. The assembly mechanisms of these liposomes remain unknown. Although several possible mechanisms can be proposed (Fig. S7), direct observation of the liposome formation process will be useful for clarifying these mechanisms (51–53). Furthermore, it is important to reduce the multilamellar liposome contamination to infinitesimally low levels.
Contamination with even a small number of multilamellar liposomes indicates that, in some cases, one has to measure the lamellarity of all liposomes to be used to validate the experimental results. If the proportion of fluorescently labeled lipids dissolved in oil is fixed constant, in principle, the fluorescence intensity of the unilamellar membrane per unit surface area I0 should be unique among different samples. Thus, the lamellarity of a single liposome could be determined from I(r) without analyzing several tens of liposomes in the same observation chamber and fitting I(r) via I0 to the plots every time, as we demonstrated here. However, it was experimentally difficult to obtain exactly the same I0 due to handling error in the preparation of lipid-oil mixtures. To overcome this problem, we confirmed that the bulk fluorescence intensity of the lipid-oil mixture Ib, which was measured by a fluorescence spectrophotometer, and the fitting parameter I0 were linearly correlated when the fluorescent lipid concentration was sufficiently low (Fig. S8). Therefore, if Ib has been measured in advance, the lamellarity of individual liposomes could be quantitatively evaluated by using I(r) and the linear relationship between I0 and Ib, without performing the cluster analysis. Recently, a new method to measure lamellarity by using differential interference contrast microscopy was proposed (54). Although this method is not straightforward, it does not require cluster analysis. Therefore, this method might be a good alternative, depending on the situation.
Conclusions
We established a method to measure the lamellarity of individual liposomes under an epifluorescence microscope. By using this method, we quantitatively verified that the inverted emulsion method efficiently generated giant unilamellar liposomes. Remarkably, lamellarity was highly robust against the lipid concentration dissolved in oil, lipid composition, and vesicle contents, including highly concentrated proteins and cell extracts. On average, 97.2% of liposomes were unilamellar under all the conditions we examined. All of these features are indispensable for liposome applications, especially designing cell models.
Recent technological developments in microfluidics have allowed us to assemble uniformly sized droplets (55) that can be transformed into uniformly sized liposomes through the oil/water interface formed inside a microfluidic device (56). By integrating on-chip sorting systems with our image-based lamellarity evaluation method, it will be possible to automatically collect uniformly sized unilamellar giant liposomes. Combination of the inverted emulsion method with fully automated sophisticated microfluidics might offer a powerful technique for mass production of desired cell model systems.
Acknowledgments
The authors thank K. Kinosita Jr. for helpful discussions, H. Eguchi, H. Kubota, K. Sato, and S. Ishii for actin preparation, and J. Takagi, K. Suzuki, and M. Tanabe for preparation of Xenopus egg extracts.
This work was supported in part by Grants-in-Aid for Young Scientists (B) and Scientific Research on Innovative Areas (M.M.) and Grants-in-Aid for Specially Promoted Research and Scientific Research (S) (S.I.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Masataka Chiba and Makito Miyazaki contributed equally to this work.
Supporting Material
Lamellarity distributions of liposomes at various lipid concentrations, compositions, and vesicle inclusions are listed. In addition, the proportion of unilamellar liposomes at each condition is compared with that of 1 mM egg PC containing 0.1% (mol/mol) rhodamine PE by Z-test and Fisher’s exact test, and the p values obtained are listed.
The time interval between the frames is 5 min. Every frame shows the maximum projection image. High salt buffer containing α-hemolysin was added to the external solution at 0 min. Actin: 10 μM. Scale bar: 5 μm.
References
- 1.Alberts B., Johnson A., Walter P. 5th ed. Garland Science; New York: 2008. Molecular Biology of the Cell. [Google Scholar]
- 2.Liu A.P., Fletcher D.A. Biology under construction: in vitro reconstitution of cellular function. Nat. Rev. Mol. Cell Biol. 2009;10:644–650. doi: 10.1038/nrm2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hotani H., Miyamoto H. Dynamic features of microtubules as visualized by dark-field microscopy. Adv. Biophys. 1990;26:135–156. doi: 10.1016/0065-227x(90)90010-q. [DOI] [PubMed] [Google Scholar]
- 4.Fygenson D.K., Marko J.F., Libchaber A. Mechanics of microtubule-based membrane extension. Phys. Rev. Lett. 1997;79:4497–4500. [Google Scholar]
- 5.Miyata H., Hotani H. Morphological changes in liposomes caused by polymerization of encapsulated actin and spontaneous formation of actin bundles. Proc. Natl. Acad. Sci. USA. 1992;89:11547–11551. doi: 10.1073/pnas.89.23.11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyata H., Nishiyama S., Kinosita K., Jr. Protrusive growth from giant liposomes driven by actin polymerization. Proc. Natl. Acad. Sci. USA. 1999;96:2048–2053. doi: 10.1073/pnas.96.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honda M., Takiguchi K., Hotani H. Morphogenesis of liposomes encapsulating actin depends on the type of actin-crosslinking. J. Mol. Biol. 1999;287:293–300. doi: 10.1006/jmbi.1999.2592. [DOI] [PubMed] [Google Scholar]
- 8.Limozin L., Sackmann E. Polymorphism of cross-linked actin networks in giant vesicles. Phys. Rev. Lett. 2002;89:168103. doi: 10.1103/PhysRevLett.89.168103. [DOI] [PubMed] [Google Scholar]
- 9.Limozin L., Bärmann M., Sackmann E. On the organization of self-assembled actin networks in giant vesicles. Eur. Phys. J. E. 2003;10:319–330. doi: 10.1140/epje/i2002-10118-9. [DOI] [PubMed] [Google Scholar]
- 10.Liu A.P., Richmond D.L., Fletcher D.A. Membrane-induced bundling of actin filaments. Nat. Phys. 2008;4:789–793. doi: 10.1038/nphys1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stachowiak J.C., Schmid E.M., Hayden C.C. Membrane bending by protein-protein crowding. Nat. Cell Biol. 2012;14:944–949. doi: 10.1038/ncb2561. [DOI] [PubMed] [Google Scholar]
- 12.Carvalho K., Tsai F.-C., Sykes C. Cell-sized liposomes reveal how actomyosin cortical tension drives shape change. Proc. Natl. Acad. Sci. USA. 2013;110:16456–16461. doi: 10.1073/pnas.1221524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka-Takiguchi Y., Kinoshita M., Takiguchi K. Septin-mediated uniform bracing of phospholipid membranes. Curr. Biol. 2009;19:140–145. doi: 10.1016/j.cub.2008.12.030. [DOI] [PubMed] [Google Scholar]
- 14.Terasawa H., Nishimura K., Yomo T. Coupling of the fusion and budding of giant phospholipid vesicles containing macromolecules. Proc. Natl. Acad. Sci. USA. 2012;109:5942–5947. doi: 10.1073/pnas.1120327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noireaux V., Libchaber A. A vesicle bioreactor as a step toward an artificial cell assembly. Proc. Natl. Acad. Sci. USA. 2004;101:17669–17674. doi: 10.1073/pnas.0408236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pontani L.-L., van der Gucht J., Sykes C. Reconstitution of an actin cortex inside a liposome. Biophys. J. 2009;96:192–198. doi: 10.1016/j.bpj.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murrell M., Pontani L.-L., Sykes C. Spreading dynamics of biomimetic actin cortices. Biophys. J. 2011;100:1400–1409. doi: 10.1016/j.bpj.2011.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murrell M.P., Voituriez R., Gardel M.L. Liposome adhesion generates traction stress. Nat. Phys. 2014;10:163–169. [Google Scholar]
- 19.Takiguchi K., Yamada A., Yoshikawa K. Entrapping desired amounts of actin filaments and molecular motor proteins in giant liposomes. Langmuir. 2008;24:11323–11326. doi: 10.1021/la802031n. [DOI] [PubMed] [Google Scholar]
- 20.Takiguchi K., Negishi M., Yoshikawa K. Transformation of actoHMM assembly confined in cell-sized liposome. Langmuir. 2011;27:11528–11535. doi: 10.1021/la2016287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinot M., Chesnel F., Gueroui Z. Effects of confinement on the self-organization of microtubules and motors. Curr. Biol. 2009;19:954–960. doi: 10.1016/j.cub.2009.04.027. [DOI] [PubMed] [Google Scholar]
- 22.Maeda Y.T., Nakadai T., Libchaber A. Assembly of MreB filaments on liposome membranes: a synthetic biology approach. ACS Synth. Biol. 2012;1:53–59. doi: 10.1021/sb200003v. [DOI] [PubMed] [Google Scholar]
- 23.Osawa M., Erickson H.P. Liposome division by a simple bacterial division machinery. Proc. Natl. Acad. Sci. USA. 2013;110:11000–11004. doi: 10.1073/pnas.1222254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Swaay D., deMello A. Microfluidic methods for forming liposomes. Lab Chip. 2013;13:752–767. doi: 10.1039/c2lc41121k. [DOI] [PubMed] [Google Scholar]
- 25.Akashi K., Miyata H., Kinosita K., Jr. Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys. J. 1996;71:3242–3250. doi: 10.1016/S0006-3495(96)79517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita Y., Oka M., Yamazaki M. A new method for the preparation of giant liposomes in high salt concentrations and growth of protein microcrystals in them. Biochim. Biophys. Acta. 2002;1561:129–134. doi: 10.1016/s0005-2736(02)00338-3. [DOI] [PubMed] [Google Scholar]
- 27.Montes L.R., Alonso A., Bagatolli L.A. Giant unilamellar vesicles electroformed from native membranes and organic lipid mixtures under physiological conditions. Biophys. J. 2007;93:3548–3554. doi: 10.1529/biophysj.107.116228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pott T., Bouvrais H., Méléard P. Giant unilamellar vesicle formation under physiologically relevant conditions. Chem. Phys. Lipids. 2008;154:115–119. doi: 10.1016/j.chemphyslip.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Tsai F.-C., Stuhrmann B., Koenderink G.H. Encapsulation of active cytoskeletal protein networks in cell-sized liposomes. Langmuir. 2011;27:10061–10071. doi: 10.1021/la201604z. [DOI] [PubMed] [Google Scholar]
- 30.Weinberger A., Tsai F.-C., Marques C. Gel-assisted formation of giant unilamellar vesicles. Biophys. J. 2013;105:154–164. doi: 10.1016/j.bpj.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funakoshi K., Suzuki H., Takeuchi S. Formation of giant lipid vesicle like compartments from a planar lipid membrane by a pulsed jet flow. J. Am. Chem. Soc. 2007;129:12608–12609. doi: 10.1021/ja074029f. [DOI] [PubMed] [Google Scholar]
- 32.Stachowiak J.C., Richmond D.L., Fletcher D.A. Unilamellar vesicle formation and encapsulation by microfluidic jetting. Proc. Natl. Acad. Sci. USA. 2008;105:4697–4702. doi: 10.1073/pnas.0710875105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stachowiak J.C., Richmond D.L., Fletcher D.A. Inkjet formation of unilamellar lipid vesicles for cell-like encapsulation. Lab Chip. 2009;9:2003–2009. doi: 10.1039/b904984c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richmond D.L., Schmid E.M., Fletcher D.A. Forming giant vesicles with controlled membrane composition, asymmetry, and contents. Proc. Natl. Acad. Sci. USA. 2011;108:9431–9436. doi: 10.1073/pnas.1016410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ota S., Yoshizawa S., Takeuchi S. Microfluidic formation of monodisperse, cell-sized, and unilamellar vesicles. Angew. Chem. Int. Ed. Engl. 2009;48:6533–6537. doi: 10.1002/anie.200902182. [DOI] [PubMed] [Google Scholar]
- 36.Pautot S., Frisken B.J., Weitz D.A. Production of unilamellar vesicles using an inverted emulsion. Langmuir. 2003;19:2870–2879. [Google Scholar]
- 37.Yanagisawa M., Iwamoto M., Oiki S. Oriented reconstitution of a membrane protein in a giant unilamellar vesicle: experimental verification with the potassium channel KcsA. J. Am. Chem. Soc. 2011;133:11774–11779. doi: 10.1021/ja2040859. [DOI] [PubMed] [Google Scholar]
- 38.Pautot S., Frisken B.J., Weitz D.A. Engineering asymmetric vesicles. Proc. Natl. Acad. Sci. USA. 2003;100:10718–10721. doi: 10.1073/pnas.1931005100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamada T., Miura Y., Takagi M. Construction of asymmetric cell-sized lipid vesicles from lipid-coated water-in-oil microdroplets. J. Phys. Chem. B. 2008;112:14678–14681. doi: 10.1021/jp807784j. [DOI] [PubMed] [Google Scholar]
- 40.Hu P.C., Li S., Malmstadt N. Microfluidic fabrication of asymmetric giant lipid vesicles. ACS Appl. Mater. Interfaces. 2011;3:1434–1440. doi: 10.1021/am101191d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki N., Miyata H., Kinosita K., Jr. Preparation of bead-tailed actin filaments: estimation of the torque produced by the sliding force in an in vitro motility assay. Biophys. J. 1996;70:401–408. doi: 10.1016/S0006-3495(96)79583-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai A., Murray A., Walczak C.E. The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods Cell Biol. 1999;61:385–412. doi: 10.1016/s0091-679x(08)61991-3. [DOI] [PubMed] [Google Scholar]
- 43.van Meer G., Voelker D.R., Feigenson G.W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song L., Hobaugh M.R., Gouaux J.E. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 45.Robertson J.D. Membrane structure. J. Cell Biol. 1981;91:189s–204s. doi: 10.1083/jcb.91.3.189s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asakura S., Oosawa F. On interaction between two bodies immersed in a solution of macromolecules. J. Chem. Phys. 1954;22:1255–1256. [Google Scholar]
- 47.Emoto K., Kobayashi T., Umeda M. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc. Natl. Acad. Sci. USA. 1996;93:12867–12872. doi: 10.1073/pnas.93.23.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macdonald P.M., Seelig J. Calcium binding to mixed phosphatidylglycerol-phosphatidylcholine bilayers as studied by deuterium nuclear magnetic resonance. Biochemistry. 1987;26:1231–1240. doi: 10.1021/bi00379a005. [DOI] [PubMed] [Google Scholar]
- 49.Ohvo-Rekilä H., Ramstedt B., Slotte J.P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 2002;41:66–97. doi: 10.1016/s0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- 50.Campillo C., Sens P., Sykes C. Unexpected membrane dynamics unveiled by membrane nanotube extrusion. Biophys. J. 2013;104:1248–1256. doi: 10.1016/j.bpj.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hase M., Yamada A., Yoshikawa K. Transport of a cell-sized phospholipid micro-container across water/oil interface. Chem. Phys. Lett. 2006;426:441–444. [Google Scholar]
- 52.Yamada A., Yamanaka T., Baigl D. Spontaneous transfer of phospholipid-coated oil-in-oil and water-in-oil micro-droplets through an oil/water interface. Langmuir. 2006;22:9824–9828. doi: 10.1021/la062221+. [DOI] [PubMed] [Google Scholar]
- 53.Ito H., Yamanaka T., Yoshikawa K. Dynamical formation of lipid bilayer vesicles from lipid-coated droplets across a planar monolayer at an oil/water interface. Soft Matter. 2013;9:9539–9547. doi: 10.1039/c3sm51766g. [DOI] [PubMed] [Google Scholar]
- 54.McPhee C.I., Zoriniants G., Borri P. Measuring the lamellarity of giant lipid vesicles with differential interference contrast microscopy. Biophys. J. 2013;105:1414–1420. doi: 10.1016/j.bpj.2013.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jimenez A.M., Roché M., Gueroui Z. Towards high throughput production of artificial egg oocytes using microfluidics. Lab Chip. 2011;11:429–434. doi: 10.1039/c0lc00046a. [DOI] [PubMed] [Google Scholar]
- 56.Matosevic S., Paegel B.M. Stepwise synthesis of giant unilamellar vesicles on a microfluidic assembly line. J. Am. Chem. Soc. 2011;133:2798–2800. doi: 10.1021/ja109137s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lamellarity distributions of liposomes at various lipid concentrations, compositions, and vesicle inclusions are listed. In addition, the proportion of unilamellar liposomes at each condition is compared with that of 1 mM egg PC containing 0.1% (mol/mol) rhodamine PE by Z-test and Fisher’s exact test, and the p values obtained are listed.
The time interval between the frames is 5 min. Every frame shows the maximum projection image. High salt buffer containing α-hemolysin was added to the external solution at 0 min. Actin: 10 μM. Scale bar: 5 μm.